

Abstract
Screening of small molecule libraries offers the potential to identify compounds that inhibit specific biological processes and, ultimately, to identify macromolecules that are important players in such processes. To date, however, most screens of small molecule libraries have focused on identification of compounds that inhibit known proteins or particular steps in a given process, and have emphasized automated primary screens. Here we have used “low tech” in vivo primary screens to identify small molecules that inhibit both cytokinesis and single cell wound repair, two complex cellular processes that possess many common features. The “diversity set”, an ordered array of 1990 compounds available from the National Cancer Institute, was screened in parallel to identify compounds that inhibit cytokinesis in D. excentricus (sand dollar) embryos and single cell wound repair in X. laevis (frog) oocytes. Two small molecules were thus identified: Sph1 and Sph2. Sph1 reduces Rho activation in wound repair and suppresses formation of the spindle midzone during cytokinesis. Sph2 also reduces Rho activation in wound repair and may inhibit cytokinesis by blocking membrane fusion. The results identify two small molecules of interest for analysis of wound repair and cytokinesis, reveal that these processes are more similar than often realized and reveal the potential power of low tech screens of small molecule libraries for analysis of complex cellular processes.
INTRODUCTION
Identification of macromolecules involved in particular cell biological processes is one of the major goals of modern cell biology. In most cases, a given macromolecule is assigned a role in a particular process as a result of either forward genetic screens, in which a particular mutant gene is determined to encode a product required for the process of interest, or reverse genetic approaches in which a particular gene product is specifically targeted for reduction or elimination. However, forward genetic approaches can only be applied to a handful of species, while reverse genetic approaches require a considerable amount of starting information. Further, both forward and reverse genetics can be problematic if a given gene product is required for multiple processes, such that its loss or depletion is lethal or otherwise prevents cells from reaching the point needed to begin the process of interest. Thus, it may be difficult to study the role of a gene product that regulates, for example, cell cycle events, if it is also involved in, say, basic metabolism. Genetic and reverse genetic approaches may also be difficult to apply to nonprotein macromolecules such as lipids, which are typically produced by the sequential action of multiple proteins and which, in turn, can serve as essential precursors for several other lipids.
The development of small molecule chemical libraries offers a potential solution to such problems. Such libraries contain thousands of different compounds, most of which are uncharacterized, but which have the potential to interact with and inhibit particular macromolecules and thus any process that is dependent on those macromolecules (Gray et al., 2001). Consequently, this approach has the potential to work in model systems that do not permit forward genetics and, unlike reverse genetics, does not require any starting information about likely players in the process being studied. The macromolecular target of any small molecule identified from a library can be identified by biochemical approaches (e.g. Lomenick et al., 2011; Titov and Liu, 2012). Even prior to such identification, specific inhibitors can be extremely useful with respect to ordering hierarchies of cell biological events. Further, because the small molecules can be applied at any time, targets that play multiple roles or which are the products of multiple gene products can nonetheless be identified.
Thus, the small molecule library screening approach holds tremendous promise for the students of cell biology. However, most of the effort to date has focused on screening small molecule libraries for inhibitors of specific proteins or specific biochemical interactions, on primary screens that can be automated, or both (e.g. Stockwell, 1999; Whitfield, 2003; Wu et al., 2003). Such approaches are quite powerful and have produced several very useful agents including blebbistatin (Straight et al., 2003) and monastrol (Mayer et al., 1999), and led to the identification of novel roles for previously identified proteins (Zhu et al., 2005). However, if small molecule libraries are to fulfill their potential, it will be necessary to show that they can be applied to complex cellular processes in intact cells assessed using nonautomated screens in that such screens still represent the most common and flexible means of phenotypic assessment.
Here we have screened a small molecule library for compounds that inhibit both single cell wound repair and cytokinesis, complex processes that have several features in common including regulation by Rho GTPases and formation of contractile arrays of actin filaments and myosin-2 (Sonnemann and Bement, 2011; Green et al., 2011) but are nevertheless generally assumed to be largely distinct. We deliberately employed primary screens that are “low tech” to assess the feasibility of the small molecule approach for situations in which automation is not possible, either due to the complexity of the process in question or limiting resources. Two small molecules were identified that inhibited both single cell wound repair and cytokinesis. The low tech primary screens were followed by subsequent “high content” screens to obtain more information about the likely targets of these inhibitors.
