

Background: ATG4 is a cysteine peptidase crucial for macroautophagy.
Results: Gene deletion mutants show that the two ATG4s of Leishmania perform distinct roles, although there is some redundancy.
Conclusion: ATG4s are not individually essential but macroautophagy, a process important in the virulence of the parasite, requires one.
Significance: Highlights the distinct roles of ATG4 isoforms and their importance for autophagy and parasite infectivity.
Keywords: Autophagy, Cysteine Protease, Leishmania, Parasite, Parasite Metabolism, Peptidases, Protease, ATG4
Abstract
Macroautophagy in Leishmania, which is important for the cellular remodeling required during differentiation, relies upon the hydrolytic activity of two ATG4 cysteine peptidases (ATG4.1 and ATG4.2). We have investigated the individual contributions of each ATG4 to Leishmania major by generating individual gene deletion mutants (Δatg4.1 and Δatg4.2); double mutants could not be generated, indicating that ATG4 activity is required for parasite viability. Both mutants were viable as promastigotes and infected macrophages in vitro and mice, but Δatg4.2 survived poorly irrespective of infection with promastigotes or amastigotes, whereas this was the case only when promastigotes of Δatg4.1 were used. Promastigotes of Δatg4.2 but not Δatg4.1 were more susceptible than wild type promastigotes to starvation and oxidative stresses, which correlated with increased reactive oxygen species levels and oxidatively damaged proteins in the cells as well as impaired mitochondrial function. The antioxidant N-acetylcysteine reversed this phenotype, reducing both basal and induced autophagy and restoring mitochondrial function, indicating a relationship between reactive oxygen species levels and autophagy. Deletion of ATG4.2 had a more dramatic effect upon autophagy than did deletion of ATG4.1. This phenotype is consistent with a reduced efficiency in the autophagic process in Δatg4.2, possibly due to ATG4.2 having a key role in removal of ATG8 from mature autophagosomes and thus facilitating delivery to the lysosomal network. These findings show that there is a level of functional redundancy between the two ATG4s, and that ATG4.2 appears to be the more important. Moreover, the low infectivity of Δatg4.2 demonstrates that autophagy is important for the virulence of the parasite.
Introduction
Macroautophagy (hereafter autophagy) is an intracellular process that sequesters cytosol and organelles within autophagosomes for delivery to and degradation within lysosomes. One main physiological significance of this process in most cell types is recycling of intracellular materials during nutrient deprivation, but in addition it plays important parts in cellular differentiation, tissue remodeling, growth control, size regulation, mitochondrial homeostasis, cellular immunity, adaptation to stresses, and unconventional protein secretion (1–6). The many steps comprising autophagy rely on a large repertoire of proteins mainly from the ATG2 family, of which more than 30 have been described. Central to the process are the ATG5-ATG12 conjugation and ATG8-lipidation pathways, which are coordinated by the activity of the ATG6-Vps34 complex (7–9).
ATG4, a cysteine peptidase of Clan CA, family C54, is a key component of the ATG8-lipidation pathway and is essential for autophagosome biogenesis. ATG4 is responsible for cleavage of ATG8 after a glycine residue close to its C terminus, facilitating conjugation of ATG8 to the lipid phosphatidylethanolamine (PE). ATG8-PE attaches to the phagophore and is involved in elongation of the phagophore and closure of the autophagosome. Subsequently, ATG4 cleaves ATG8 from the PE on the outside of the autophagosome membrane, the reverse of the initial conjugation reaction (10–13), which allows interaction and fusion of the autophagosome with the endosomal-lysosomal compartments (14–16). The conjugation and deconjugation activities of ATG4 are crucial for autophagosome genesis and function.
Mammals and plants have multiple orthologues of the single yeast ATG4. There appears to be some functional redundancy (17) but the four ATG4 orthologues in humans have distinct roles in autophagosome genesis and cell survival, which is reflected in their preferred substrates among the six ATG8 orthologues. ATG4B has activity toward all of the ATG8 homologues and is the key regulator for autophagy in humans, its absence has severe consequences in the cell (18–21). ATG4D is predominantly involved in mitophagy and apoptosis (22, 23), whereas ATG4C has restricted substrate specificity and is only required for survival during prolonged stress (19).
Human ATG4A and ATG4B are redox regulated, they are the target for direct but reversible oxidation of a regulatory cysteine residue by reactive oxygen species (ROS), which inactivates the deconjugation activity (24, 25). The catalytic cysteine of ATG4 that processes ATG8 prior to its conjugation to PE is not susceptible to ROS oxidation. Its continued activity ensures that the lipidated ATG8 is formed and, in the absence of the deconjugation activity of ATG4, accumulates within the cell and is available for increased autophagosome biogenesis under conditions of oxidative stress and starvation. Moreover, ROS can up-regulate the expression of ATG4 in a JNK signaling-dependent manner to ensure that an adequate amount of processed ATG8 is available for autophagosome biogenesis after autophagic induction in MEF-7 cells (26). This up-regulation of ATG4 expression can be abrogated by either JNK knockdown or addition of the ROS scavenger N-acetylcysteine (NAC). The relationship between ROS and JNK activation was established previously (27, 28), but this link with ATG4 demonstrates a clear correlation between ROS production and the induction of autophagy. Oxidative stress caused by high ROS levels causes extensive and irreversible structural changes to proteins and organelles within cells (29, 30), thus the induction of autophagy is a mechanism to maintain quality control, cellular homeostasis, and cellular energetic balance (24). Further evidence of this link comes from the findings that autophagy-deficient mutants have high ROS levels. Human ATG4C and ATG4D are not redox regulated in the same way and neither are the ATG4s of yeast and plants, suggesting that these latter organisms must deal with ROS stress differently.
Leishmania, the causative agent of important diseases known as the leishmaniases, which are prevalent in many tropical and subtropical regions, carries out autophagy, comprising two conjugation pathways similar to yeast and higher eukaryotes, which is important for cellular differentiation, mitochondrial function, and phospholipid homeostasis (31–33). It contains two genes encoding ATG4, the two enzymes are designated ATG4.1 and ATG4.2 and share only 19% identity (33). They have differential activity toward the unusual four classes of ATG8 of the parasite (32). ATG4.1 preferentially processes ATG8, ATG8B, and ATG8C, whereas ATG4.2 preferentially cleaves ATG8A (32). This suggests that the two forms of ATG4 have differing roles in the parasite.
The main aim of this study was to determine whether the two ATG4s are individually essential for Leishmania major and to elucidate the effects that removal of each ATG4 gene has on the parasite. Our approach using targeted gene deletion has also enabled us to obtain insights into the roles of each enzyme, including the parts that they may play in dealing with ROS, the relationship between ROS and autophagy in Leishmania, and the importance of the ATG4s for parasite survival and infectivity.
EXPERIMENTAL PROCEDURES
Parasite Handling Procedures
Promastigotes of L. major (MHOM/IL/80/Friedlin, designated WT for this study) were grown in modified Eagle's medium (designated complete HOMEM medium) with 10% (v/v) heat-inactivated fetal calf serum at 25 °C, as described previously (34). Parasite numbers were estimated using an improved Neubauer hemocytometer. In this report, except when stated otherwise, early log, mid-log, and early stationary phases of promastigote growth correspond to ∼5 × 105, 5 × 106, and 9 × 106 parasites ml−1, respectively. The following antibiotics were added to the cultures of the Δatg4 mutants and the derived cell lines as follows: hygromycin B (Sigma) at 50 μg ml−1; phleomycin (Cayla, France) at 10 μg ml−1, puromycin (Calbiochem) at 10 μg ml−1; blasticidin S (Calbiochem) at 10 μg ml−1; and neomycin (G418, Geneticin, Invitrogen) at 25 μg ml−1.
Metacyclogenesis and Infectivity of Leishmania to Macrophages and Mice
Metacyclic promastigotes in L. major stationary phase cultures were assessed by using either the peanut agglutinin method (35) or Western blot analysis using antibodies to the metacyclic promastigote-specific protein, HASPB (see below).
Infectivity of Leishmania lines to macrophages was determined using peritoneal macrophages from CD1 mice resuspended in RPMI media (Sigma) at 5 × 105 ml−1 and left to adhere onto coverslips overnight. These cells were infected with stationary phase promastigote cultures or amastigotes harvested from mice footpad lesions at a ratio of ∼2 promastigotes or 0.5 amastigotes per macrophage and incubated for up to 5 days at 32 °C in 5% CO2, 95% air. Non-phagocytosed promastigotes or amastigotes were removed after 24 h by washing four times with RPMI. Parasite abundance within the macrophages after 1 and 5 days was determined after the cells were fixed in methanol and stained with Giemsa for 10 min.
