

Background: Sur1-NCCa-ATP channels implicated in acute CNS injury are hypothesized to be formed by co-association of Sur1 and a nonselective cation channel.
Results: Sur1 and Trpm4 form heteromers that exhibit pharmacological properties of Sur1 and biophysical properties of Trpm4.
Conclusion: Sur1 and Trpm4 co-assemble to form the unique Sur1-Trpm4 channel.
Significance: Identification of Sur1-Trpm4 channels has broad implications in acute CNS injuries.
Keywords: ABC Transporter, Brain, Ion Channels, Membrane Biophysics, Multifunctional Protein, Neurobiology, Neuroprotection, Sur1, Trpm4
Abstract
The sulfonylurea receptor 1 (Sur1)-NCCa-ATP channel plays a central role in necrotic cell death in central nervous system (CNS) injury, including ischemic stroke, and traumatic brain and spinal cord injury. Here, we show that Sur1-NCCa-ATP channels are formed by co-assembly of Sur1 and transient receptor potential melastatin 4 (Trpm4). Co-expression of Sur1 and Trpm4 yielded Sur1-Trpm4 heteromers, as shown in experiments with Förster resonance energy transfer (FRET) and co-immunoprecipitation. Co-expression of Sur1 and Trpm4 also yielded functional Sur1-Trpm4 channels with biophysical properties of Trpm4 and pharmacological properties of Sur1. Co-assembly with Sur1 doubled the affinity of Trpm4 for calmodulin and doubled its sensitivity to intracellular calcium. Experiments with FRET and co-immunoprecipitation showed de novo appearance of Sur1-Trpm4 heteromers after spinal cord injury in rats. Our findings depart from the long-held view of an exclusive association between Sur1 and KATP channels and reveal an unexpected molecular partnership with far-ranging implications for CNS injury.
Introduction
Transient receptor potential melastatin 4 (Trpm4)2 is a member of a large superfamily of membrane proteins that includes 28 nonselective cation channels, most of which conduct both monovalent and divalent cations, including Ca2+ (1–3). Trpm4 is exceptional in that it is one of only two ion channels known in the mammalian genome that conduct monovalent cations exclusively and nonselectively (4). Because it is activated by intracellular Ca2+, and because its activation causes cell depolarization that reduces the inward electrical driving force for Ca2+, activation of Trpm4 provides negative feedback that decreases Ca2+ influx (4, 5).
Sulfonylurea receptor 1 (Sur1) is among the most thoroughly studied proteins involved in ion channel formation (6–9). Sur1 is a member of the large superfamily of ATP-binding cassette proteins, most of which hydrolyze ATP to transport solutes across biological membranes. Among ATP-binding cassette proteins, Sur1 is atypical in that it performs no known transport function, but undergoes heteromultimeric assembly with the ATP-sensitive potassium selective channel, Kir6.2, to form KATP channels. Sur1 is the target of sulfonylurea drugs used for decades to treat diabetes mellitus type 2.
Emerging evidence indicates that Sur1 is transcriptionally up-regulated de novo in several types of injury involving the central nervous system (CNS), including cerebral ischemia, traumatic brain injury, subarachnoid hemorrhage, germinal matrix hemorrhage, and spinal cord injury (10). Surprisingly, in these conditions, up-regulation of Sur1 is not accompanied by up-regulation of Kir6.2, but is associated instead with up-regulation of a novel, nonselective cation (NC) channel that is regulated by intracellular Ca2+ and ATP, as well as by Sur1: the so-called Sur1-regulated NCCa-ATP channel. The role of Sur1 in regulating this channel is now well established, but the molecular identity of the pore-forming subunit has not been determined.
Here, we show that the direct co-association of Sur1 with Trpm4 gives rise to a novel ion channel complex: the Sur1-Trpm4 channel. The identification of Sur1-Trpm4 channels has broad implications in multiple types of acute CNS injuries.
EXPERIMENTAL PROCEDURES
Molecular Biology
The recombinant proteins used in this study are listed in Table 1. To construct expression plasmids for Citrine (Ci)- and Cerulean (Ce)-fused proteins, cDNA sequences of Citrine or Cerulean were amplified by PCR and inserted into pECE-FLAG-Sur1, pMyc-Trpm4, pMyc-Kir6.2, and pMyc-Kir2.1 at the N or C terminus of each protein. Two alanine molecules were inserted between the individual full-length proteins and the fluorescent proteins to give steric flexibility. To construct Myc epitope-fused expression plasmids of mouse Kir6.2, mouse Kir2.1, mouse Trpm4, and human Hif1α, each cDNA sequence was cloned into an expression vector, pCMV-Tag3C (Stratagene, Grand Island, NY). To construct an expression plasmid encoding a fusion protein of Sur1-Trpm4, the cDNA sequence of Trpm4 was amplified and cloned into pcDNA-His6-Sur1 at the C-terminal end of His6-Sur1. An 8-amino acid-long glycine linker, GGGSGGGA, was used to connect the two proteins to provide flexibility between their interacting domains. To make a bicistronic expression vector, the resulting cDNA sequence encoding the Sur1-Trpm4 fusion protein was cloned into pEF1α-IRES-AcGFP1 (Clontech). To construct an expression vector of Sur1 with the hygromycin B-resistant gene (pHygroB-Sur1), the cDNA sequence of the hygromycin B-resistant gene was amplified by PCR and inserted into the pcDNA-His6-Sur1. All plasmids constructed by PCR amplification were verified by sequencing prior to transfection. Transfections were performed using Lipofectamine 2000 (Invitrogen).
TABLE 1.
Recombinant proteins used in this study
| Plasmida | Protein | Host vector |
|---|---|---|
| pSur1-Ce | FLAG-Sur1-Cerulean | pECE |
| pCi-Trpm4 | Citrine-Myc-Trpm4 | pCMV-Tag3C |
| pTrpm4-Ci | Myc-Trpm4-Citrine | pCMV-Tag3C |
| pCi-Kir6.2 | Citrine-Myc-Kir6.2 | pCMV-Tag3C |
| pCi-Kir2.1 | Citrine-Myc-Kir2.1 | pCMV-Tag3C |
| pcDNA-His6-Sur1b | His6-Sur1 | pcDNA3.1 |
| pECE-FLAG-Sur1c | FLAG-Sur1 | pECE |
| pMyc-Trpm4 | Myc-Trpm4 | pCMV-Tag3C |
| pMyc-Kir6.2 | Myc-Kir6.2 | pCMV-Tag3C |
| pMyc-Hif1α | Myc-Hif1α | pCMV-Tag3C |
| pTrpm4-EGFPd | Trpm4-EGFP | pEGFP-N1 |
| pIRES-Sur1-Trpm4 | His6-Sur1-Trpm4 | pEF1α-IRES-AcGFP1 |
| pHygroB-Sur1 | His6-Sur1 | pcDNA-His6-Sur1 |
a Fusion to the N terminus of Sur1 or Trpm4 is indicated by listing the adduct first; fusion to the C terminus of Sur1 or Trpm4 is indicated by listing the adduct second.
c Gift of Dr. Show-Ling Shyng, Oregon Health and Science University, Portland, OR.
d See Gerzanich et al. (28).
Cell Culture, Development of Stable Cell Line, and Transfection
COS-7 and HEK-293 cells were maintained in Dulbecco's modified Eagle's medium with 4.5 and 1.0 g/liter glucose (Invitrogen), respectively. Rat insulinoma RIN-m5F cells (ATCC, Manassas, VA) were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium. All culture media were supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin.
To develop stable cell lines that express constitutively high level of Sur1, HEK-293 cells were transfected with the pHygroB-Sur1 plasmid, and colonies were selected from a culture medium containing 200 μg/ml hygromycin B. Transfections were performed using Lipofectamine 2000 (Invitrogen). Expression of Sur1 from the selected cell lines was confirmed by immunolabeling and immunoblot (see Fig. 8).
FIGURE 8.