MATERIALS AND METHODS
Compounds for screen and phenotypic analysis
The small molecule library used for this screen was the National Cancer Institute Developmental Therapeutics Program (NCI DTP) Diversity Set (I; NCI/NIH DTP, Bethesda, MD, USA; http://dtp.cancer.gov/branches/dscb/diversity_explanation.html). Additional aliquots of compounds used in this study were obtained from the NCI DTP and were kept as 50mM stocks in DMSO. The compounds from the set were not pooled but rather screened individually.
Xenopus laevis oocyte preparation and wound healing screen
Xenopus oocytes were harvested by surgery of adult female frogs, incubated in 1x Barth’s Solution (Yu and Bement, 2007), and selected for pigment quality and size. Follicle coats were removed by treating with 5mg/mL collagenase (Invitrogen, Carlsbad, CA) for one hour and manually removing the follicle layer. For the wound healing screen, two to three oocytes were incubated in each well of a 96 well plate containing a 50 μM solution of each library compound in 0.5% DMSO in 1x Barth’s and incubated for 1–2 hours at 16°C; control wells contained 0.5% DMSO alone in 1x Barth’s. Following incubation, 5–10 wounds to the plasma membrane of the animal hemisphere were made using a 150 μM-wide glass capillary needle. Cells were incubated overnight at 16°C and observed the following day. Compounds were determined to inhibit or not inhibit wound healing as indicated by the presence of membrane bubbles, cytoplasm in the surrounding medium, and/or pigment displacement by observation under a dissecting microscope. For the toxicity assay, oocytes were prepared as above and incubated in 50 μM solutions of compounds in 0.5% DMSO in 1x Barth’s (with controls in 0.5% DMSO alone in 1x Barth’s) overnight at 16°C. Cells were observed the following day for cytoplasm leakage and pigment displacement.
Dendraster excentricus preparation, cytokinesis screen and DIC microscopy
Gametes were harvested from D. excentricus adults by injecting 1ml 0.55 mM KCl into the coelomic cavity and collecting gametes in sea water (SW). For fertilization, 0.5 ml diluted sperm and 2 ml eggs were mixed in 50 ml seawater. After 10 min, fertilization envelopes were removed by passing zygotes through a 130 μm sieve, allowed to recover for at least 30 min, and then 40 μL (40–80 embryos) were added to wells of 96-well plates containing a final concentration of 50 μM for each compound tested. Embryos were incubated in 96-well plates on a sea table and scored every hour for four hours for failures in cytokinesis. To assess multinuclearity in treated D. excentricus embryos, zygotes were prepared as above and at 16-cell stage, Hoechst 33342 was added to the medium and embryos were observed using a widefield fluorescence microscope. To image dividing embryos, D. exentrocus zygotes were cultured in SW or 50 μM compound in 0.1% DMSO in SW. Zygotes were mounted onto glass slides with coverslips using silicon grease and 18 × 18 mm coverslips. Images were collected with an Olympus BX51 using a Point Grey Grasshopper 2 camera.
Xenopus laevis embryo preparation and stereomicroscopy
Xenopus eggs were harvested and fertilized as in Miller and Bement, 2009. Embryos at 4-cell stage were cultured in 0.1x MMR or 50 μM compound in 0.1% DMSO in 0.1x MMR in a Petri dish fitted with 1 mm × 1 mm plastic mesh (Spectrum Laboratories, Rancho Dominguez, CA, USA) immediately prior to imaging. Images were collected using a Nikon SMZ-1500 stereoscope with a Hamamatsu CCD camera an analyzed by Metamorph and ImageJ.