The infectivity to mice was determined using groups of 5 mice that were inoculated subcutaneously within a footpad with 5 × 105 stationary phase L. major promastigotes or amastigotes, harvested from mice footpad lesions, suspended in 200 μl of PBS, pH 7.4. The thickness of the lesion in infected footpads was measured using a caliper over a 5–12-week period.
Amastigote Isolation from Infected Mice and Transformation to Promastigotes
Amastigotes were excised, into cold PBS containing 50 μg ml−1 of gentamycin (Sigma), from footpad lesions of mice, inoculated 5 weeks earlier with 5 × 105 stationary phase promastigotes, after removal of the skin. The lesion material was ground in a glass tissue grinder and centrifuged at 150 × g for 1 min at 4 °C to remove the large debris. The supernatant was then centrifuged at 1700 × g for 15 min and the subsequent pellet was resuspended and treated as required by the design of the experiment.
The number of L. major amastigotes in footpad lesions of infected mice was determined using the limiting dilution assay as described previously (36). Briefly, amastigotes of L. major were excised from footpads and resuspended in 10 ml of complete HOMEM medium with 50 μg ml−1 of gentamycin. The parasite suspension was serially diluted in duplicate flasks, incubated for 5 days at 26 °C, and then inspected daily for parasite growth. For quantifying the morphological forms in these cultures, parasites within the culture flasks were classified according to the following criteria: amastigotes, ovoid to round but lacking an emergent flagellum, or promastigotes, elongated bodies with a flagellum equal to or longer than the cell body length. Other parasites were designated as intermediate forms. A minimum of 200 cells was examined and the resulting differential counts were expressed as percentages.
Western Blot Analyses
Parasites were harvested at 1,000 × g for 10 min, washed twice in PBS, and the pellets were either used immediately or stored at −20 °C. Parasite lysates used for this analysis were produced by resuspension of parasite pellets in lysis buffer, comprising 0.25 m sucrose, 0.25% Triton X-100, 10 mm EDTA, and a mixture of peptidase inhibitors (10 μm E-64, 2 mm 1,10-phenanthroline, 4 μm pepstatin A, and 1 mm phenylmethylsulfonyl fluoride). After a 10-min incubation in ice, lysates were centrifuged at 15,700 × g for 30 min at 4 °C, and the resulting supernatant (designated soluble fraction) was retained for analysis. Protein concentrations were determined according to the Bradford procedure (Bio-Rad) using bovine serum albumin as the protein standard.
Western blot analysis was performed on L. major lysates prepared as described above. The primary anti-GFP (Abcam Ltd.) and the secondary anti-rabbit antibodies were used at a 1:2,000 (v/v) and 1:5,000 (v/v) dilutions, respectively. The primary anti-His (Invitrogen) and the secondary anti-mouse antibodies were used at 1:1,000 (v/v) and 1:50,000 (v/v), respectively. To assess metacyclogenesis, the metacyclic promastigote-specific proteins SHERP and HASPB were monitored using anti-SHERP and anti-HASPB antibodies (produced in rabbit) at a dilution of 1:5,000 (v/v). The antibodies were kindly provided by Prof. Deborah Smith (University of York). Loading controls used the anti-TbEF1α antibody (Upstate) at a dilution of 1:5,000 (v/v). The West-Pico chemiluminescence detection system (Pierce) was used to visualize antigens.
Generation of L. major ATG4 Null Mutants and Re-expressing Cell Lines
To generate the ATG4.1 null mutant, the 1014-bp 5′ flank fragment of ATG4.1 (LmjF32.3890) was amplified by PCR from L. major genomic DNA with primers NT164 (5′-AAG CTT CCT CGT CAC CTT CCT CGA TGA CAG GCG GGT-3′) and NT165 (5′-GTC GAC TCC GGG GGT GCA GAC GGG GCT GTA G-3′), digested with HindIII and SalI, and inserted into the HindIII/SalI-digested pGL345-HYG (37) to give pGL345ATG4.1-HYG5′. The 3′ flank fragment was amplified by PCR with primers NT166 (5′-CCC GGG TCG TAG CGC CAG GAC GCT GAT CGT CAT AGC-3′) and NT167 (5′-AGA TCT ATC ACG CTG CAC AGT ATC ACC CGG T-3′). The resulting 1008-bp fragment was digested with SmaI and BglII and cloned into the same restriction sites in the pGL345ATG4.1-HYG5′ plasmid to give pGL345ATG4.1HYG5′3′. The cassette used for transfection of L. major promastigotes was released by HindIII/BglII digestion. The pGL345ATG4.1BSD5′3′ plasmid used for replacement of the second allele was generated from plasmid pGL345ATG4.1HYG5′3′ by replacing the SpeI/BamHI ORF of the hygromycin resistance gene with a SpeI/BamHI ORF of the blasticidin S-deaminase gene. A population of parasites resistance to hygromycin was generated after transfection with pGL345ATG4.1HYG5′3′ (see below). This population was used for a second round of transfection with the pGL345ATG4.1BSD5′3′ construct. Two blasticidin-resistant clones from independent events, designated Δatg4.1a and Δatg4.1b, were selected for analysis. The generation of the Δatg4.2 line has been described previously (33).
To generate the lines re-expressing ATG4.1, the ATG4.1 gene modified with a polyhistidine tag at the C-terminal end (to verify protein expression) was inserted in the pRIB-Pur bearing the puromycin resistance gene (38). PCR primers NT158 (5′-CTC GAG ATG GGC ACG AAC GCC AAA GTG GCA GAG-3′) and NT159 (5′-ATC GAT CTA ATG ATG ATG ATG GCT CGG TGG AGA AGA GAT TGA ATT-3′) produced the polyhistidine-tagged version of ATG4.1 containing BglII and BamHI sites, respectively. The 1077-bp fragment was digested by BglII/BamHI and cloned into pRIB-Pur vector previously digested with the same restriction enzymes to give the pRIB-PurATG4.1 plasmid. The integration cassette from this plasmid was excised by digestion with PacI and PmeI before electroporation of L. major promastigotes. This fragment possesses the 5′ and 3′ flanking regions for chromosomal integration at the ribosomal locus. Proteins at this locus are constitutively expressed in both promastigote and amastigote life cycle forms (39). This cell line was designated Δatg4.1::ATG4.1. The pRIB-PurATG4.2 plasmid was made similarly using the polyhistidine ATG4.2 but was amplified using primers NT160 (5′-CTC GAG ATG GCT CCG CTC CGT GCA AGA TT-3′) and NT161 (5′-AGA TCT TCA ATG ATG ATG ATG ATG ATG ATG ATC CAG ATA CTC CCA-3′). The resultant cell line was designated Δatg4.2::ATG4.2. The creation of cell lines expressing GFP-ATG8 and ATG4.2 have been described previously (33).
L. major promastigotes were electroporated (NucleofectorTM) with 15 μg of DNA of the extra chromosomal constructs, namely pN-GFP-ATG8 and pN-ATG4.2, and 30 μg of the pRIB-PurATG4.1 or pRIB-PurATG4.2 cassettes. Transfectants were selected with the appropriate antibiotic at the concentrations detailed above. All PCR assays were carried out in a GeneAmp 9600 PCR system (PerkinElmer Life Sciences) for 30 cycles of denaturation (94 °C, 15 s), annealing (65 °C, 15 s), and extension (72 °C, 2 min). For the attempts to generate the ATG4 double null, the pGL345ATG4.1NEO5′3′ and pGL345ATG4.1BLE5′3′ plasmids were generated from plasmid pGL345ATG4.1HYG5′3′ by replacing the SpeI/BamHI ORF of the hygromycin resistance gene with SpeI/BamHI ORFs of the neomycin resistance and bleomycin resistance genes, respectively.
Southern Blot Analyses
Genomic DNA was extracted according to standard procedures (40). DNA (10 μg) was digested with XhoI, fractionated by agarose gel electrophoresis, nicked, denatured, neutralized, and blotted onto HybondTM-N+ membrane (Amersham Biosciences) by capillary transfer. The probe was prepared from a 1014-bp HindIII/SalI 5′ flank fragment excised from the T-vector (Promega) using a random priming kit (Prime-It; Stratagene) and purified on Microspin S-200 HR columns (Amersham Biosciences). Filters was hydridized overnight at 65 °C with a [α-32P]dATP-labeled 5′ flank fragment probe in Church-Gilbert hybridization solution. Filters were washed under high stringency (15 min at 65 °C with 2× SSC, 0.1% SDS and twice with 0.2× SSC, 0.1% SDS) and exposed to x-ray film (Konica Medical Film).