Co-expression with Sur1 increases the sensitivity of Trpm4 to Ca2+. A, immunolabeling for Sur1 in His6-Sur1-overexpressing HEK-293 (Sur1-HEK-293) cells, showing prominent expression in all cells (right panel) as compared with nontransfected HEK-293 cells (left panel). The nuclei were labeled with 4′-6-diamidino-2-phenylindole (DAPI; blue). B, immunoblots for Sur1, Trpm4, and CaM from HEK-293 cells or Sur1-HEK-293 cells transiently transfected to express Myc-Trpm4. Note that the abundance of calmodulin associated with Myc-Trpm4 (CaM Co-IP) is greater in cells co-expressing Sur1 and Trpm4 (lanes 2 and 4; replicate experiments) as compared with cells expressing Trpm4 alone (lanes 1 and 3). Sur1 was immunoblotted using total lysate, and was detected using anti-Sur1-a antibody. Myc-Trpm4 was immunoisolated using anti-Myc antibody, and immunoisolated proteins were detected using anti-Trpm4-a antibody and anti-CaM antibody. Bar graph: densitometric analysis showing the mean (±S.E.) abundance of calmodulin co-immunoprecipitated with Trpm4, normalized to the total amount of Trpm4, in the absence and presence of Sur1 co-expression; n = 6; **, p < 0.01. C, single channel recordings of inside-out patches from HEK-293 cells or Sur1-HEK-293 cells transiently transfected to express Ci-Myc-Trpm4; patches were pulled in a bath solution containing 1 μm Ca2+ to record base-line channel activity, after which the bath solution was changed to one containing 2 mm Ca2+ to maximally activate all channels. The patches were studied using the voltage clamp protocol depicted. Bar graph, mean (±S.E.) open channel probability of Trpm4, observed at +100 mV and at −100 mV, in 1 μm Ca2+ relative to that in 2 mm Ca2+, in cells expressing Trpm4 alone or co-expressing Sur1 and Trpm4. Charge carrier, Cs+; **, p < 0.01.
Rat Model of Spinal Cord Injury
This study was approved by the Institutional Animal Care and Use Committee of the University of Maryland. A spinal cord injury with hemicervical cord contusion at the seventh cervical vertebra (C7) was performed in rats as described (13). Spinal cords were harvested at 6 h after injury for experiments with co-immunoprecipitation and at 24 h after injury for experiments with Förster resonance energy transfer (FRET) analysis.
Antibodies
The anti-Sur1 and anti-Trpm4 antibodies used in this study are listed in Table 2. Anti-Myc and anti-HSC70 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-FLAG antibody and anti-calmodulin antibody were purchased from Cell Signaling Technology (Beverly, MA). Two different sources of both anti-Sur1 and anti-Trpm4 antibodies were developed for this study (Table 2). To construct a bacterial expression vector encoding the intracellular nucleotide-binding domain 1 of Sur1 fused with hexahistidine, the corresponding region of the rat Sur1 cDNA sequence (amino acids 598–965 of NP_037171) was cloned into pQE30 (Qiagen, Gaithersburg, MD). The recombinant protein was purified by using a Ni+-nitrilotriacetic acid-agarose column and used to raise antibodies in rabbit and in goat, which was performed by a commercial service (Covance, Princeton, NJ). Using the same method, anti-Trpm4 antibodies targeting the N-terminal intracellular domain of mouse Trpm4, corresponding to amino acids 1–612 (NP_780339), were developed in rabbit and in chicken.
TABLE 2.
Anti-Sur1 and anti-Trpm4 antibodies used in this study
sc, Santa Cruz Biotechnology.
| Name | Host species | Source |
|---|---|---|
| Anti-Sur1-a | Rabbit | Custom |
| Anti-Sur1-b | Goat | Custom |
| Anti-Trpm4-a | Rabbit | Custom |
| Anti-Trpm4-b | Chicken | Custom |
| Anti-Trpm4-c | Rabbit | sc-67125 |
| Anti-Trpm4-d | Goat | sc-27539 |
| Anti-Trpm4-e | Goat | sc-27540 |
The specificity of the antibodies described above was validated using lysates from the appropriate Sur1 or Trpm4 expression systems and spinal cord tissues from wild-type, Trpm4−/−, and Abcc8−/− mice. Anti-Sur1-b antibody and anti-Trpm4-b antibody were used to immunoisolate Sur1 and Trpm4, respectively, from the various lysates. Subsequently, in one experiment, immunoblots of the immunoisolated proteins were performed using anti-Sur1-a antibody and anti-Trpm4-a antibody, respectively (see Fig. 9). In a parallel experiment, electrophoresis gels of the immunoisolated proteins were developed with Coomassie Blue stain, the protein bands at the appropriate molecular masses were isolated, and peptide fragments were analyzed by mass spectrometry (Taplin Mass Spectrometry Facility, Harvard Medical School, Boston, MA), which confirmed the identity of Sur1 and Trpm4.
FIGURE 9.
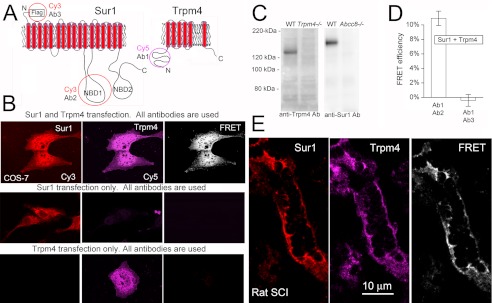
FRET analysis of Sur1-Trpm4 heteromers in spinal cord tissue after injury. A, schematic diagrams of Sur1 and Trpm4 proteins, showing the positions of epitopes for various antibodies (Ab) that were studied. B, COS-7 cells co-expressing FLAG-Sur1 and Myc-Trpm4, or expressing either one alone, as indicated, were incubated with both anti-Sur1-a and anti-Trpm4-e antibodies followed by both anti-rabbit Cy3-conjugated and anti-goat Cy5-conjugated secondary antibodies. Left and middle columns, fluorescence images of the two chromophores; right column, FRET images of the two chromophores. Results shown are representative of 6 replicates. Note that primary antibodies against Sur1 and Trpm4 do not cross-react. C, spinal cord tissue lysates were obtained from wild-type (WT), Trpm4−/−, and Abcc8−/− mice, as indicated. Anti-Sur1-b antibody and anti-Trpm4-b antibody were used to immunoisolate Sur1 and Trpm4, respectively, from the various tissue lysates; immunoisolated proteins were detected using anti-Sur1-a antibody (anti-Sur1 Ab) and anti-Trpm4-a antibody (anti-Trpm4 Ab). Note that primary antibodies against Sur1 and Trpm4 do not cross-react. D, average FRET efficiencies at steady state for the experiment in B using anti-Trpm4-e antibody (Ab1), detected with anti-goat Cy5-conjugated antibody versus intracellularly directed anti-Sur1-a antibody (Ab2) or extracellularly directed anti-FLAG antibody (Ab3), detected with Cy3-conjugated antibody, in COS-7 cells transfected to express FLAG-Sur1 and Myc-Trpm4; n = 58–64. E, immunolabeling and FRET images of a microvessel 24 h after spinal cord injury. Immunolabeling as in B; results shown are representative of 4 replicates.
FRET
COS-7 cells were transfected with plasmids for Cerulean- and Citrine-fused Sur1 and Trpm4, with Kir6.2 and Kir2.1 used as positive and negative controls, respectively. Preliminary experiments showed that transfection using a plasmid ratio of 16:1 for FLAG-Sur1-Cerulean:Citrine-Myc-Trpm4 yielded good FRET signals. Cells were studied 24–48 h after transfection. Cells were fixed with 4% formaldehyde for 20 min, and FRET fluorescence imaging was analyzed as described below. FRET experiments were also performed using fluorophore-conjugated secondary antibodies. These experiments were carried out using COS-7 cells transfected with FLAG-Sur1 and Myc-Trpm4 plasmids, with a plasmid ratio of 16:1 for Sur1:Trpm4, and using rat spinal cord tissues after traumatic injury, with remote spinal cord tissues from the same rats used as negative controls. Cells were studied 24–48 h after transfection, and tissues were studied 24 h after trauma. Immunolabeling was carried out as described (14). Briefly, cells and tissues were incubated with a mixture of primary antibodies overnight at 4 °C. The primary antibodies were anti-Trpm4-e, directed against the intracellular N terminus of Trpm4, and anti-Sur1-a, directed against the first intracellular nucleotide-binding domain (NBD1) of Sur1 or, as a negative control in COS-7 cells, rabbit anti-FLAG (Cell Signaling Technology), directed against the extracellular FLAG fused to the N terminus of Sur1. After washing with PBS, the sections and the cells were incubated with a mixture of donkey anti-rabbit IgG conjugated with Cy3 and mouse anti-goat IgG conjugated with Cy5 for 1 h.
FRET fluorescence imaging was analyzed on a commercial laser scanning microscope combination system, LSM510 Meta (Zeiss, Jena, Germany) with an Axiovert inverted microscope. The following settings were used throughout the experiments: (i) for Cerulean: excitation, 458 nm; detection, 480–520 nm; (ii) for Citrine: excitation, 514 nm; detection, 525–580 nm; (iii) for Cy3: excitation, 543 nm; detection, 565–615 nm; and (iv) for Cy5: excitation, 633 nm; detection, 650–704 nm. The images were captured and processed with the LSM Image Examiner software (Zeiss), with an integration time of 5 ms/pixel.