Confocal imaging of Xenopus wound healing
Probes for F-actin (mCherry-utr1-261; Burkel et al., 2007) and active Rho and Active Cdc42 (eGFP-rGBD and eGFP-wGBD and mRFP-wGBD, respectively; Benink and Bement, 2005) were transcribed using an SP6 in vitro translation kit (Ambion, Austin, TX, USA). mRNA was injected into Xenopus oocytes to a final concentration of 10–20 ng/ml 16–24 h prior to imaging. Confocal images were collected with a 63x 1.4 NA objective on a Zeiss Axiovert 100M (Carl Zeiss Microimaging USA, Thornwood, NY, USA) microscope equipped with the Bio-Rad Lasersharp Confocal Package (Bio-Rad Laboratories, Hercules, CA, USA). For wound healing imaging, six 0.4 μm z-sections were collected for each 6s time point. For wound-induced exocytosis imaging, a single z-section was collected every 2s. Oocytes were wounded with a pulse from a Micropoint nitrogen pumped dye laser (Laser Science, Inc., Franklin, MA, USA). 4-D image rendering was performed using Volocity (Improvision, PerkinElmer, Waltham, MA, USA) and image analysis and quantifications were performed using ImageJ. For all confocal experiments, oocytes were incubated in control or compound treatment conditions for 90–120 min prior to imaging.
Confocal imaging of microtubules and MgcRacGAP in D. excentricus cytokinesis
To detect microtubules, mRNA encoding 2XmCherry-EMTB (von Dassow et al., 2009) was prepared as above and microinjected into embryos at the 1 cell stage at a needle concentration of 200 ng/μl. To directly detect midzones, sea urchin MgcRacGAP was amplified from cDNA and ligated to DNA encoding 3XGFP. mRNA from this construct was prepared as above and microinjected into one cell embryos at a needle concentration of 25 ng/μl. Microinjections were performed as described in von Dassow et al. (2009). Samples were imaged on an Olympus FV-1000 using a 60X 1.42 NA water-immersion objective.
RESULTS
Workflow
To identify inhibitors of cytokinesis and single cell wound repair, the “Diversity Set”— a small molecule library of 1990 compounds—was obtained from the National Cancer Institute and first used in preliminary experiments to identify appropriate starting concentrations of compounds. In preliminary experiments, one plate of the library was used to determine a screening concentration that resulted in 1–5% of the compounds causing inhibition of cytokinesis or single cell wound healing (i.e. a 1–5% “hit rate”), based on the goal of avoiding false negatives while still producing a manageable number of hits. A concentration of 50 μM produced the desired hit rate for both wound healing and cytokinesis; this concentration was therefore employed for the primary screens.
The overall screen workflow is shown in Figure 1. The 1990 small molecules of the diversity set were first screened independently for inhibition of frog (X. laevis) oocyte wound healing and sand dollar (D. excentricus) embryonic cytokinesis; these primary screens produced 57 and 60 hits respectively. A total of 21 “overlap compounds”— i.e. small molecules that inhibited both cytokinesis and wound healing — were identified (Fig. 1). These compounds were then subjected to tests for toxicity and inhibition of ATP production to determine the specificity of the hits. Small molecules that were either toxic or inhibited ATP production were excluded from further analysis. This winnowing processes left two hits: NSC2805 and NSC292596, the chemical structures of which are shown in Figure 1. Based on their ability to inhibit cytokinesis and single cell wound repair, two processes that entail formation and closure of rings of F-actin and myosin-2, we provisionally dubbed these two compounds sphinctostatin 1 and sphinctostatin 2 (Sph1 and Sph2).
Figure 1. Schematic showing overview of chemical screen and structure of the two compounds identified by it.

A. In the primary screen, 1990 small molecules from the diversity kit were screened in parallel for their ability to inhibit wound healing in Xenopus oocytes or cytokinesis in sand dollar embryos. In the secondary screen, compounds that inhibited both processes were then then subjected to toxicity and ATP production screens. Two small molecules--NSC2805 and NSC292596 were positive in the primary screen and negative in the secondary screen. Numbers indicate hits/total compounds. B. Structures of NSC2805 and NSC292596.