Monitoring and Quantification of Autophagy
For live imaging, promastigotes of WT[pN-ATG8], Δatg4.1[pN-ATG8], Δatg4.1::ATG4.1[pN-ATG8], Δatg4.2[pN-ATG8], and Δatg4.2::ATG4.2[pN-ATG8] in HOMEM growth medium were mounted on coverslips and observed by fluorescence microscopy equipped with a FITC filter set and images were captured. The occurrence of GFP-ATG8-containing puncta (putative autophagosomes) within these cells was assessed in at least 100 cells from 3 independent experiments.
Use of H2DCF-DA, Alamar Blue, MitoTracker Red, and MitoTracker Green
For assessing the levels of ROS in the promastigotes, cells at 1 × 107 promastigotes ml−1 were harvested by centrifugation, washed once in serum-free HOMEM, and 2 × 106 cells in 200 μl were incubated with 0.1 mm H2DCF-DA (Molecular Probes) for 2 h at 26 °C and fluorescence was measured using a microtiter plate reader (excitation 380–420 nm, barrier filter 520 nm). The Alamar Blue assay was used to evaluate metabolic activity and cell viability, 4 × 106 promastigotes of WT and Δatg4 lines at log phase of growth were subjected to starvation conditions (PBS) for up to 8 h at 26 °C or normal growth conditions. Alamar Blue (resazurine salt, Sigma) was added to a final concentration of 0.0125% for the final hour of incubation and its reduction was measured using the fluorescence microtiter plate reader (excitation 550 nm, barrier filter 590 nm). For assessing mitochondrial morphology and mitochondrial membrane potential of the respective lines, 107 promastigotes were incubated with either 0.2 μm MitoTracker GreenTM (Invitrogen) or 0.1 μm MitoTracker Red CMXRos (Invitrogen), respectively, for 30 min at 26 °C. Promastigotes were then washed in PBS and either assessed for fluorescence (of MitoTracker Red at excitation 579 nm, barrier filter 599 nm; of MitoTracker Green at excitation 490 nm, barrier filter 516 nm) using a microtiter plate reader or mounted on glass slides for live imaging analysis by fluorescence microscopy using a Nikon TE2000S inverted microscope equipped with FITC and mCherry filter sets. Images were processed using IPlabs 3.7 image processing software (BD Biosciences Bioimaging). For assessing the relationship between autophagy, ROS and mitochondrial morphology and function, NAC at 2.5 or 5 mm was included in some experiments.
Detection of Oxidized Proteins
Carbonylated proteins were detected with an Oxyblot protein oxidation detection kit (Chemicon International Ltd.) according to the manufacturer's protocol. Briefly, promastigote lysates containing 50 μg of protein were incubated with 2,4-dinitrophenylhydrazine for 20 min at room temperature, resolved by SDS-PAGE, and detected by Western blot analysis with anti-2,4-dinitrophenol rabbit antibodies (diluted at 1:200).
Scanning Electron Microscopy (SE) Analyses
Parasites isolated from mice were fixed with 2.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, for 40 min and dehydrated. Samples were dried prior to mounting on an aluminum stub using either silver paint or double stick carbon tape. Samples were then introduced into the chamber of the sputter coater and coated with a very thin film of gold/palladium before SE examination. Body lengths of WT and transgenic lines were measured using the ESI Vision 3.2 Image analysis software (Olympus Soft Imaging Solutions). A minimum of 200 cells was examined for each sample.
Data Processing
Experimental data from macrophage infections, mice infectivity, Alamar Blue, MitoTracker Red, MitoTracker Green, and H2DCF-DA assays were pooled for comparison using unpaired t tests. A p value of <0.05 was used as the level of significance. Counts of puncta from ATG-tagged proteins and the measurements of body lengths of promastigotes were performed on at least 100 cells and analyzed statistically as described above.
RESULTS
Targeted Deletion of the L. major ATG4 Genes
We have described previously the generation of the ATG4.2 null mutant (Δatg4.2) and an add-back line (Δatg4.2::ATG4.2) and how analysis of the mutants demonstrated that the cysteine peptidase ATG4.2 is important for differentiation between developmental forms of Leishmania (33). To facilitate a comparison between the contributions of the two ATG4s in the biology of the parasite, we generated a ATG4.1 gene deficient cell line (Fig. 1A). The two independent clones obtained (designated Δatg4.1a and Δatg4.1b) were confirmed by Southern blot analysis to lack the ATG4.1 gene and have the correct integration of the replacement cassette (Fig. 1B). Hybridization of XhoI-digested genomic DNA with a 5′-flank probe revealed a 7.6-kb product corresponding to the ATG4.1 locus of the WT parasites, which is absent in the Δatg4.1 lines. In these, a DNA fragment of ∼5.0 kb is visible, consistent with correct integration of the HYG and BSD drug resistance markers in the ATG4.1 locus. As a control, ATG4.1 modified with a C-terminal His6 tag was re-expressed in the Δatg4.1 lines (to give Δatg4.1::ATG4.1). Successful expression of ATG4.1 was confirmed by detection of a 43-kDa protein, which was absent from lysates of WT and Δatg4.1 promastigotes, by Western blot analysis with anti-His antibody (Fig. 1C). Subsequent phenotypic analyses were performed on both Δatg4.1 clones, but as they behaved similarly data are presented for Δatg4.1a only (hereafter referred to as Δatg4.1). Although the Δatg4.1 and Δatg4.2 null mutants were readily obtained by two successive rounds of targeting their endogenous loci, multiple attempts to generate a line that lacked both ATG4.1 and ATG4.2 genes were unsuccessful. Single allele deletions of the second ATG4 gene in both Δatg4.1 and Δatg4.2 lines were successful (to give Δatg4.2/atg4.1−/+ and Δatg4.1/atg4.2−/+, respectively), but the second round of allele replacement using these lines resulted in trisomic parasite lines in which both selectable markers had integrated into the appropriate endogenous locus but there appeared an additional copy of the endogenous gene. As both of these loci had been successfully targeted in generating the Δatg4.1 and Δatg4.2 mutants, the results of these genetic manipulation attempts suggest strongly that the parasite requires at least one copy of either ATG4.1 or ATG4.2.
FIGURE 1.

Generation of the ATG4.1 null mutant. A, schematic representation of the ATG4.1 locus and the plasmid constructs used for gene replacement. The ATG4.1 and antibiotic resistance genes are shown as arrows, flanking DNA sequences are shown as boxes. Restriction enzymes used for the different constructs and the expected sizes of the XhoI fragments are shown. 5′-DHFR and 3′-DHFR, dihydrofolate reductase flanking regions; BSD, blasticidin resistance gene; HYG, hygromycin resistance gene. B, Southern blot analysis of genomic DNA digested with XhoI, separated on a 1% agarose gel, blotted onto a nylon membrane, and hybridized with a 32P-labeled DNA probe corresponding to the 5′-flanking region of ATG4.2. C, Western blot analysis using the α-His antibodies of extracts from early stationary phase promastigotes. EF1α served as a loading control.
L. major Mutants Lacking ATG4s Form GFP-ATG8-containing Puncta
Δatg4.1 and Δatg4.2 both grew indistinguishably from WT as promastigotes in vitro. GFP-ATG8 has been validated as a molecular marker for autophagy in Leishmania and has been used to show that autophagy increases during differentiation between life cycle stages with the abundance of autophagosomes in promastigotes being greatest during metacyclogenesis (33, 41). Thus we used this marker protein with the mutant lines to investigate the involvement of the two ATG4s in autophagy. The lines were transfected with pN-GFP-ATG8 and the occurrence of GFP-labeled puncta was monitored (Fig. 2A). The Δatg4.1 lines (designated Δatg4.1[pN-ATG8] and Δatg4.1::ATG4.1[pN-ATG8]) at the mid-logarithmic phase of growth produced one to two GFP-labeled autophagosomes per cell, which was similar to WT cells. In contrast Δatg4.2[pN-ATG8] had multiple puncta. With Δatg4.1, the proportion of cells bearing the GFP-labeled structures peaked at day 7 (when metacyclogenesis was maximal), similarly to the situation with WT promastigotes although the number of cells with puncta was lower (Fig. 2B). The proportion of cells bearing GFP-labeled structures returned to WT levels in Δatg4.1::ATG4.1[pN-ATG8]. In both cases, the proportion of cells with GFP-labeled puncta subsequently declined to low levels as stationary phase was reached and metacyclogenesis ended. In contrast, Δatg4.2[pN-ATG8] had a higher proportion of cells with autophagosomes at early logarithmic phase of growth, a proportion that increased as the logarithmic phase progressed. Most notably, the cells retained puncta at stationary phase of growth. Δatg4.2 promastigotes also were characterized by the presence of multiple puncta (Fig. 2A), which were restored to WT numbers in Δatg4.2::ATG4.2[pN-ATG8]. Western blot analysis monitored the proportion of cytosolic and lipid-associated forms of ATG8 (GFP-ATG8* and GFP-ATG8-PE, respectively). All the cell lines at stationary phase of growth contained GFP-ATG8-PE, but the Δatg4.2[pN-ATG8] line had a significantly higher proportion of GFP-ATG8-PE relative to GFP-ATG8* (Fig. 2C), which is consistent with the accumulation of autophagosomes as observed by fluorescence microscopy.