FRET efficiency was determined by measuring fluorescence before and after photobleaching (15). The FRET acceptors (Citrine or Cy5) were selectively photobleached by repeatedly scanning regions of interest with the maximum intensity of the laser until the fluorescence of the acceptors decreased to less than 30% of the original fluorescence. The fluorescence emission from the FRET donors (Cerulean or Cy3) and the acceptors were measured sequentially. The FRET efficiency (%) was calculated as E = (IDA − IDB)/IDA × 100, where IDA and IDB represent the steady-state donor intensity after and before bleaching, respectively. The FRET efficiency was assessed in a minimum of 30 different regions of interest for a single experimental set.
Immunoprecipitation and Immunoblot Analyses
COS-7 cells were transfected with plasmids for FLAG-Sur1 and Myc-Trpm4, with Myc-Kir6.2 and Myc-Hif1α used as positive and negative controls, respectively. In different experiments, we performed transfections using different plasmid ratios, as indicated, for Sur1:Trpm4. Cells were studied 24–48 h after transfection. Tissues from spinal cord-injured rats were studied 6 h after trauma.
Total lysates from COS-7 cells transfected with various combinations of plasmids and from spinal cord tissues were prepared in lysis buffer (1.5 mm KH2PO4, 8 mm Na2HPO4 (pH 7.3), 3 mm KCl, 137 mm NaCl, and 1% Triton X-100) with freshly added protease inhibitor mixture (Roche Applied Science). To immunoprecipitate Sur1, Trpm4, or Myc-fused proteins from the lysates, samples were incubated with anti-Sur1-b antibody, anti-Trpm4-b antibody, or anti-Myc antibody from mouse (Santa Cruz Biotechnology) overnight at 4 °C. The immune complex formed was isolated by protein G-Sepharose for antibodies from goat and mouse, or anti-chicken IgY antibody agarose for the chicken antibody, and washed three times with the lysis buffer. To remove N-linked glycans, the immunoprecipitated samples were further incubated with peptide:N-glycosidase F (New England Biolabs, Ipswich, MA) for 1 h at 37 °C. To elute the proteins, the beads were resuspended in 2× NuPAGE LDS sample buffer (Invitrogen), and boiled for 5 min. The resulting samples were examined by immunoblot analysis.
For the co-expression experiments, proteins were detected using anti-Sur1-a, anti-Trpm4-a, anti-Myc (Santa Cruz Biotechnology), anti-calmodulin (Cell Signaling Technology), and anti-HSC70 (Santa Cruz Biotechnology) antibodies. For the spinal cord experiments, proteins were detected using anti-Sur1-a, anti-Trpm4-a, anti-Trpm4-b, anti-Trpm4-c, and anti-Trpm4-d antibodies.
Surface Biotinylation Assay
COS-7 cells transfected with various combinations of plasmids, as indicated, were washed with ice-cold Dulbecco's PBS and incubated with 0.5 mg/ml EZ-Link Sulfo-NHS-Biotin (Pierce) dissolved in Dulbecco's PBS for 30 min at 4 °C. The biotinylation reaction was quenched by washing with Tris-buffered saline (50 mm Tris (pH 7.4) and 150 mm NaCl) for 5 min. Total lysates were prepared from the cells in the lysis buffer (see above) for 1 h at 4 °C and cleared by centrifugation. Biotinylated proteins were isolated by NeutrAvidin-agarose beads (Pierce), washed three times with the cell lysis buffer, resuspended in 2× NuPAGE LDS sample buffer (Invitrogen), and boiled for 5 min. Immunoblot analysis of HSC70 was performed to validate the preparation of the biotinylated proteins.
Patch Clamp Electrophysiology
COS-7 cells were co-transfected with plasmids for FLAG-Sur1 and Myc-Trpm4 (along with pEGFP; experiment of Fig. 5), or FLAG-Sur1-Ce and Ci-Myc-Trpm4 (experiments of Figs. 6 and 7). COS-7 cells were transfected with the plasmid for the Sur1-Trpm4 fusion protein (experiment of Fig. 6). HEK-293 and Sur1-HEK-293 cells were transfected with the plasmid for Ci-Myc-Trpm4 (experiment of Fig. 8). Fluorescent cells were studied at room temperature 48–72 h after transfection, as described (16, 17). Membrane currents were recorded in intact cells using the nystatin-perforated whole-cell configuration (18) and in cell-free isolated membrane patches using the inside-out configuration (19). For whole-cell macroscopic recordings, we used a nystatin-perforated patch technique with a bath solution containing (in mm): 145 CsCl, 1 CaCl2, 1 MgCl2, 32.5 HEPES, 12.5 glucose, pH 7.4. The pipette solution contained (in mm): 145 CsCl, 8 MgCl2, and 10 HEPES, pH 7.2. For inside-out patch recording, we used a bath solution containing (in mm): 145 CsCl, 1.5 CaCl2, 1 MgCl2, 5 EGTA, 32.5 HEPES, 12.5 glucose, pH 7.4. The pipette contained (in mm): 145 CsCl, 1 MgCl2, 0.2 CaCl2, 5 EGTA, 10 HEPES, pH 7.3. For experiments requiring low concentration of free Ca2+ in the bath solution, a Ca2+-EGTA buffered solution was employed, and free [Ca2+] was calculated using the program WEBMAXC v2.10.
FIGURE 5.

Co-expression with Sur1 endows Trpm4 with sensitivity to the Sur1 activator, diazoxide. A, single channel recordings (left) and single channel conductances (right) from inside-out patches from COS-7 cells expressing Myc-Trpm4 alone or co-expressing FLAG-Sur1 and Myc-Trpm4. Conditions were as follows: 11–15 patches per condition; Ca2+, 1 μm; ATP, 0; charge carrier, Cs+. h.p., holding potential; pS, picosiemens. B, representative whole-cell membrane currents in COS-7 cells co-expressing FLAG-Sur1 and Myc-Trpm4, or expressing Myc-Trpm4 alone, as indicated, before and after exposure to diazoxide and, for Trpm4 alone, after ATP depletion. Left and right, currents shown at low (left) and high (right) temporal resolution during ramp pulses (−100 to +100 mV in 500 ms, repeated every 15 s; holding potential, −50 mV). CTR, control cells without transfection. C, magnitude (mean ± S.E.) of the inward current at −50 mV activated by diazoxide (left) or by ATP depletion (right) during experiments performed as in B, in control cells without transfection (CTR), cells expressing Myc-Trpm4 alone, FLAG-Sur1 alone or co-expressing FLAG-Sur1 and Myc-Trpm4; n denotes number of cells tested. The currents were activated by diazoxide (100 μm) or by an ATP-depleting mixture (sodium azide (NaAz), 1 mm plus 2-deoxyglucose (2DG), 10 mm). Charge carrier, Cs+; **, p < 0.01; for all experiments. Sur1:Trpm4 plasmid ratio, 2.3:1.
FIGURE 6.

Co-expression with Sur1 endows Trpm4 with sensitivity to the Sur1 inhibitor, glibenclamide. A, representative whole-cell current from a COS-7 cell co-expressing FLAG-Sur1-Ce and Ci-Myc-Trpm4; the current was activated by A23187 (10 μm) and was inhibited by subsequent application of glibenclamide (1 μm). B, concentration-response data (mean ± S.E., filled squares) for glibenclamide inhibition in COS-7 cells co-expressing FLAG-Sur1-Ce and Ci-Myc-Trpm4 were obtained from whole-cell recordings and were fitted with a standard logistic equation with IC50 = 850 nm; 5–7 cells at each concentration. The empty circle shows the inhibitory effect of 1 μm glibenclamide in cells that expressed Ci-Myc-Trpm4 alone. Whole-cell currents were activated by A23187 (10 μm). Charge carrier, Cs+; HP, holding potential. Sur1:Trpm4 plasmid ratio, 12:1. C, single channel activity in a responsive inside-out patch from a COS-7 cell co-expressing FLAG-Sur1-Ce and Ci-Myc-Trpm4, before and after application of glibenclamide (5 μm), shown at low and high (box insets) temporal resolution. Conditions were as follows: Ca2+, 1 μm; ATP, 0; charge carrier, Cs+. D, immunoblots of total lysate from COS-7 cells expressing His6-Sur1 alone (lane 2), Myc-Trpm4 alone (lane 3), or the His6-Sur1-Trpm4 fusion protein (lane 4), labeled with anti-Sur1 antibody-a (left panel) or anti-Trpm4-a antibody (right panel); also shown is surface expression (Biotin) of Trpm4 alone (lane 5) and of the Sur1-Trpm4 fusion protein (lane 6). Surface expression was assessed using biotinylation of surface proteins; results shown are representative of 4 replicates. E, single channel activity in an inside-out patch from a COS-7 cell expressing the His6-Sur1-Trpm4 fusion protein, before and after application of glibenclamide (5 μm). Conditions were as follows: Ca2+, 1 μm; ATP, 0; charge carrier, Cs+. F, the effect of glibenclamide (1 or 5 μm) on open channel probability (nPo) at 20 s and at 1 min after application of glibenclamide to COS-7 cells expressing Ci-Myc-Trpm4 alone (empty bars), co-expressing FLAG-Sur1-Ce and Ci-Myc-Trpm4 (gray bars), or expressing the His6-Sur1-Trpm4 fusion protein (FP, black bars); 3–5 patches for each condition. G, concentration-response data (mean ± S.E.) for steady-state glibenclamide inhibition in COS-7 cells expressing Ci-Myc-Trpm4 alone (empty circles), co-expressing FLAG-Sur1-Ce and Ci-Myc-Trpm4 (gray circles), or expressing the His6-Sur1-Trpm4 fusion protein (black circles). The same patches as in F plus additional patches for Ci-Myc-Trpm4 alone were used.