Screen details
In the primary wound healing screen, follicle-free X. laevis oocytes were incubated for one hour at RT in 96 well plates, the wells of which contained the library compounds dissolved in Barth’s solution. DMSO, the vehicle for the library compounds, was used as a control. Oocytes were then manually punctured with a glass needle and transferred to a 16°C incubator overnight. The following day, each well was inspected using a dissecting microscope. In controls, wounds healed completely with only small aggregations of pigment left to reveal the wound site (Fig. 2). Failed healing was revealed by the presence of cytoplasm leaking from the oocytes into the medium and/or large bulges extending from the oocyte surface (Fig. 2 and not shown). Compounds that resulted in more than half the oocytes showing healing failure were judged to be positive hits and were confirmed by repeating the assay. Using this approach it was possible for one person to screen ~100 compounds/day.
Figure 2. Oocyte wound healing is inhibited by micromolar concentrations of Sph1 and Sph2.

A. Brightfield micrographs of oocytes wounded in presence of DMSO (negative control; 0.1%) cytochalasin (positive control; 20 μM) or Sph1 or Sph2 (100 μM). Failed healing revealed by cytoplasm leaking from oocytes. B. Dose response analysis of healing in DMSO (0.2%) or indicated concentrations (in μM) of Sph1 or Sph2. n ≥ 10 for each concentration tested.
In the primary cytokinesis screen, freshly fertilized D. excentricus eggs were stripped of their fertilization envelopes, allowed to recover for 30 min and then transferred to 96 well plates containing the library compounds dissolved in sea water, or, as controls, DMSO or sea water alone. The 96 well plates were then transferred to a sea table (12–14°C) for further incubation. The progression of cell division was monitored every 60 min for the next four hours using a dissecting microscope. Cytokinesis failure was revealed by failure of fertilized eggs to undergo furrowing and/or by regression of furrows after initial formation (not shown but see below). Compounds that resulted in more than half of embryos failing cytokinesis were scored as positive hits and were confirmed by repeating the assay. Using this approach it was possible for one person to screen ~200 compounds/day. To ensure that cytokinesis failure did not result from cell cycle arrest, the continuation of mitosis was assessed by monitoring the number of nuclei and spindle asters at later times after fertilization (not shown but see below). Agents that resulted in complete cell cycle arrest (rather than inhibition of cytokinesis with continued nuclear cycling) were excluded from further analysis.
For both screens, several considerations were found to be important. First, the number of cells added to each well had to be limited (2–3 oocytes/well for wound healing, ~40–80 embryos for cytokinesis); higher numbers of cells often resulted in markedly less inhibition. Second, repeated freeze-thaw cycles of the small molecules significantly reduced the potency of many of the drugs and was therefore avoided. Third, having all of a given primary screen done by a single individual helped maintain the consistency of the results. Fourth, carefully testing the health of different batches of oocytes or eggs prior to screening saved considerable time and avoided excess freeze thaw cycles for the libraries. Fifth, consistency in transferring roughly equal numbers of oocytes and eggs to wells required practice and even so, it was important to check to make sure that all wells had sufficient numbers of cells immediately after transfer. Sixth, the time of drug application strongly influenced the effects of the drugs. That is, application immediately after stripping of the fertilized eggs greatly increased the potency of the cytokinetic inhibitors but also increased the extent of cytokinesis failure in controls. Conversely, application of drugs after the second or third cleavage greatly reduced the efficacy of several agents that inhibited cytokinesis, including Sph2.
The overlap compounds from the two primary screens were subjected to two additional screens. In the toxicity screen, X. laevis oocytes were incubated overnight in the compounds without wounding. The following day, oocytes were inspected using a dissecting microscope. Agents that resulted in oocytes that displayed signs of ill health — mottling of pigment or leaking of cytoplasm were judged to be toxic and eliminated from further analysis. Those overlap compounds that did not display toxicity were next assessed for their effects on ATP production and those which resulted in more than 25% reduction in cellular ATP levels after 1 hour were excluded from further analysis. One unanticipated result came from the above analysis: several of the compounds that had no obvious effects in the basic toxicity screen nonetheless resulted in considerable ATP depletion (not shown) in spite of the fact that the toxicity assay entailed an overnight incubation. Thus, a toxicity assay such as the one described above is not necessarily sufficient to detect ATP significant depletion.