FIGURE 2.

Autophagosome formation in promastigotes of the Δatg4 mutants. A, occurrence of GFP-ATG8 puncta in L. major promastigotes in normal medium. The arrows indicate autophagosomes. Scale bar, 10 μm. B, the proportion of GFP-ATG8-expressing promastigotes with autophagosomes was assessed during growth in vitro. Data are mean ± S.D. from three independent experiments. Asterisks denote values statistically different from WT cells, p < 0.05. C, Western blot analysis of GFP-ATG8-PE. Promastigote cell extracts expressing GFP-ATG8 were separated by SDS-PAGE in the presence of 6 m urea and analyzed by Western blotting with an anti-GFP antibody. The faster migrating, lipidated bands are labeled GFP-ATG8-PE, whereas the un-lipidated bands migrating more slowly are labeled GFP-ATG8*. D, the occurrence of GFP-ATG8-labeled autophagosomes resulting from starvation of early log stage promastigotes in PBS for 2 h with or without added NAC. Data are mean ± S.D. from three independent experiments. E, occurrence of GFP-ATG8A puncta in starved L. major promastigotes. The arrows indicate autophagosomes. Scale bar, 10 μm. F, the occurrence of GFP-ATG8A-labeled autophagosomes resulting from starvation of early log stage promastigotes in PBS with or without added NAC. Note the discontinuity in the ordinate axis.
We have shown previously that autophagy in L. major is induced by starvation (33), so we quantified the number of autophagosomes in the mutants after starvation (Fig. 2D). The proportion of cells with GFP-ATG8 puncta increased dramatically in WT, Δatg4.1, Δatg4.1::ATG4.1, and Δatg4.2::ATG4.2 promastigotes. In contrast, the proportion of Δatg4.2 promastigotes with GFP-ATG8 puncta was initially relatively high and this only increased a little under starvation conditions.
The Requirement for the Different ATG4s in Autophagy Involving ATG8A
Previously, we showed that the preferred substrate for ATG4.2 was ATG8A (32). To investigate further the relative roles of ATG4.1 and ATG4.2 in the biogenesis of ATG8A-containing puncta, Δatg4.1[pN-ATG8A] and Δatg4.2[pN-ATG8A] lines were generated, incubated in starvation medium, and monitored for the formation of ATG8A-containing puncta (Fig. 2E). Δatg4.1[pN-ATG8A] produced multiple GFP-ATG8A-labeled structures per cell, similar to WT promastigotes. In contrast Δatg4.2[pN-ATG8A] produced no such puncta, as described previously (32). The proportion of promastigotes with GFP-ATG8A puncta under starvation conditions was ∼90% in WT, Δatg4.1, Δatg4.1::ATG4.1, and Δatg4.2::ATG4.2 (Fig. 2F), a great increase from 2 to 5% in non-starved cells. No puncta were detected in Δatg4.2 promastigotes, strongly supporting the hypothesis that ATG8A-dependent puncta formation is totally dependent upon ATG4.2.
Metacyclogenesis and Infectivity of the Δatg4 Mutants
We reported previously that Δatg4.2 promastigotes are defective in metacyclogenesis (33), thus we investigated whether Δatg4.1 has a similar defect. Δatg4.1 promastigotes differ from Δatg4.2 promastigotes in that they apparently differentiated fully to metacyclic promastigotes, as judged by the lectin peanut agglutinin assay (Fig. 3A) and the presence of the metacyclic-specific markers, SHERP and HASPB (Fig. 3B); the process seemed delayed and there was a significant difference in the number of metacyclic promastigotes at day 7 between the WT parasites and Δatg4.1 (p < 0.05), but by day 10 there was no difference (Fig. 3A).
FIGURE 3.

Metacylogenesis and infectivity of L. major ATG4 null mutants. A, the percentage of metacyclic promastigotes in stationary phase populations of promastigotes was determined by the peanut lectin agglutination method. Values shown are the mean ± S.D. from three independent experiments. *, data for Δatg4.1 differed significantly from those for WT promastigotes at day 7 (p < 0.01). B, extracts of 107 stationary phase promastigotes at day 7 (D7) or day 10 (D10) were assessed for metacyclic-specific proteins by Western blot analysis with anti-SHERP and anti-HASPB antibody. Cysteine synthase (CS) served as a loading control. C and D, mouse peritoneal macrophages were infected in vitro with stationary phase promastigotes or amastigotes isolated from lesions in BALB/c mice. Cells were assessed at 1 and 5 days post-infection for the percentage of infected macrophages. Data are mean ± S.D. and are representative of three independent experiments. *, infection level of ATG4 null mutant lines differed significantly from those of WT parasites (p < 0.05). E–H, BALB/c mice were infected with 2 × 107 promastigotes or amastigotes and footpad lesion size was monitored weekly. Data are mean ± S.D. of five mice. *, infection level of ATG4 null mutant lines differed significantly from those of WT or the re-expressing line (p < 0.05).
We compared the two mutants for infectivity to macrophages in vitro and mice. Stationary phase promastigotes of both Δatg4.1 and Δatg4.2 infected macrophages in vitro in similar numbers to WT parasites but each of them survived poorly over 5 days with many macrophages clearing the infection (Fig. 3C). In contrast, amastigotes of Δatg4.1, purified from mice, were as infective as WT amastigotes and proliferated equally well (Fig. 3D). Amastigotes of Δatg4.2 also infected macrophages well but survived poorly over 5 days (Fig. 3D, right panel).
Analysis of the infectivity of Δatg4.1 and Δatg4.2 lines to BALB/c mice using promastigotes and amastigotes yielded results that correlated well with those from the in vitro analysis using macrophages (Fig. 3, E-H). Lesions resulting from inoculation of stationary phase Δatg4.1 and Δatg4.2 promastigotes were slower growing than those of WT parasites, this was reversed by re-expression of the genes (Figs. 3, E and G). In contrast, inoculation of Δatg4.1 amastigotes generated lesions similar to those of WT amastigotes (Fig. 3F), whereas inoculation of Δatg4.2 amastigotes resulted in slower growing lesions (Fig. 3H). Lesion development with amastigotes of Δatg4.2::ATG4.2 was at an intermediate level between WT and Δatg4.2 (Fig. 3H). These findings suggest that both ATG4s play some role in facilitating infection of mice by promastigotes, and that ATG4.2 is also important in the survival and multiplication in mice of inoculated amastigotes, whereas ATG4.1 is not.
Morphology of Δatg4.2 in Mice
The finding that inoculation of Δatg4.2 amastigotes into BALB/c resulted in slower growing lesions than WT parasites prompted us to investigate whether the lesions were equivalent in terms of parasite numbers per volume. The results showed that the number of amastigotes contained within lesions of comparable width was significantly lower for Δatg4.2 than with WT and Δatg4.1 (p < 0.05; Fig. 4A). Scanning electron microscopy (SE) analysis of Δatg4.2 purified from lesions showed that they had a longer body length on average than WT amastigotes (p < 0.05; Fig. 4, B and C), a difference largely reversed by re-expression of ATG4.2 in Δatg4.2::ATG4.2. Δatg4.1 amastigotes were not significantly different in size from WT amastigotes (p > 0.05; data not shown).
FIGURE 4.