FIGURE 7.

Co-expression with Sur1 changes the sensitivity of Trpm4 to Mg2+. A, single channel recordings of inside-out patches from COS-7 cells expressing Ci-Myc-Trpm4 alone or co-expressing FLAG-Sur1-Ce and Ci-Myc-Trpm4, as indicated. Patches were exposed first to Na2-ATP (100 μm), then to Mg2+-ATP (100 μm), and then to 0 ATP. All solutions contained Ca2+ (1 μm). B, bar graph showing the mean (±S.E.) open channel probability (nPo) relative to that in Na2-ATP for the three conditions in A, as indicated. Conditions were as follows: charge carrier, Cs+; 10 patches per condition. Sur1:Trpm4 plasmid ratio, 12:1; *, p < 0.05; **, p < 0.01.
RESULTS
Sur1 and Trpm4 Co-associate
FRET
We performed intermolecular FRET analyses to evaluate the physical interaction between Sur1 and Trpm4. Cerulean and Citrine fluorophores were fused to Sur1 and Trpm4 at either the N terminus or the C terminus. C terminus-fused Sur1 was co-expressed with C terminus-fused Trpm4 or with N terminus-fused Trpm4. N terminus-fused Kir6.2 and N terminus-fused Kir2.1 served as positive and negative controls, respectively (Fig. 1A). FRET imaging microscopy was used to detect protein-protein interactions in single cells. FRET was detected when Sur1 was co-expressed with the positive control, Kir6.2, as well as with N terminus-fused Trpm4, but not with the negative control, Kir2.1 (Fig. 1B). FRET was not observed between C terminus-fused Sur1 and C terminus-fused Trpm4 (Fig. 1B), as found previously (20), although the two did co-associate, as found in co-immunoprecipitation experiments (Fig. 1E).
FIGURE 1.

Evidence from FRET that Sur1 and Trpm4 co-associate. A, schematic diagram of Sur1 and Trpm4 proteins, showing the positions of Ci and Ce fluorescent tags. B, fluorescence images (left and middle columns) of each fluorophore and FRET signal (right column) for COS-7 cells expressing FLAG-Sur1-Ce and Ci-Myc-Kir6.2 (positive control), FLAG-Sur1-Ce and Ci-Myc-Kir2.1 (negative control), FLAG-Sur1-Ce and Ci-Myc-Trpm4, and FLAG-Sur1-Ce and Myc-Trmp4-Ci; FRET within the cell membrane is indicated (arrows); results shown are representative of 3–7 replicates. C and D, average FRET efficiency as a function of time (C) and at steady state (D) for the four conditions in B; n = 32–45; Sur1:Trpm4 plasmid ratio, 16:1. Error bars indicate mean ± S.E. E, immunoblot of COS-7 cells expressing FLAG-Sur1-Ce and Ci-Myc-Trpm4, or FLAG-Sur1-Ce and Myc-Trmp4-Ci, as indicated. Immunoisolation was performed using anti-Sur1-b antibody (anti-Sur1-b Ab) or anti-Trpm4-b antibody (anti-Trpm4-b Ab), as indicated, and proteins were detected with anti-Sur1-a antibody. Sur1:Trpm4 plasmid ratio, 13:1.
Co-expression of Sur1 with N terminus-fused Trpm4 gave rise to FRET that localized, in part, to the endoplasmic reticulum, consistent with heteromeric assembly of Sur1 and Trpm4 within this organelle (Fig. 1B). In most cells, FRET was discernible as a contour line demarcating the edge of the cell, consistent with plasma membrane localization of Sur1-Trpm4 heteromers (Fig. 1B, arrows). The time course of FRET efficiency in a typical experiment measured at the plasma membrane during photobleaching and the average steady-state FRET efficiency measured at the plasma membrane in multiple experiments are shown in Fig. 1, C and D. At steady state, efficiencies of 9–10% were recorded for both Kir6.2 and N terminus-fused Trpm4 as compared with values of ∼0% for Kir2.1 and C terminus-fused Trpm4. These findings are consistent with an intrinsic capacity for stable co-association of Sur1 and Trpm4.
Co-immunoprecipitation
We performed co-immunoprecipitation experiments to further evaluate the interaction between Sur1 and Trpm4. FLAG-Sur1 was co-expressed with Myc-Trpm4. Myc-Kir6.2 and Myc-Hif1α were used as positive and negative controls, respectively. Immunoblot of total lysates using anti-Myc antibody showed the expected bands for each condition (Fig. 2A), including four bands representing Kir6.2 monomers and multimers (multiples of ∼38 kDa) (21, 22), a single band for Hif1α, and two closely spaced bands for Trpm4 (135 and 147 kDa) (23), with the more slowly migrating form, which represents highly glycosylated (HG) Trpm4 (see below), being dominant. After co-immunoprecipitation using anti-Sur1 antibody, immunoblot again showed the four bands expected for the positive control, Kir6.2, and no band for the negative control, Hif1α. Two bands for Trpm4 were again identified, but unlike with total lysate, now the faster migrating form, which represents core-glycosylated (CG) Trpm4 (see below), was dominant (Fig. 2A).
FIGURE 2.

Evidence from co-immunoprecipitation (Co-IP) that Sur1 and Trpm4 co-associate. A and B, immunoblots of COS-7 cells expressing FLAG-Sur1 and either Myc-Kir6.2 (positive control) or Myc-Hif1α (negative control) or Myc-Trpm4. Left and right, immunoblots of total lysate (left) and after Co-IP (right), with immunoisolation performed using anti-Sur1-a antibody and protein detected with anti-Myc antibody (A) or with immunoisolation performed using anti-Myc antibody and protein detected with anti-Sur1-a antibody (B). HG and CG, highly glycosylated and core-glycosylated Trpm4, respectively. Results shown are representative of 7 replicates for Sur1-Trpm4 and Sur1-Kir6.2 and 3 replicates for Hif1α. Sur1:Trpm4 plasmid ratio, 1:1.
In the same series of experiments, co-immunoprecipitation using anti-Myc antibody yielded Sur1 with the positive control, Kir6.2, no band for the negative control, Hif1α, and a prominent band with Trpm4 (Fig. 2B). The finding that Sur1 and Trpm4 co-associated, as illustrated in Fig. 2, A and B, with immunoprecipitation of Sur1 and immunoprecipitation of Trpm4, was replicated in seven independent co-expression experiments, with Sur1:Trpm4 plasmid ratios of 1:1, 3:1, and 9:1.
In the heterologous expression system that we studied, the surface expression of Sur1 depended on Trpm4 expression in a complex way. First, when both were co-expressed, the surface expression of Sur1 was inversely related to the amount of Trpm4 expressed. Optimal surface expression of Sur1 was observed with low levels of Trpm4, whereas an overabundance of Trpm4 was associated with reduced surface expression of Sur1 (Fig. 3A). One potential explanation for this was competition between plasmids.
FIGURE 3.

Surface expression of Sur1 requires Trpm4, but is reduced by overexpression of Trpm4. A, immunoblots of COS-7 cells co-expressing His6-Sur1 and Trpm4-EGFP using different amounts and different ratios of plasmids, as indicated. Left four lanes, immunoblots for Sur1 and Trpm4 of total lysates; Right four lanes, immunoblots for surface expressed protein. Surface expression of Sur1 was assessed using biotinylation of surface proteins, with HSC70 used as a negative control for surface proteins. Note that overexpression of Trpm4 yields a less favorable ratio of Sur1 to Trpm4 at the surface (asterisks). Results shown are representative of 3 replicates; immunoblot was performed using anti-Sur1-a antibody and anti-Trpm4-a antibody. B, immunoblot of COS-7 cells expressing His6-Sur1 with and without co-expression of Myc-Trpm4, as indicated. Left two lanes, total expression of Sur1; right two lanes, surface expression of Sur1; surface expression was assessed as in A. Note that Trpm4 expression is required for appreciable surface expression of Sur1; immunoblot was performed using anti-Sur1-a antibody. Sur1:Trpm4 plasmid ratio, 8:1. C, densitometric analysis of the experiment in B showing the surface-to-total expression ratio (mean ± S.E.) for Sur1 without and with co-expression of Trpm4; n = 3. For comparison, the surface-to-total expression ratio for Sur1 in insulinoma cells (Ins) is also shown. n = 2; **, p < 0.01. D, immunoblots of COS-7 cells expressing Myc-Trpm4 with and without co-expression of His6-Sur1, as indicated. Left two lanes, total expression of Trpm4; right two lanes, surface expression of Trpm4; surface expression was assessed as in A. Note that Sur1 expression has no effect on surface expression of Trpm4. Results shown are representative of 4 replicates; immunoblot was performed using anti-Trpm4-a antibody. Sur1:Trpm4 plasmid ratio, 2:1.