Characterization of wound healing inhibition by Sph1 and Sph2
Two compounds, Sph1 and Sph2, inhibited both single cell wound repair and cytokinesis and also passed the specificity screens. To gain further insight into the modes of actions of these compounds, we first analyzed their effects on wound healing in more detail. Dose response experiments revealed that both Sph1 and Sph2 inhibited wound repair in a dose-dependent manner and were effective in the micromolar (Sph2) to low micromolar (Sph1) range (Fig. 2). However, the two drugs produced different phenotypes in the wound assay: Sph1 resulted in extensive leakage of the cytoplasm and death following overnight incubation after wounding while Sph2 resulted in large bulges at the wound site but less leakage and cell death (Fig. 2).
Wounding of oocytes elicits accumulation of actin filaments and myosin-2 around wounds (Mandato and Bement, 2001) that occurs as a result of local activation of Rho and Cdc42 in concentric zones that encircle the wound (Benink and Bement, 2005). The effects of Sph1 and Sph2 on Rho and Cdc42 were therefore assessed, using DMSO as a control. Neither Sph1 nor Sph2 inhibited Cdc42 activation around wounds relative to DMSO (Fig. 3), nor did they have any obvious effect on resting levels of Rho activity as assessed via GFP-rGBD (Fig. 3). In contrast, both agents sharply reduced Rho activation around wounds, as judged by both inspection (Fig. 3A) and quantification (Fig. 3B, 3C), with Sph1 being more effective than Sph2. Not surprisingly, given the effects of these agents on Rho, while neither agent had any apparent effect on the level of cortical F-actin in resting oocytes, both the accumulation of actin around wounds and the rate of contractile ring closure was reduced by Sph1 and Sph2 relative to DMSO (not shown).
Figure 3. Sph1 and Sph2 reduce wound-induced Rho activation.

A. Fluorescent images from 4D movie series showing distribution of active Rho (green) and active Cdc42 (red) at increasing times after wounding. Time in min:sec; negative number indicates time prior to wounding. B. Quantification of Rho (top panel) and Cdc42 (bottom panel) activity in presence of DMSO or Sph1. Values represent average intensity of fluorescence signal in the zone surrounding the wound divided by average intensity of signal prior to wounding (t = −00:06). Results presented as mean ± S.D.; n > 6. C. Quantification of Rho (top panel) and Cdc42 (bottom panel) activity in presence of DMSO or Sph2. Values represent average intensity of fluorescence signal in the zone surrounding the wound divided by average intensity of signal prior to wounding (t = −00:06). Results presented as mean ± S.D.; n ≥ 6.
Characterization of cytokinesis inhibition by Sph1 and Sph2
We next sought to characterize the features of cytokinesis inhibition by Sph1 and Sph2. Dose response curves were run with D. excentricus at two different temperatures, 12–14°C (sea table) and 22–25°C (room temperature) to reflect the fact that these organisms have a wide temperature tolerance for development and the fact that subsequent live imaging experiments were performed at RTo (see below). At 12–14°C, both Sph1 and Sph2 were effective down to the low micromolar range (Fig. 4A). At 22–25°C, Sph1 was even more potent, showing effects down to the mid-nanomolar range, while the potency of Sph2 were not obviously altered (Fig. 4B).
Figure 4. Sand dollar cytokinesis is inhibited by submicromolar concentrations of Sph1 and Sph2.