Morphology and transformation of amastigotes of Δatg4.2. A, parasite load in footpad lesions of infected BALB/c mice. The lesions from five mice were dissected when they reached 4 mm in thickness, the tissue was disrupted in PBS, and the number of parasites was determined microscopically. The lines indicate the means, which for WT and Δatg4.1 are significantly different from Δatg4.2, p < 0.05. B, scanning electron micrographs. Bar represents 4 μm. C, histogram of body lengths of amastigotes isolated from lesions in mice. The abscissa shows body length ranges in μm; the ordinate indicates percent of cells counted. *, data for Δatg4.2 differed significantly from those for WT parasites in all class intervals (p < 0.05). D, transformation of amastigotes isolated from BALB/c mice. The data show the disappearance of amastigotes (A) and the appearance of promastigotes (P) over 7 days.
Transformation of the parasites extracted from lesions in mice back to promastigotes was assessed by inoculating them into 5 ml of medium at the cell density of 2 × 105 ml−1. Under these conditions, Δatg4.2 parasites differentiated into promastigotes more quickly than the WT and Δatg4.1 parasites (Fig. 4D).
Use of the Δatg4 Mutants to Study the Relationship between ROS Levels and Autophagy in Leishmania
The relationship between ROS levels and induction of autophagy known for mammalian cells prompted us to study whether there is a correlation between ROS levels and the abundance of autophagosomes in Leishmania. The approaches we adopted were measuring ROS levels in the mutants, correlating these with autophagosome generation, and measuring the sensitivity of the autophagy-compromised mutants to oxidative stress. We also modulated the ROS levels in promastigotes through the addition of the antioxidant NAC (known to scavenge H2O2, hydroxyl radical, and superoxide within cells and to help maintain a reduced intracellular environment (42)), and monitored how this affected autophagy.
NAC added to the starvation buffer inhibited the biogenesis of GFP-ATG8-containing puncta in all cell lines except Δatg4.2 promastigotes (Fig. 2D). The proportion of Δatg4.2 promastigotes with GFP-ATG8-containing autophagosomes was not statistically different whether or not NAC was added (Fig. 2D). These data show an apparent correlation between increased ROS levels and number of GFP-ATG8 puncta in starved promastigotes (except for Δatg4.2) and are consistent with autophagy being induced by ROS as reported for other cells (24, 25, 43). However, the accumulation of GFP-ATG8 containing puncta in Δatg4.2 even when ROS levels had been diminished using NAC confirm that this mutant has dysfunctional autophagic machinery.
The addition of NAC also abrogated the formation of the GFP-ATG8A structures (Fig. 2F). These data suggest that ATG8A-dependent puncta formation is similar to autophagy involving ATG8 in being induced by ROS.
Increased ROS Levels and Oxidatively Damaged Proteins in Δatg4
To elucidate the parts played by each L. major ATG4 in any autophagic response to counter ROS, we measured the levels of ROS in normal and starved promastigotes of the various lines using 2,7-dichlorofluorescin (H2DCF-DA)(24, 25, 44, 45). This revealed that ROS levels in Δatg4.2 were 2-fold higher (p < 0.01) than in WT promastigotes at early log phase (day 2), whereas those in Δatg4.1 were similar to WT (Fig. 5A, compare with Fig. 2B). ROS levels were increased 40-fold in WT promastigotes during a 2-h starvation, a similar scale of increase also occurred in the two null mutant lines (Fig. 5A). Addition of NAC at 2.5 mm to the starvation medium suppressed ROS levels at 2 h very greatly in promastigotes of all lines except Δatg4.2 (Fig. 5A). More NAC (5 mm) was required to produce an equivalent reduction of ROS in Δatg4.2 (Fig. 5A).
FIGURE 5.
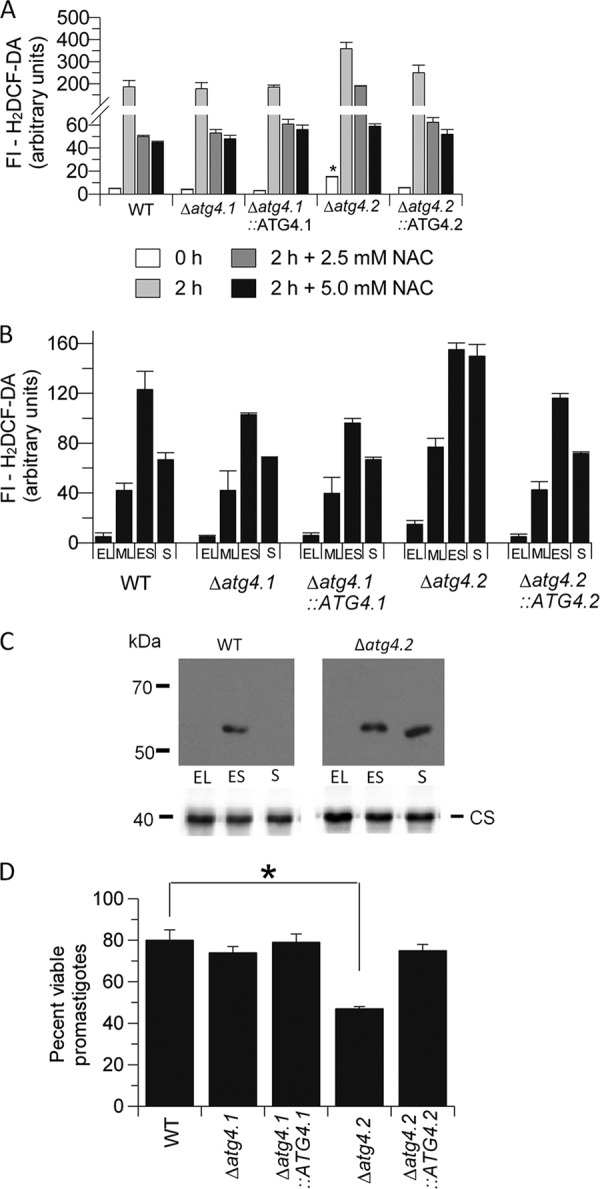
Increased ROS levels and oxidatively damaged proteins in Δatg4.2 promastigotes. A, ROS levels, measured using 50 mm H2DCF-DA, in early log promastigotes incubated for 2 h at 27 °C in PBS with or without NAC. Note the discontinuity in the ordinate axis. Data are mean ± S.D. from independent experiments. *, ROS levels in Δatg4.2 were significantly higher than in WT promastigotes, p < 0.001. B, ROS levels in promastigotes at early log (EL), mid log (ML), early stationary (ES), and stationary (S) phases of growth (days 2, 4, 6, and 9, respectively). C, occurrence of carbonylated protein in extracts of promastigotes at the growth phases detailed in B. Cysteine synthase (CS) served as a loading control. D, log phase promastigotes were starved for 2 h in PBS and then assessed for the proportion of viable cells compared with WT promastigotes in complete HOMEM medium using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide colorimetric assay. *, denotes statistically significant difference, p < 0.05.
ROS levels during in vitro growth of promastigotes were also assessed to determine whether there was any apparent relationship between these and the occurrence of autophagy. The levels in WT promastigotes increased as early log phase progressed to mid log phase and early stationary phase but then declined at stationary phase (Fig. 5B, time points equivalent to days 2, 4, 6, and 9 in Fig. 2). Thus the period at which ROS levels were highest coincided with the time when autophagy was also elevated. The same profile occurred with Δatg4.1 and Δatg4.1::ATG4.1. However, the situation was different in the Δatg4.2 line, DCF-DA fluorescence intensity was 2-fold higher than its WT counterpart at early log phase, increased as log phase progressed, and was maintained at this high level during stationary phase. This phenotype was restored to WT levels in the Δatg4.2::ATG4.2 re-expressor.
One consequence of elevated ROS in a cell is the unwanted oxidation of proteins, which can be removed via autophagy. Protein carbonyl content has been shown to be a biomarker for protein oxidation (46), thus we investigated the presence of carbonylated proteins in parasite extracts by derivatization with 2,4-dinitrophenylhydrazine. The stable dinitrophenylhydrazone product was assessed by Western blot analysis. Just a single protein of ∼60 kDa was detected in extracts of WT promastigotes, as previously reported (47, 48), and only in those of promastigotes at early stationary phase of growth when ROS levels are highest (Fig. 5C). However, the same analysis with Δatg4.2 yielded the equivalent protein also in extracts of stationary phase promastigotes (Fig. 5C). Thus for this mutant there is an apparent correlation between elevated ROS levels, protein carbonylation, and restricted autophagic flux.