We also examined the surface expression of Sur1, with versus without Trpm4 co-expression. Under these conditions, the surface expression of Sur1 was completely dependent on co-expression of Trpm4 (Fig. 3B), as is observed with Kir6.2 (24, 25). The result of this experiment indicated that competition between plasmids could not account for the finding of Fig. 3A, that overabundant expression of Trpm4 leads to less surface expression of Sur1. The requirement for Trpm4 for surface expression of Sur1 suggested that, by co-associating, Trpm4 apparently can shield the endoplasmic reticulum retention signal of Sur1 to promote its trafficking to the plasma membrane. Under optimal conditions of Sur1 and Trpm4 co-expression, the ratio of surface-to-total expression of Sur1 was similar to levels in insulinoma cells (Fig. 3C).
Trpm4 forms functional membrane channels by itself (1, 4). As expected, the surface expression of Trpm4 was not affected by co-expression of Sur1 (Fig. 3D).
Glycosylation
Two forms of Trpm4 were recognized in total lysates of COS-7 cells, a CG form and an HG form (Figs. 2A and 4A). The one that co-associated with Sur1 was predominantly the CG form (Fig. 2A).
FIGURE 4.

Co-associated Sur1 and Trpm4 are predominantly core-glycosylated. A, immunoblot for Trpm4 of total lysate from COS-7 cells expressing Myc-Trpm4, before (left lane) and after (right lane) deglycosylation with peptide:N-glycosidase F (DG). Note the occurrence of both HG (147 kDa) and CG (135 kDa) forms of Trpm4. Immunoblot was performed using anti-Trpm4-a antibody; results shown are representative of 7 replicates. B, immunoblot for Sur1 of total lysate from COS-7 cells co-expressing FLAG-Sur1 and Myc-Kir6.2, or FLAG-Sur1 and Myc-Trpm4, as indicated, before (three left lanes) and after (far right lane) deglycosylation with peptide:N-glycosidase F (DG). Note the occurrence of both highly glycosylated (HG; 165 kDa) and core-glycosylated (CG; 150 kDa) forms of Sur1 when co-expressed with Kir6.2, but the occurrence of only the CG form of Sur1 when co-expressed with Trpm4. Immunoblot was performed using anti-Sur1-a antibody; results shown are representative of 3–7 replicates. Sur1:Trpm4 plasmid ratio, 1:1. C, immunoblot of surface Sur1 from COS-7 cells co-expressing His6-Sur1 and Myc-Kir6.2, or His6-Sur1 and Myc-Trpm4, as indicated. The third lane is of total lysate after deglycosylation with peptide:N-glycosidase F (DG). Note that with Kir6.2, both glycosylation forms are present at the surface, whereas with Trpm4, the CG form of Sur1 is dominant at the surface. Surface expression of Sur1 was assessed using biotinylation of surface proteins; immunoblot was performed using anti-Sur1-a antibody. Sur1:Trpm4 plasmid ratio, 1.5:1.
Sur1 also is found in CG and HG forms (21, 26, 27). When co-expressed with Kir6.2, both forms of Sur1 were identified (150 and 165 kDa) in total lysate (Fig. 4B), but when co-expressed with Trpm4, CG Sur1 was dominant in total lysate (Figs. 2B and 4B). We also examined the glycosylation state of surface Sur1. When co-expressed with Kir6.2, both glycosylation forms of Sur1 were found on the surface (Fig. 4C) (21, 26), but when co-expressed with Trpm4, only CG Sur1 was identified at the surface (Fig. 4C).
Functional Implications of Sur1 Trpm4 Co-assembly
Sur1-Trpm4 Channels
We used patch clamp electrophysiology to determine whether Sur1-Trpm4 heteromers would co-assemble to form functional Sur1-Trpm4 channel complexes with predicted biophysical and pharmacological properties (16, 17). Cesium was used as the charge carrier to block all K+ channels, including KATP channels. Either expression of Trpm4 alone or co-expression of Sur1 and Trpm4 gave rise to membrane currents with biophysical properties characteristic of Trpm4 (2, 5, 28); in both cases, single channel recordings of inside-out patches demonstrated permeability to Cs+, with a single channel conductance of 23–25 picosiemens (Fig. 5A). Also, in cells expressing Trpm4 alone, as well as cells co-expressing Sur1 and Trpm4, whole-cell membrane currents were activated by depleting ATP (Fig. 5, B and C).
We used whole-cell recordings to study the response to the Sur1 activator, diazoxide (29). In cells that co-expressed Sur1 and Trpm4, diazoxide activated an inward Cs+ current at physiological potentials that reversed near 0 mV (Fig. 5B). Diazoxide was without effect in cells expressing either Sur1 alone or Trpm4 alone (Fig. 5, B and C). Because diazoxide has no effect on Trpm4 alone, channel activation by diazoxide under conditions of Sur1 and Trpm4 co-expression is consistent with the presence of functional Sur1-Trpm4 channel complexes at the cell membrane.
We used whole-cell recordings to study the response to the Sur1 inhibitor, glibenclamide (30, 31). Whole-cell currents were activated by increasing the intracellular Ca2+ concentration, which yielded an inward Cs+ current that was indistinguishable from that activated by diazoxide or ATP depletion. In cells that co-expressed Sur1 and Trpm4, this current was blocked by glibenclamide in a concentration-dependent manner (Fig. 6, A and B). The apparent potency of glibenclamide in the Sur1-Trpm4 co-expression system (IC50, 850 nm) was less than that reported for native Sur1-Trpm4 channels (IC50, 48 nm) (17), but was significantly greater than that for Trpm4 alone (5% block with 1 μm, Fig. 6B; 70% block with 100 μm (32)).
We examined the effect of glibenclamide on inside-out patches from cells that co-expressed Sur1 and Trpm4. In some patches, glibenclamide (1 or 5 μm) had little or no effect on a 25-picosiemen Ca2+-sensitive conductance (not shown). However, in “responsive” patches, the addition of glibenclamide (1 or 5 μm) resulted in >90% channel inhibition (Fig. 6, C and G). These experiments also showed that in responsive patches, glibenclamide was not acting as an open channel blocker, but instead reduced the open channel probability (Fig. 6C). A reduction in open channel probability, as reported for sulfonylurea drugs with native Sur1-Trpm4 channels (17), as well as with KATP channels (33), is consistent with an effect via the known molecular interaction between glibenclamide and Sur1 (30).
We hypothesized that the lower potency of glibenclamide blockade observed in the whole-cell experiments, as well as the absence of block in some inside-out patches, might be due to the presence at the membrane not only of Sur1-Trpm4 channels, which are expected to be highly sensitive to glibenclamide, but also of nonassociated Trpm4 channels, which are relatively insensitive to glibenclamide. To examine this hypothesis, we constructed a Sur1-Trpm4 fusion protein, similar to that reported for Sur1-Kir6.2 (21). When expressed by itself, the Sur1-Trpm4 fusion protein would preclude nonassociated Trpm4 from being present at the membrane. The fusion protein of Sur1 (150 kDa) and Trpm4 (135 and 147 kDa, CG and HG, respectively (Fig. 4, A and B)) had the expected mass of ∼290 kDa and was identified using both anti-Sur1 and anti-Trpm4 antibodies (Fig. 6D). Notably, the Sur1-Trpm4 fusion protein also was identified at the surface membrane (Fig. 6D).
The application of glibenclamide to inside-out patches with the Sur1-Trpm4 fusion protein resulted in an initial brief transient increase in open channel probability that was followed by a stable reduction in open probability (Fig. 6, E–G). In patches with the fusion protein, 1 or 5 μm glibenclamide reduced the open channel probability >90% (Fig. 6G), similar to the effect observed in responsive patches from the co-expression system (Fig. 6G) and similar to that observed with native channels (17). Notably, a transient increase in open probability was never observed with Sur1-Trpm4 channels in the co-expression system (Fig. 6, C and F) or with native channels, leading us to believe that the transient increase in open probability observed with the fusion protein may have been due to the linker that we used, which may have impaired the transition between conformational states. Regardless, the finding that “forced co-association” between Sur1 and Trpm4 yielded a functional channel complex with pharmacological properties of Sur1 and biophysical properties of Trpm4 gave further evidence that “free co-association” could lead to functional Sur1-Trpm4 channels and, moreover, indicated that 1:1 stoichiometry between Sur1 and Trpm4 is appropriate for the formation of functional channels.