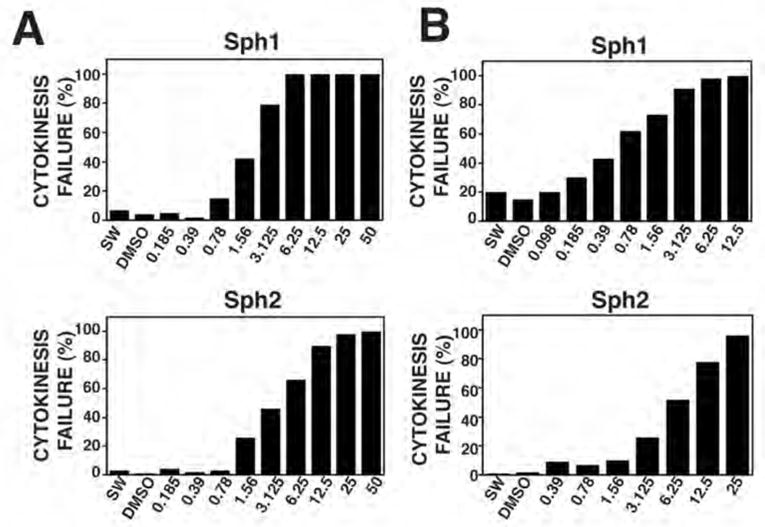
A. Dose response analysis of healing at 14°C in sea water (SW) or sea water containing DMSO (0.2%) or indicated concentrations (in μM) of Sph1 (top panel) or Sph2 (bottom panel). B. Dose response analysis of cytokinesis at RT in sea sater (SW) or sea water containing DMSO (0.2%) or indicated concentrations (in μM) of Sph1 (top panel) or Sph2 (bottom panel). n ≥ 80 for each concentration tested.
To better understand the effects of Sph1 and Sph2 on cytokinesis, embryos of D. excentricus or X. laevis were subject to time lapse imaging following treatment with these agents or, as a control, DMSO (Fig. 5). Fig. 5A provides single frames taken from time lapse movies of sand dollar zygotes beginning just at the onset of anaphase and ending prior to the point when the next round of division is about to commence. Neither Sph1 nor Sph2 prevented aster formation or enlargement (Fig. 5A; arrows), indicating that the metaphase-anaphase transition was not inhibited. However, later in the cell cycle, problems were clearly evident: The asters in Sph1-treated embryos did not appear to separate completely, and a shallow furrow formed but failed to ingress further. The asters in Sph2-treated embryos separated normally and the furrow ingresses quite far but then regressed. Fig. 5B provides single frames taken from time lapse movies of sand dollar embryos beginning at anaphase onset and ending after several cell cycles. At a point when controls had successfully cleaved several times, Sph1- and Sph2-treated embryos contained multiple asters trapped in a common cytoplasm (Fig. 5B, arrows). Likewise, Sph1-and Sph2-treated embryos contained multiple nuclei in a common cytoplasm, as assessed by Hoechst staining (not shown). Fig. 5C shows early cell divisions in X. laevis embryos. Sph1 treatment results in furrows that regress fail to ingress deeply (arrows) while Sph2 treatment results in tearing of the cortex in regions flanking the furrows (double arrows).
Figure 5. Sph1 and Sph2 produce distinct cytokinetic phenotypes.

A. Light micrographs from time lapse movies showing first mitosis in sand dollar embryos treated with DMSO (Con; 0.1%), Sph1 (4 μM) or Sph2 (5 μM). Images begin with start of anaphase with asters indicated by arrows. Time in min:sec. B. Light micrographs from time lapse movies showing several mitoses in sand dollar embryos treated with DMSO (Con; 0.1%), Sph1 (3 μM) or Sph2 (8 μM). Arrows at 01:20:00 time point in Sph1 and Sph2 samples show multiple asters in common cytoplasm. Time in hour:min:sec. C. Light micrographs from time lapse movies showing first early mitoses in Xenopus embryos treated with DMSO (Con; 0.1%), Sph1 (50 μM) or Sph2 (50 μM). Arrows in Sph1, 30:00 time point show furrows that subsequently regress. Double arrows in Sph2 53:00 show regions of membrane tearing that flank closing cleavage furrow. Time in min:sec and hour:min:sec.