Δatg4.2 Promastigotes Are Less Able to Withstand Starvation and Oxidative Stresses
Autophagy generally has a role in protecting against starvation and other stresses, thus we tested the hypothesis that deletion of ATG4 would hinder autophagic flux and make promastigotes of L. major more susceptible to stresses. Δatg4.2 promastigotes were indeed less able to withstand starvation in PBS, with viability, as assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, after 2 h starvation being 42% less than that of WT, Δatg4.1, Δatg4.1::ATG4.1, and Δatg4.2::ATG4.2 promastigotes under the same conditions (Fig. 5D). Viability of the WT itself was reduced to ∼80% compared with that when in growth medium.
The sensitivity of the various lines to exogenous hydrogen peroxide was also assessed. The results show that Δatg4.2 promastigotes are more susceptible than the other lines, with an IC50 of 0.21 ± 0.02 mm compared with IC50 values of around 0.5 mm for the other lines. These findings suggest that deletion of ATG4.1 does not interfere with the ability of the promastigote to withstand these stresses, whereas removal of ATG4.2 does have a detrimental effect.
Δatg4.2 Promastigotes Have a Dysfunctional Mitochondrion
There is increasing evidence that general autophagy is required for the maintenance of mitochondrial homeostasis, and we have recently shown this to be so for Leishmania by generating mutants lacking ATG5 (31). Thus we investigated whether removal of the individual ATG4s from L. major had an impact upon the mitochondrion of the parasite. MitoTracker Green staining, which is used to assess total mitochondrial mass, showed that Δatg4.1 promastigotes were similar to WT promastigotes and also that more than three-quarters of Δatg4.2 promastigotes at stationary phase of growth contained a reticulate mitochondrion characteristic of WT promastigotes (Fig. 6A). The remainder (∼20%) of Δatg4.2 promastigotes had collapsed or fragmented mitochondrial morphology. However, there was a similar overall intensity of the fluorescence in all the cell lines, consistent with there being little difference in mitochondrial mass and this remained the same in the presence of 5 mm NAC (data not shown). A much lower fluorescence intensity was observed with MitoTracker Red (which assesses mitochondrial membrane potential) in Δatg4.2 compared with WT promastigotes (Fig. 6B, closed bars; p < 0.05), suggesting that mitochondrial function was impaired in this null mutant. No such difference was noted for the other mutants (Fig. 6B, closed bars). Addition of NAC to normal medium greatly increased the mitochondrial membrane potential of Δatg4.2 promastigotes by 4 h (Fig. 6B, open bars) and also suppressed ROS levels (Fig. 6C, open bars) although the mitochondrial network was still disrupted in 15 ± 1% of promastigotes at this time point (Fig. 6E). Prolonged incubation in normal medium containing NAC for up to 48 h maintained ROS at relatively low levels (Fig. 6C, open bars) and the mitochondrial membrane potential close to but significantly lower than that of WT promastigotes (Fig. 6, compare D with B) and there was also a significant decrease in the proportion of cells with a defective mitochondrion (to 6 ± 1%, Fig. 6E). These results suggest that the lack of ATG4.2 and consequent impaired autophagic flux is responsible for the increased levels of ROS and that these lead to impaired mitochondrial homeostasis and function.
FIGURE 6.

Evidence for a dysfunctional mitochondrion in Δatg4.2 but not Δatg4.1. A, the types of mitochondrial morphology observed by fluorescence microscopy in promastigotes stained with MitoTracker Green. Scale bar, 10 μm. B, the fluorescent intensity from MitoTracker Red, at 0.1 μm, of 2 × 106 promastigotes in normal medium after 4 h incubation at 26 °C with and without NAC. Data are mean ± S.D. from three independent experiments. * and ** denote statistically significant differences between, respectively, WT and Δatg4.2 and Δatg4.2 with and without NAC. C–E, effect of addition of NAC to Δatg4.2 promastigotes at 2 × 106 ml−1 in normal medium over 48 h. C, ROS levels measured using 50 mm H2DCF-DA. D, fluorescent intensity from MTR. E, the proportion of Δatg4.2 promastigotes with an abnormal mitochondrial morphology.
DISCUSSION
This study aimed to elucidate the roles of the two ATG4 enzymes in L. major by generating gene deletion mutants. Both individual null mutants were generated successfully using promastigotes and their phenotypes were subsequently analyzed. The main defects observed for Δatg4.1 were the fewer promastigotes that contained GFP-ATG8-containing autophagosomes (Fig. 2B) and poorer ability of promastigotes to successfully infect macrophages in vitro (Fig. 3, C and D) and in mice (Fig. 3, E-H), even though they appeared to undergo metacyclogenesis in vitro similarly to, although slightly slower than, WT parasites (Fig. 3, A and B). The capacity of this mutant to produce autophagosomes, albeit less abundantly than the WT parasites, implies some redundancy in function between the two ATG4s. We have previously shown that although ATG4.2 has higher activity toward the ATG8A group of paralogues, it can hydrolyze ATG8 in vitro after prolonged incubation (32) and this is supported by the observation in this study of GFP-ATG8-containing puncta in mutants lacking ATG4.1. However, puncta containing ATG8B and ATG8C, also preferred substrates for ATG4.1 (32), were not formed in Δatg4.1 (data not shown). Thus the viability of Δatg4.1 shows that these proteins and the puncta that they form do not have an essential role in the parasite. The reduced ability of promastigotes of Δatg4.1 to infect macrophages and mice may reflect a reduction in flux through the autophagic pathway and so, perhaps, a reduced capacity for differentiation between life cycle forms. Thus these data are consistent with the contributions of ATG4.1 to autophagy being significant but not essential for Leishmania development.
The deletion of ATG4.2 had more profound effects upon the parasite. The lack of ATG4.2 markedly reduces the ability of the parasite, whether as promastigotes and amastigotes, to infect and survive in both macrophages in vitro (Fig. 3, C and D) and also mice (Fig. 3, E–H). As Δatg4.2 had a significantly higher amount of lipidated ATG8 (Fig. 2C) and a higher number of GFP-ATG8-containing puncta at stationary phase of growth (Fig. 2B), we conclude that the lower virulence of this mutant is due primarily to a block in the delivery of autophagosomes to the lysosomal network, probably due to poor cleavage of ATG8 from the mature autophagosomes, and thus a reduction in flux through the ATG8-dependent autophagic pathway. To further characterize the identity of the higher number of GFP-ATG8-containing puncta in Δatg4.2 at stationary phase of growth, we transfected the line with mC-ATG5, which also labels autophagosomes (31). These analyses showed that most of the puncta were labeled with GFP-ATG8 only, very rarely was labeling observed with mC-ATG5 (data not shown). As ATG5 is released from nascent autophagosomes before they mature, this is entirely consistent with our idea that the additional puncta in Δatg4.2 are mature autophagosomes. Unfortunately there are no other markers for autophagosomes currently available for use with Leishmania. However, electron microscopic analyses also revealed that structures typical of autophagosomes (as exemplified in Ref. 41) were more abundant in Δatg4.2 than in Δatg4.1 or WT parasites (data not shown).
The significance of the deconjugation of ATG8-PE by ATG4 has been described previously for yeast (49). In this organism, with just a single ATG4 gene, the expression of a mutated Atg8 (truncated at the glycine required for conjugation to PE, which avoids the need for the initial processing by ATG4) in an atg4-deletion mutant produced a low autophagic efficiency but high levels of ATG8-PE and increased numbers of autophagosomes in the cytosol. These results with yeast and also now Leishmania Δatg4.2 confirm that ATG4s have delipidation activity that is required at the late stage of the autophagic cycle. This conclusion is further supported by an inhibitory effect on autophagosome occurrence of overexpressing ATG4.2 in Leishmania promastigotes (32) and HsAtg4b in human cells (49). We have also shown previously that recombinant ATG4.2 can cleave ATG8-PE and release ATG8 (32), which adds further support for the important role of ATG4.2 at this stage of the autophagic process.
The increase in number of autophagosomes resulting from deletion of ATG4.2 contrasts with our finding (detailed in Ref. 41) that inhibiting lysosomal peptidases caused an increase in undigested autophagosomal material within the lysosomal network, an effect not observed in this current study, but not an increased number of mature autophagosomes. These differing effects resulting from the two interventions are consistent with the role we postulate for ATG4.2 in autophagy in Leishmania.