Mg2+-ATP
Co-assembly of Sur1 with Trpm4 is predicted to confer sensitivity to Mg2+-ATP. With the KATP channel, ATP interacts directly with Kir6.2 to inhibit the channel, but in the presence of Mg2+, ATP activates the channel through an interaction with the nucleotide-binding folds of Sur1 (7, 35). Like Kir6.2, Trpm4 is directly inhibited by ATP (4). We sought to determine whether coupling with Sur1 would impart upon Trpm4 sensitivity to activation by Mg2+-ATP.
In the absence of Mg2+, and with a low concentration of ATP, the open channel probability was modest and was indistinguishable in patches with Trpm4 alone versus patches with Sur1-Trpm4 channels (Fig. 7). The addition of Mg2+, without changing the concentration of ATP, resulted in a doubling of the open probability for patches with Trpm4 alone (2), but resulted in a 10-fold increase in the open probability for patches with Sur1-Trpm4 channels (Fig. 7). More striking, when ATP was removed, patches with Trpm4 alone showed minimal change in open probability, whereas patches with Sur1-Trpm4 channels exhibited a 30-fold increase in open probability (Fig. 7).
Calmodulin
The experiment with Mg2+-ATP, which was carried out in the presence of a constant concentration of Ca2+ (1 μm), suggested that the apparent sensitivity of Trpm4 to intracellular Ca2+ might be increased by co-association with Sur1. The intrinsic sensitivity of Trpm4 to Ca2+ is mediated by an interaction between calmodulin (CaM) and four CaM-binding sites located at the C terminus of Trpm4 (2). We performed co-immunoprecipitation experiments to assess the effect of Sur1 on the Trpm4-CaM interaction. We studied HEK-293 cells versus a stable line of HEK-293 cells that constitutively overexpresses Sur1 (hereafter, Sur1-HEK-293 cells) (Fig. 8, A and B). By using these two cell lines, both could be treated identically with transient transfection of one plasmid to express Trpm4, avoiding the potential imbalance introduced by co-transfection. In cells that expressed Sur1, the apparent affinity of Trpm4 for CaM was double that observed in cells that did not express Sur1 (Fig. 8B).
We assessed the functional implication of an increase in apparent affinity for CaM using HEK-293 versus Sur1-HEK-293 cells. To circumvent problems with desensitization of Trpm4 (2), channel activity was measured immediately after forming inside-out patches in a bath solution containing 1 μm Ca2+, and activity was normalized to subsequent activity at the saturating concentration of 2 mm Ca2+ (2). Patches from cells expressing Trpm4 alone typically showed activity from 1–3 channels simultaneously, whereas patches from cells expressing both Sur1 and Trpm4 typically showed simultaneous activity from twice this number of channels (Fig. 8C). Normalized currents in patches from cells expressing both Sur1 and Trpm4 were twice as large as those in patches from cells expressing Trpm4 alone (Fig. 8C).
Sur1-Trpm4 Heteromers in Spinal Cord Injury
In spinal cord injury, Sur1 and Trpm4 are transcriptionally up-regulated and play critical pathological roles (13, 28, 36). The phenotype observed after spinal cord injury in Abcc8−/− mice is exactly the same as in Trpm4−/− mice, with both showing equivalent protection from the autodestructive process termed “progressive hemorrhagic necrosis,” which is marked by secondary hemorrhage due to fragmentation of microvessels. These observations led to the hypothesis that Sur1 and Trpm4 co-associate in vivo following traumatic CNS injury.
To extend FRET imaging microscopy to tissues after spinal cord injury, we developed an antibody-based method using Cy3- and Cy5-conjugated secondary antibodies as the chromophores (Fig. 9, A–D). Control experiments were carried out on cultured cells that co-expressed Sur1 and Trpm4, using primary antibodies directed against the intracellular N terminus of Trpm4 and against the intracellular nucleotide-binding domain (NBD1) of Sur1 or, as a negative control, against the FLAG-fused extracellular N terminus of Sur1 (Fig. 9A). At steady state, a FRET efficiency of ∼11% was recorded when using the two intracellularly directed antibodies as compared with a value ∼0% when the antibody against Sur1 was directed to the extracellular epitope (Fig. 9D).
We used this antibody-based FRET technique to evaluate rat spinal cord tissues before and after injury. In uninjured spinal cord, Sur1 and Trpm4 immunolabeling was minimal (28, 36), and FRET signals were absent (not shown). However, 24 h after spinal cord injury, Sur1 and Trpm4 immunolabeling was prominent, immunolabeling for Sur1 and Trpm4 co-localized, and FRET signals were detected in various cellular structures, including microvessels (Fig. 9E).
We also performed co-immunoprecipitation experiments to evaluate tissues after spinal cord injury. Immunoprecipitation showed abundant Sur1 and Trpm4 (Fig. 10, A and B). Co-immunoprecipitation using anti-Trpm4 antibody yielded Sur1, and co-immunoprecipitation using anti-Sur1 antibody yielded Trpm4 (Fig. 10, A and B). In these experiments, we used four different anti-Trpm4 antibodies to detect Trpm4 after immunoisolation with anti-Sur1 antibody, and all four detected the same band, two examples of which are shown in Fig. 10. Importantly, co-immunoprecipitation using anti-Sur1 antibody yielded Trpm4 only after spinal cord injury, not in uninjured spinal cord (Fig. 10, D and E). Together, these findings with co-immunoprecipitation and FRET are consistent with co-assembly of Sur1 and Trpm4 in vivo following spinal cord injury.
FIGURE 10.

Co-immunoprecipitation of Sur1-Trpm4 heteromers from spinal cord tissue after injury. A, immunoblot using anti-Sur1-a antibody of protein immunoisolated from insulinoma cells (Ins, positive control) and from spinal cord 6 h after injury using anti-Sur1-b antibody (SCI IP) or anti-Trpm4-b antibody (SCI Co-IP), and protein immunoisolated from insulinoma cells after treatment with peptide:N-glycosidase F (Ins DG). B, immunoblot using anti-Trpm4-a antibody of total lysate from COS-7 cells expressing Myc-Trpm4 (PC, positive control), of protein immunoisolated from small intestine (SmI, positive control) and spinal cord 6 h after injury using anti-Trpm4-b antibody (SCI IP) or anti-Sur1-b antibody (SCI Co-IP), and protein immunoisolated from small intestine after treatment with peptide:N-glycosidase F (SmI DG). C, immunoblot using anti-Trpm4-d antibody of protein immunoisolated from spinal cord 6 h after injury using anti-Trpm4-b antibody (SCI IP) or anti-Sur1-b antibody (SCI Co-IP), before and after treatment with peptide:N-glycosidase F (DG). Similar results were obtained using anti-Trpm4-b and antiTrpm4-c antibodies for detection. D, immunoblot using anti-Trpm4-a antibody of protein immunoisolated from uninjured spinal cord (CTR) or spinal cord 6 h after injury (SCI) using anti-Trpm4-b antibody (IP) or anti-Sur1-b antibody (Co-IP). Note that co-associated Sur1-Trpm4 is found only after spinal cord injury. E, densitometric analysis showing the mean (±S.E.) abundance of Trpm4 associated with Sur1 in the uninjured spinal cord versus 6 h after spinal cord injury (SCI) (n = 3; **, p < 0.01).
DISCUSSION
The principal finding of the present study is that Sur1 and Trpm4 have an intrinsic capacity for stable co-association, as shown by our experiments with co-immunoprecipitation, as well as with FRET based on N terminus-, but not C terminus-fused Trpm4. This intrinsic capacity for co-association resembles the association between Sur1 and Kir6.2 in forming Sur1-regulated KATP (Sur1-Kir6.2) channels, but important differences are evident. Sur1-regulated KATP channels are constitutively expressed, both in pancreatic β cells and in neurons, but to date, Sur1-Trpm4 channels have been identified in neurons, astrocytes, and endothelial cells only under conditions of CNS injury, where they are transcriptionally up-regulated during the acute phase after injury (10).
Native Kir6.2 possesses an endoplasmic reticulum retention signal that precludes its independent trafficking to the cell membrane without Sur1 (9). Trpm4 has no such constraint and thus readily traffics by itself to the cell membrane to form functional channels (1, 4). Our findings are in accord with these observations, showing that co-expression of Sur1 with Trpm4 did not influence trafficking of Trpm4 to the cell membrane, but it facilitated trafficking of Sur1 to the cell membrane. Many details remain to be resolved about the association between Sur1 and Trpm4, but the observation that the endoplasmic reticulum retention signal on Sur1 apparently was shielded by co-association with Trpm4 suggests that the domain(s) of Sur1 that interacts with Trpm4 may be similar to that which interacts with Kir6.2.