A block in autophagy in Δatg4.2 would also account for other observed phenotypic characteristics of Δatg4.2, including the reduced ability to maintain viability during oxidant and starvation stresses and a lower ability to induce autophagy when nutrients were limiting. It is also likely that such reduced flux would hinder cellular differentiation of the parasite and so could explain why the forms of the parasite isolated from mice were abnormally large and differentiated more rapidly to promastigotes, they were not fully differentiated to amastigotes even in a mammal (Fig. 4). Similar findings have been reported for mouse fibroblast cells, which when defective in autophagy were unable to reduce their size in response to starvation (50), and Arabidopsis, where mutants lacking the two ATG4s (named atg4a4b-1) were unable to form autophagosomes under nutrient-limiting conditions (51).
ATG4.2 of Leishmania also has a high activity toward ATG8A, the paralogue of ATG8 that forms puncta primarily under starvation conditions (32). ATG4.1 does not cleave ATG8A, which explains why ATG8A-labeled puncta were not formed in Δatg4.2 (Fig. 2, F and G). This inability could play some part in determining the phenotype of this null line, but as yet the full significance of the ATG8A-containing putative autophagosomes is unknown.
Autophagy plays a part in protecting against ROS-mediated damage of proteins and organelles in eukaryotic cells (52), and its absence results in elevated ROS levels and consequently accumulation of oxidized proteins, a lower mitochondrial membrane potential and dysfunctional mitochondria (52, 53). Moreover, autophagy can be induced by elevated ROS (24–26). Our investigation of the relationship between ROS and autophagy in Leishmania has shown that ROS levels do vary as Leishmania promastigotes grow in vitro with, interestingly, higher levels occurring during the phase when metacyclogenesis occurs and autophagy is elevated (Fig. 5B). Δatg4.2 had higher levels of ROS than WT throughout this growth, with notably higher levels at stationary phase when the numbers of autophagosomes are also markedly elevated. This is consistent again with autophagic flux being reduced in this mutant and a role for autophagy in dealing with ROS stress, hypotheses that can be tested in future studies.
There was also a prominent depolarization of the mitochondrion and some fragmentation in Δatg4.2 (Fig. 6). We have recently shown that mutants lacking ATG5, which do not produce autophagosomes, have an even more markedly dysfunctional mitochondrion with an increased mitochondrial mass (31), thus the data for Δatg4.2 are consistent with this. There is increasing evidence for a close inter-relationship between autophagy and mitochondrial homeostasis (54–56) and these data with Leishmania Δatg4.2 support this.
Our experiments using the antioxidant NAC in parasites starved for 2 h showed that this agent resulted in lower ROS levels in all lines (Fig. 5A) and also to decreased production of autophagosomes containing ATG8 and ATG8A (Fig. 2) and restoration of mitochondrial integrity (Fig. 6). This is consistent with ROS being an important regulator of autophagy in Leishmania and also supports ROS as a mediator of mitochondrion damage, although direct evidence needs to be obtained in future studies to confirm this.
In human cells, ROS modulates the autophagic pathway during amino acid starvation (25). This appears to occur through ROS inhibiting the deconjugation activity of ATG4, causing ATG8-PE to accumulate at the pre-autophagosomal membranes. This fosters autophagosome biogenesis, and so autophagy to remove more rapidly oxidized proteins and damaged organelles (24, 45). This mechanism cannot operate in Leishmania, however, as neither Leishmania ATG4 is affected directly by ROS, as demonstrated by the lack of inhibition by 5 mm H2O2 of the hydrolytic activities of recombinant ATG4.1 and ATG4.2 toward their preferred substrates, His-ATG8-HA and His-ATG8A-HA, respectively (data not shown). Yeast also similarly differs from mammalian cells in this way and this organism has a regulatory mechanism in which ATG4 is blocked from ATG8-PE in nascent autophagosomes by the ATG12-ATG5 complex, ATG18 and ATG21 (8). It remains to be determined whether such a blockage functions in Leishmania but we have shown that the parasite does have the ATG5-ATG12 conjugation pathway (31) and a gene encoding ATG18 is present in the L. major genome (www.geneDB.org, LmjF29.1575). However, there is not currently strong evidence for a gene encoding ATG21 and so if such inhibition occurs the precise mechanism is likely to differ from that in yeast.
Our results overall show that the ATG4s of L. major are different, there is some redundancy in function between them especially in autophagosome biogenesis, but that ATG4.2 would appear to be more important than ATG4.1 for the later stages of the autophagic pathway when ATG8 is cleaved from the autophagosomes. Neither is individually essential, and the finding that cells lacking both ATG4.1 and ATG4.2 could not be obtained, even though both loci were demonstrated to be targetable by the gene deletion constructs, suggests that ATG4 performs essential roles in Leishmania. The activity of the enzymes toward not only ATG8 but also ATG8A, both of which function in autophagy in some conditions, makes autophagy in Leishmania an interesting system to investigate in terms of the roles of the individual ATG4 proteins.
The results presented also support the idea that autophagy is important for differentiation of the parasite, and especially in enabling promastigotes to infect mammals. Importantly, however, ATG4.2 appears to have a constitutive role in the growth of the parasite in mammals. This is an important finding that opens up many new avenues for further investigation on the roles of autophagy in the Leishmania-mammal interaction.
Acknowledgment
We thank Laurence Tetley (Glasgow) for the SE analyses.
This work was supported Medical Research Council Grant G0700127 and core funding from Wellcome Trust Grant 085349.
- ATG/ATG
- autophagy related gene/protein
- H2DCF-DA
- 2′7′-dichlorodihydrofluorescein diacetate
- PE
- phosphatidylethanolamine
- ROS
- reactive oxygen species
- NAC
- N-acetylcysteine
- SE
- scanning electron microscopy.
REFERENCES
- 1. Deretic V. (2011) Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol. Rev. 240, 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goldman S. J., Zhang Y., Jin S. (2011) Autophagic degradation of mitochondria in white adipose tissue differentiation. Antioxid. Redox Signal. 14, 1971–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Manjithaya R., Subramani S. (2011) Autophagy. A broad role in unconventional protein secretion? Trends Cell Biol. 21, 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mörck C., Pilon M. (2006) C. elegans feeding defective mutants have shorter body lengths and increased autophagy. BMC Dev. Biol. 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mörck C., Pilon M. (2007) Caloric restriction and autophagy in Caenorhabditis elegans. Autophagy 3, 51–53 [DOI] [PubMed] [Google Scholar]
- 6. Youle R. J., Narendra D. P. (2011) Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee J. A., Beigneux A., Ahmad S. T., Young S. G., Gao F. B. (2007) ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 17, 1561–1567 [DOI] [PubMed] [Google Scholar]
- 8. Nair U., Cao Y., Xie Z., Klionsky D. J. (2010) Roles of the lipid-binding motifs of Atg18 and Atg21 in the cytoplasm to vacuole targeting pathway and autophagy. J. Biol. Chem. 285, 11476–11488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanida I. (2011) Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 14, 2201–2214 [DOI] [PubMed] [Google Scholar]
- 10. Kimura S., Noda T., Yoshimori T. (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 [DOI] [PubMed] [Google Scholar]
- 11. Kirisako T., Baba M., Ishihara N., Miyazawa K., Ohsumi M., Yoshimori T., Noda T., Ohsumi Y. (1999) Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 147, 435–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kirisako T., Ichimura Y., Okada H., Kabeya Y., Mizushima N., Yoshimori T., Ohsumi M., Takao T., Noda T., Ohsumi Y. (2000) The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 151, 263–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xie Z., Nair U., Klionsky D. J. (2008) Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 19, 3290–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Geng J., Nair U., Yasumura-Yorimitsu K., Klionsky D. J. (2010) Post-Golgi Sec proteins are required for autophagy in Saccharomyces cerevisiae. Mol. Biol. Cell 21, 2257–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kimura S., Noda T., Yoshimori T. (2008) Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 33, 109–122 [DOI] [PubMed] [Google Scholar]