Several factors likely account for the relatively selective conditions that we found were required for demonstrating surface expression of Sur1-Trpm4 channels in a heterologous co-expression system, including possible differences in transfection efficiency for different plasmids, differences in protein turnover rates, and the ability of Trpm4 to traffic to the cell membrane without Sur1. We studied different expression conditions, including Sur1:Trpm4 plasmid ratios ranging from 1:1 to 16:1. Although co-immunoprecipitation from total lysate was observed under all conditions, higher ratios of Sur1:Trpm4 were required to observe FRET and to obtain surface expression of Sur1. Overabundant expression of Trpm4 led to a dominance of Trpm4 at the cell membrane that was associated with reduced surface expression of Sur1. These observations likely account for the failure to demonstrate Sur1-Trpm4 channels in a recently published study (20). In that study, the expression conditions used were such that Trpm4 currents were very large. Our data suggest that under such conditions, the likelihood of detecting Sur1-Trpm4 channels would be low. The other major finding of the study by Sala-Rabanal et al. (20) was that FRET does not occur when fluorescent tags are placed on the C terminus of both Sur1 and Trpm4. Our data corroborated this finding, but expanded upon it by showing that Sur1 with C terminus-fused Cerulean and Trpm4 with C terminus-fused Citrine do co-associate, as shown in co-immunoprecipitation experiments. However, when Cerulean was fused to the C terminus of Sur1 and Citrine was fused to the N terminus of Trpm4, we detected excellent FRET signals. These findings, combined with the findings from our co-immunoprecipitation experiments, confirm the intrinsic capacity for stable co-association between Sur1 and Trpm4.
The ability of Trpm4 to traffic to the cell membrane without Sur1 also complicated our experiments on glibenclamide blockade. Even when using conditions that we found to be optimal for detecting Sur1-Trpm4 channels in the co-expression system, we encountered situations that were best explained by hypothesizing that both Sur1-Trpm4 heteromers and Trpm4 homomers were present at the cell membrane. In one series of experiments, we studied channel blockade using whole-cell recordings in which channels were activated by increasing the intracellular Ca2+ concentration. Although both Sur1-associated and nonassociated Trpm4 channels are activated by Ca2+, the application of a modest concentration of glibenclamide (1–5 μm) would be expected to affect predominantly the Sur1-Trpm4 heteromers, not the Trpm4 homomers, which are relatively insensitive to glibenclamide. In another series of experiments, we studied channel blockade using inside-out patches. In some cases, we found no effect of glibenclamide, whereas in other cases, we found a potent blocking effect. When the problem of nonassociated Trpm4 was eliminated by studying the Sur1-Trpm4 fusion protein, we found the expected high sensitivity to glibenclamide, with >90% block with 1–5 μm glibenclamide, as reported for native channels (17), and significantly greater than the ∼5% block produced by the same concentrations of glibenclamide with Trpm4 alone. Because the Sur1 activator, diazoxide, is without effect on Trpm4, co-expression experiments in which closed channels were activated using diazoxide were not confounded by nonassociated Trpm4. An important question for future experiments will be to determine how it is in nature that Sur1-associated Trpm4 is favored over unassociated Trpm4 in conditions of CNS injury, as implied by the high sensitivity to glibenclamide (IC50, 48 nm) reported for native Sur1-Trpm4 channels in astrocytes from hypoxic gliotic capsule and neurons from ischemic cortex (17, 37).
The favored glycosylation states for the subunits of KATP and Sur1-Trpm4 channels appear to be different. Kir6.2 lacks N-linked glycosylation sites, but Sur1 (21, 26) and Trpm4 (38) are found in two glycosylation states, core-glycosylated and highly glycosylated. With KATP channels, co-association of Sur1 with Kir6.2 promotes glycosylation of Sur1 (21), and glycosylation is required for efficient surface expression (26). By contrast, with Sur1-Trpm4 channels, co-association of Sur1 with Trpm4 appears to favor noncomplex glycosylation. Co-immunoprecipitation of Sur1 and Trpm4 revealed predominantly core-glycosylated forms of both proteins, not only in the heterologous COS-7 co-expression system but also in tissues from spinal cord injury. Because complex glycosylation occurs in the Golgi apparatus, the finding that co-associated Sur1 and Trpm4 are only core-glycosylated suggests that they associate in the endoplasmic reticulum, before transit to the Golgi, and that co-association hinders complex glycosylation of the assembled Sur1-Trpm4 channel.
Our patch clamp experiments demonstrated that co-assembly of Sur1 with Trpm4 has important functional implications. By co-assembling with Sur1, Trpm4 gained sensitivity to diazoxide and to sulfonylurea drugs such as glibenclamide. Also, Trpm4 doubled its apparent affinity for CaM, resulting in a doubling of its sensitivity to physiological concentrations of intracellular Ca2+. Given that the primary identified function of Trpm4 is to act as negative feedback on Ca2+ influx (4, 5), we speculate that the increase in sensitivity to Ca2+ may be the principal adaptive advantage conveyed by the association with Sur1. To date, Sur1 coupling with Trpm4 has been identified only under conditions of acute CNS injury, suggesting that de novo up-regulation of Sur1-Trpm4 channels may serve a role in protecting cells from the excess Ca2+ influx frequently encountered under pathological conditions involving the CNS.
The co-association of Trpm4 with Sur1 also confers sensitivity to Mg2+-ATP. As compared with Trpm4 alone, exposure of Sur1-Trpm4 channels to Mg2+ greatly amplified the effect of a subsequent decrease in the intracellular ATP concentration. This effect of Sur1 coupling may be of biological significance in CNS ischemia/hypoxia or other conditions associated with severe ATP depletion, wherein the unchecked influx of monovalent cations resulting from ATP depletion would predispose to oncotic cell swelling and necrotic cell death (28, 34, 37). This specific effect of coupling Sur1 and Trpm4 may account for the observations that pharmacological inhibition or gene suppression of either Sur1 or Trpm4 is highly protective in a variety of CNS injuries (10); the presence of either Sur1 or Trpm4 alone is not harmful, but when they co-associate, there is a much greater likelihood that ATP depletion will have a rapid, deadly effect on the cell.
Acknowledgments
We thank Dr. Joseph Bryan, Pacific Northwest Diabetes Research Institute, Seattle, WA, for providing the hexahistidine-fused hamster Sur1 expression plasmid, pcDNA-His6-Sur1; Dr. Show-Ling Shyng, Oregon Health and Science University, Portland, OR, for providing the FLAG epitope fused hamster Sur1 expression plasmid, pECE-FLAG-Sur1; Dr. L. Eric Huang, University of Utah, Salt Lake City, UT, for providing the human Hif1α plasmid, pHA-Hif1α; and Dr. Thomas Blanpied, University of Maryland, Baltimore, MD, for providing the Citrine and Cerulean expression plasmids, Citrine-1 and Cerulean-1. We are grateful to Dr. Zhihua Geng for purifying proteins used for antibody production; to Dr. Svetlana Ivanova for immunolabeling tissues; and to Dr. Orest Tsymbalyuk for performing surgeries for spinal cord injury.
This work was supported, in whole or in part, by National Institutes of Health Grants NS060801 and NS061808 (NINDS) and HL082517 (NHLBI) (to J. M. S.) and NS061934 and NS072501 (NINDS) (to V. G.). This work was also supported by grants from the Department of Veterans Affairs (Baltimore, MD), the Department of the Army (Grant W81XWH 1010898), and the Christopher and Dana Reeve Foundation (to J. M. S.). J. M. S. holds a United States patent (Number 7,285,574), “A novel non-selective cation channel in neural cells and methods for treating brain swelling.” J. M. S. is a member of the scientific advisory board and holds shares in Remedy Pharmaceuticals. No support, direct or indirect, was provided to J. M. S., or for this project, by Remedy Pharmaceuticals.
- Trpm4
- transient receptor potential melastatin 4
- Sur1
- sulfonylurea receptor 1
- Ci
- Citrine
- Ce
- Cerulean
- IP
- immunoprecipitation
- Co-IP
- co-immunoprecipitation
- CG
- core-glycosylated
- HG
- highly glycosylated
- CaM
- calmodulin.
REFERENCES
- 1. Launay P., Fleig A., Perraud A. L., Scharenberg A. M., Penner R., Kinet J. P. (2002) TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 109, 397–407 [DOI] [PubMed] [Google Scholar]
- 2. Nilius B., Prenen J., Tang J., Wang C., Owsianik G., Janssens A., Voets T., Zhu M. X. (2005) Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. J. Biol. Chem. 280, 6423–6433 [DOI] [PubMed] [Google Scholar]
- 3. Song M. Y., Yuan J. X. (2010) Introduction to TRP channels: structure, function, and regulation. Adv. Exp. Med. Biol. 661, 99–108 [DOI] [PubMed] [Google Scholar]
- 4. Vennekens R., Nilius B. (2007) Insights into TRPM4 function, regulation and physiological role. Handb. Exp. Pharmacol. 269–285 [DOI] [PubMed] [Google Scholar]
- 5. Guinamard R., Sallé L., Simard C. (2011) The non-selective monovalent cationic channels TRPM4 and TRPM5. Adv. Exp. Med. Biol. 704, 147–171 [DOI] [PubMed] [Google Scholar]
- 6. Aguilar-Bryan L., Clement J. P., 4th, Gonzalez G., Kunjilwar K., Babenko A., Bryan J. (1998) Toward understanding the assembly and structure of KATP channels. Physiol. Rev. 78, 227–245 [DOI] [PubMed] [Google Scholar]
- 7. Nichols C. G. (2006) KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476 [DOI] [PubMed] [Google Scholar]
- 8. Bryan J., Muñoz A., Zhang X., Düfer M., Drews G., Krippeit-Drews P., Aguilar-Bryan L. (2007) ABCC8 and ABCC9: ABC transporters that regulate K+ channels. Pflugers Arch. 453, 703–718 [DOI] [PubMed] [Google Scholar]
- 9. Aittoniemi J., Fotinou C., Craig T. J., de Wet H., Proks P., Ashcroft F. M. (2009) Review. SUR1: a unique ATP-binding cassette protein that functions as an ion channel regulator. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 257–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simard J. M., Woo S. K., Schwartzbauer G. T., Gerzanich V. (2012) Sulfonylurea receptor 1 in central nervous system injury: a focused review. J. Cereb. Blood Flow Metab. 32, 1699–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nichols C. G., Shyng S. L., Nestorowicz A., Glaser B., Clement J. P., 4th, Gonzalez G., Aguilar-Bryan L., Permutt M. A., Bryan J. (1996) Adenosine diphosphate as an intracellular regulator of insulin secretion. Science 272, 1785–1787 [DOI] [PubMed] [Google Scholar]
- 12. Winkler M., Lutz R., Russ U., Quast U., Bryan J. (2009) Analysis of two KCNJ11 neonatal diabetes mutations, V59G and V59A, and the analogous KCNJ8 I60G substitution: differences between the channel subtypes formed with SUR1. J. Biol. Chem. 284, 6752–6762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simard J. M., Woo S. K., Norenberg M. D., Tosun C., Chen Z., Ivanova S., Tsymbalyuk O., Bryan J., Landsman D., Gerzanich V. (2010) Brief suppression of Abcc8 prevents autodestruction of spinal cord after trauma. Sci. Transl. Med. 2, 28ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. König P., Krasteva G., Tag C., König I. R., Arens C., Kummer W. (2006) FRET-CLSM and double-labeling indirect immunofluorescence to detect close association of proteins in tissue sections. Lab. Invest. 86, 853–864 [DOI] [PubMed] [Google Scholar]
- 15. Hyun Y. M., Chung H. L., McGrath J. L., Waugh R. E., Kim M. (2009) Activated integrin VLA-4 localizes to the lamellipodia and mediates T cell migration on VCAM-1. J. Immunol. 183, 359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen M., Simard J. M. (2001) Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J. Neurosci. 21, 6512–6521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen M., Dong Y., Simard J. M. (2003) Functional coupling between sulfonylurea receptor type 1 and a nonselective cation channel in reactive astrocytes from adult rat brain. J. Neurosci. 23, 8568–8577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horn R., Marty A. (1988) Muscarinic activation of ionic currents measured by a new whole-cell recording method. J. Gen. Physiol 92, 145–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hamill O. P., Marty A., Neher E., Sakmann B., Sigworth F. J. (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391, 85–100 [DOI] [PubMed] [Google Scholar]
- 20. Sala-Rabanal M., Wang S., Nichols C. G. (2012) On potential interactions between non-selective cation channel TRPM4 and sulfonylurea receptor SUR1. J. Biol. Chem. 287, 8746–8756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clement J. P., 4th, Kunjilwar K., Gonzalez G., Schwanstecher M., Panten U., Aguilar-Bryan L., Bryan J. (1997) Association and stoichiometry of KATP channel subunits. Neuron 18, 827–838 [DOI] [PubMed] [Google Scholar]
- 22. Yoshida H., Feig J. E., Morrissey A., Ghiu I. A., Artman M., Coetzee W. A. (2004) KATP channels of primary human coronary artery endothelial cells consist of a heteromultimeric complex of Kir6.1, Kir6.2, and SUR2B subunits. J. Mol. Cell Cardiol. 37, 857–869 [DOI] [PubMed] [Google Scholar]
- 23. Meissner M., Obmann V. C., Hoschke M., Link S., Jung M., Held G., Philipp S. E., Zimmermann R., Flockerzi V. (2011) Chapter 6: Lessons of Studying TRP Channels with Antibodies. in: TRP Channels (Zhu M. X., ed) pp. 135–148 CRC Press, Boca Raton, FL: [PubMed] [Google Scholar]
- 24. Sharma N., Crane A., Clement J. P., 4th, Gonzalez G., Babenko A. P., Bryan J., Aguilar-Bryan L. (1999) The C terminus of SUR1 is required for trafficking of KATP channels. J. Biol. Chem. 274, 20628–20632 [DOI] [PubMed] [Google Scholar]
- 25. Zerangue N., Schwappach B., Jan Y. N., Jan L. Y. (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron 22, 537–548 [DOI] [PubMed] [Google Scholar]
- 26. Conti L. R., Radeke C. M., Vandenberg C. A. (2002) Membrane targeting of ATP-sensitive potassium channel: effects of glycosylation on surface expression. J. Biol. Chem. 277, 25416–25422 [DOI] [PubMed] [Google Scholar]
- 27. Aguilar-Bryan L., Bryan J. (1999) Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 20, 101–135 [DOI] [PubMed] [Google Scholar]
- 28. Gerzanich V., Woo S. K., Vennekens R., Tsymbalyuk O., Ivanova S., Ivanov A., Geng Z., Chen Z., Nilius B., Flockerzi V., Freichel M., Simard J. M. (2009) De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat. Med. 15, 185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shyng S., Ferrigni T., Nichols C. G. (1997) Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J. Gen. Physiol 110, 643–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mikhailov M. V., Mikhailova E. A., Ashcroft S. J. (2001) Molecular structure of the glibenclamide binding site of the β-cell KATP channel. FEBS Lett. 499, 154–160 [DOI] [PubMed] [Google Scholar]
- 31. Vila-Carriles W. H., Zhao G., Bryan J. (2007) Defining a binding pocket for sulfonylureas in ATP-sensitive potassium channels. FASEB J. 21, 18–25 [DOI] [PubMed] [Google Scholar]
- 32. Demion M., Bois P., Launay P., Guinamard R. (2007) TRPM4, a Ca2+-activated nonselective cation channel in mouse sino-atrial node cells. Cardiovasc. Res. 73, 531–538 [DOI] [PubMed] [Google Scholar]
- 33. Gillis K. D., Gee W. M., Hammoud A., McDaniel M. L., Falke L. C., Misler S. (1989) Effects of sulfonamides on a metabolite-regulated ATPi-sensitive K+ channel in rat pancreatic B-cells. Am. J. Physiol. 257, C1119–C1127 [DOI] [PubMed] [Google Scholar]
- 34. Simard J. M., Woo S. K., Gerzanich V. (2012) Transient receptor potential melastatin 4 and cell death. Pflugers Arch. 464, 573–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tucker S. J., Gribble F. M., Zhao C., Trapp S., Ashcroft F. M. (1997) Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature 387, 179–183 [DOI] [PubMed] [Google Scholar]
- 36. Simard J. M., Tsymbalyuk O., Ivanov A., Ivanova S., Bhatta S., Geng Z., Woo S. K., Gerzanich V. (2007) Endothelial sulfonylurea receptor 1-regulated NCCa-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. J. Clin. Invest. 117, 2105–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simard J. M., Chen M., Tarasov K. V., Bhatta S., Ivanova S., Melnitchenko L., Tsymbalyuk N., West G. A., Gerzanich V. (2006) Newly expressed SUR1-regulated NCCa-ATP channel mediates cerebral edema after ischemic stroke. Nat. Med. 12, 433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu H., El Zein L., Kruse M., Guinamard R., Beckmann A., Bozio A., Kurtbay G., Mégarbané A., Ohmert I., Blaysat G., Villain E., Pongs O., Bouvagnet P. (2010) Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ. Cardiovasc. Genet. 3, 374–385 [DOI] [PubMed] [Google Scholar]