- 16. Köchl R., Hu X. W., Chan E. Y., Tooze S. A. (2006) Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 7, 129–145 [DOI] [PubMed] [Google Scholar]
- 17. Mariño G., Uría J. A., Puente X. S., Quesada V., Bordallo J., López-Otín C. (2003) Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J. Biol. Chem. 278, 3671–3678 [DOI] [PubMed] [Google Scholar]
- 18. Read R., Savelieva K., Baker K., Hansen G., Vogel P. (2011) Histopathological and neurological features of Atg4b knockout mice. Vet. Pathol. 48, 486–494 [DOI] [PubMed] [Google Scholar]
- 19. Mariño G., Salvador-Montoliu N., Fueyo A., Knecht E., Mizushima N., López-Otín C. (2007) Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J. Biol. Chem. 282, 18573–18583 [DOI] [PubMed] [Google Scholar]
- 20. Mariño G., Fernández A. F., Cabrera S., Lundberg Y. W., Cabanillas R., Rodríguez F., Salvador-Montoliu N., Vega J. A., Germanà A., Fueyo A., Freije J. M., López-Otín C. (2010) Autophagy is essential for mouse sense of balance. J. Clin. Invest. 120, 2331–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cabrera S., Mariño G., Fernández A. F., López-Otín C. (2010) Autophagy, proteases and the sense of balance. Autophagy 6, 961–963 [DOI] [PubMed] [Google Scholar]
- 22. Betin V. M., Lane J. D. (2009) Atg4D at the interface between autophagy and apoptosis. Autophagy 5, 1057–1059 [DOI] [PubMed] [Google Scholar]
- 23. Betin V. M., Lane J. D. (2009) Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 122, 2554–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scherz-Shouval R., Elazar Z. (2011) Regulation of autophagy by ROS. Physiology and pathology. Trends Biochem. Sci. 36, 30–38 [DOI] [PubMed] [Google Scholar]
- 25. Scherz-Shouval R., Shvets E., Fass E., Shorer H., Gil L., Elazar Z. (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26, 1749–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Y., Luo Q., Yuan L., Miao C., Mu X., Xiao W., Li J., Sun T., Ma E. (2012) JNK-dependent Atg4 up-regulation mediates asperphenamate derivative BBP-induced autophagy in MCF-7 cells. Toxicol. Appl. Pharmacol. 263, 21–31 [DOI] [PubMed] [Google Scholar]
- 27. Wong C. H., Iskandar K. B., Yadav S. K., Hirpara J. L., Loh T., Pervaiz S. (2010) Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS One 5, e9996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boya P., González-Polo R. A., Casares N., Perfettini J. L., Dessen P., Larochette N., Métivier D., Meley D., Souquere S., Yoshimori T., Pierron G., Codogno P., Kroemer G. (2005) Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 25, 1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim I., Rodriguez-Enriquez S., Lemasters J. J. (2007) Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 462, 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lemasters J. J., Nieminen A. L., Qian T., Trost L. C., Elmore S. P., Nishimura Y., Crowe R. A., Cascio W. E., Bradham C. A., Brenner D. A., Herman B. (1998) The mitochondrial permeability transition in cell death. A common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta 1366, 177–196 [DOI] [PubMed] [Google Scholar]
- 31. Williams R. A., Smith T. K., Cull B., Mottram J. C., Coombs G. H. (2012) ATG5 is essential for ATG8-dependent autophagy and mitochondrial homeostasis in Leishmania major. PLoS Pathog. 8, e1002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Williams R. A., Woods K. L., Juliano L., Mottram J. C., Coombs G. H. (2009) Characterization of unusual families of ATG8-like proteins and ATG12 in the protozoan parasite Leishmania major. Autophagy 5, 159–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Besteiro S., Williams R. A., Morrison L. S., Coombs G. H., Mottram J. C. (2006) Endosome sorting and autophagy are essential for differentiation and virulence of Leishmania major. J. Biol. Chem. 281, 11384–11396 [DOI] [PubMed] [Google Scholar]
- 34. Williams R. A., Kelly S. M., Mottram J. C., Coombs G. H. (2003) 3-Mercaptopyruvate sulfurtransferase of Leishmania contains an unusual C-terminal extension and is involved in thioredoxin and antioxidant metabolism. J. Biol. Chem. 278, 1480–1486 [DOI] [PubMed] [Google Scholar]
- 35. Sacks D. L., Hieny S., Sher A. (1985) Identification of cell surface carbohydrate and antigenic changes between noninfective and infective developmental stages of Leishmania major promastigotes. J. Immunol. 135, 564–569 [PubMed] [Google Scholar]
- 36. Afonso L. C., Scott P. (1993) Immune responses associated with susceptibility of C57BL/10 mice to Leishmania amazonensis. Infect. Immun. 61, 2952–2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mottram J. C., McCready B. P., Brown K. G., Grant K. M. (1996) Gene disruptions indicate an essential function for the LmmCRK1 cdc2-related kinase of Leishmania mexicana. Mol. Microbiol. 22, 573–583 [DOI] [PubMed] [Google Scholar]
- 38. Garami A., Ilg T. (2001) Disruption of mannose activation in Leishmania mexicana. GDP-mannose pyrophosphorylase is required for virulence, but not for viability. EMBO J. 20, 3657–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Misslitz A., Mottram J. C., Overath P., Aebischer T. (2000) Targeted integration into a rRNA locus results in uniform and high level expression of transgenes in Leishmania amastigotes. Mol. Biochem. Parasitol. 107, 251–261 [DOI] [PubMed] [Google Scholar]
- 40. Medina-Acosta E., Cross G. A. (1993) Rapid isolation of DNA from trypanosomatid protozoa using a simple “mini-prep” procedure. Mol. Biochem. Parasitol. 59, 327–329 [DOI] [PubMed] [Google Scholar]
- 41. Williams R. A., Tetley L., Mottram J. C., Coombs G. H. (2006) Cysteine peptidases CPA and CPB are vital for autophagy and differentiation in Leishmania mexicana. Mol. Microbiol. 61, 655–674 [DOI] [PubMed] [Google Scholar]
- 42. Dolai S., Pal S., Yadav R. K., Adak S. (2011) Endoplasmic reticulum stress-induced apoptosis in Leishmania through Ca2+-dependent and caspase-independent mechanism. J. Biol. Chem. 286, 13638–13646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Azad M. B., Chen Y., Gibson S. B. (2009) Regulation of autophagy by reactive oxygen species (ROS). Implications for cancer progression and treatment. Antioxid. Redox Signal. 11, 777–790 [DOI] [PubMed] [Google Scholar]
- 44. Scherz-Shouval R., Elazar Z. (2007) ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 17, 422–427 [DOI] [PubMed] [Google Scholar]
- 45. Scherz-Shouval R., Shvets E., Elazar Z. (2007) Oxidation as a post-translational modification that regulates autophagy. Autophagy 3, 371–373 [DOI] [PubMed] [Google Scholar]
- 46. Dalle-Donne I., Rossi R., Giustarini D., Gagliano N., Di Simplicio P., Colombo R., Milzani A. (2002) Methionine oxidation as a major cause of the functional impairment of oxidized actin. Free Radic. Biol. Med. 32, 927–937 [DOI] [PubMed] [Google Scholar]
- 47. Bhattacharya A., Biswas A., Das P. K. (2008) Role of intracellular cAMP in differentiation-coupled induction of resistance against oxidative damage in Leishmania donovani. Free Radic. Biol. Med. 44, 779–794 [DOI] [PubMed] [Google Scholar]
- 48. Dolai S., Yadav R. K., Pal S., Adak S. (2009) Overexpression of mitochondrial Leishmania major ascorbate peroxidase enhances tolerance to oxidative stress-induced programmed cell death and protein damage. Eukaryot. Cell 8, 1721–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tanida I., Ueno T., Kominami E. (2004) LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36, 2503–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hosokawa N., Hara Y., Mizushima N. (2006) Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett. 580, 2623–2629 [DOI] [PubMed] [Google Scholar]
- 51. Yoshimoto K., Hanaoka H., Sato S., Kato T., Tabata S., Noda T., Ohsumi Y. (2004) Processing of ATG8s, ubiquitin-like proteins, and their deconjugation by ATG4s are essential for plant autophagy. Plant Cell 16, 2967–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krebiehl G., Ruckerbauer S., Burbulla L. F., Kieper N., Maurer B., Waak J., Wolburg H., Gizatullina Z., Gellerich F. N., Woitalla D., Riess O., Kahle P. J., Proikas-Cezanne T., Krüger R. (2010) Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson's disease-associated protein DJ-1. PLoS One 5, e9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schapira A. H. (2008) Mitochondrial dysfunction in neurodegenerative diseases. Neurochem. Res. 33, 2502–2509 [DOI] [PubMed] [Google Scholar]
- 54. Graef M., Nunnari J. (2011) A role for mitochondria in autophagy regulation. Autophagy 7, 1245–1246 [DOI] [PubMed] [Google Scholar]
- 55. Mortensen M., Watson A. S., Simon A. K. (2011) Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy 7, 1069–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Okamoto K., Kondo-Okamoto N. (2012) Mitochondria and autophagy. Critical interplay between the two homeostats. Biochim. Biophys. Acta 1820, 595–600 [DOI] [PubMed] [Google Scholar]