We next employed confocal fluorescence microscopy to follow the details of cytokinesis inhibition. Because expression of fluorescent probes from microinjected mRNA required several hours (von Dassow et al., 2009) this approach could not be employed with Sph2, as this agent was less effective after the first cell division (see above). Thus, we focused our efforts on Sph1. Live imaging of microtubules using fluorescent EMTB (von Dassow et al., 2009) showed that Sph1 had no obvious effect on formation of astral microtubules after anaphase onset nor on the overall timing of events after anaphase, as judged by the reformation of the nuclei (Fig. 6A). In contrast, midzone microtubules, which form between the separating spindle poles, were far less abundant in Sph1 treated embryos (Fig. 6A).
Figure 6. Sph1 inhibits midzone formation.

A. Fluorescence micrographs from time lapse imaging of microtubules in control (Con) and Sph1 (10 μM) treated sand dollar embryos at anaphase onset. Both samples have numerous astral microtubules but only the control shows robust microtubules between the spindle poles. Arrows (04:00 in control; 05:00 in Sph1) show reforming nuclei. B. Fluorescence micrographs from time lapse imaging of microtubules (red) and MgcRacGAP (green) in control (Con) and Sph1 (5 μM) treated sand dollar embryos. Top rows, double labeling; bottom rows, MgcRacGAP only. Control shows recruitment of MgcRacGAP to midzone (arrow); Sph1-treated sample fails to recruit MgcRacGAP to midzone. Double arrow shows midbody from previous division; asterisks show MgcRacGAP accumulating in reforming nuclei.
As an alternative approach to assess the effect of Sph1 on spindle midzones, the distribution of sea urchin MgcRacGAP, tagged with 3XGFP, was followed in controls and Sph1-treated embryos. MgcRacGAP is a well-known marker of the spindle midzone (White and Glotzer, 2012). In controls, 3XGFP-MgcRacGAP clearly accumulated between separating asters after anaphase onset (arrow, Fig. 6B). In contrast, when Sph1 was applied ~15 min prior to anaphase onset, 3XGFP-MgcRacGAP fails to accumulate between the separating spindle poles, in spite of the fact that it is clearly evident in the midbody from the previous cell division (ie the division that occurred prior to Sph1 application; double arrows, Fig. 6B) and in the reforming nuclei after failed cytokinesis (asterisks, Fig. 6B). Thus, Sph1 inhibits formation of the spindle midzone.
DISCUSSION
In this study, we have identified two small molecules, provisionally dubbed sphinctostatin 1 and sphinctostatin 2 (Sph1 and Sph2), that impair both single cell wound repair and cytokinesis. The molecular targets of these agents remain to be identified, but based on the following considerations it appears likely that they are specific and their targets are not too far upstream from the core processes that control cytokinesis and single cell wound repair: First, Sph1 and Sph2 are not simply toxic, based on their effects on oocytes after overnight incubation. Second, in contrast to most of the other initial hits in the screen, these agents do not result in ATP depletion. Third, the effects of the two small molecules are concentration-dependent, as expected for agents with a specific target. Fourth, neither of these agents results in apparent F-actin and microtubule disassembly in resting cells at the concentrations required to inhibit wound repair and cytokinesis. Fifth, they do not cause cell cycle arrest, indicating that they do not inhibit protein synthesis since this is an essential requirement for cell cycle progression.
Target identification and comparison of the effects of Sph1 and Sp2 on other, related targets will be necessary to determine the relative specificity of these agents. Nonetheless, the range of concentrations required to perturb wound healing in oocytes is considerably less than agents such as the myosin-2 inhibitor blebbistatin or the Rho kinase inhibitor Y23753 (Burkel et al., 2012) and comparable to agents such as nocodazole (Benink and Bement, 2005) or taxol (Mandato and Bement, 2003), possibly reflecting the relative impermeability of Xenopus oocytes to many pharmacological agents. Similarly, the range of concentrations required to inhibit cytokinesis are less than trichostatin A (von Dassow et al., 2009), but comparable to nocodazole (Foe and von Dassow, 2009). These observations indicate that further studies designed to identify the targets of Sph1 and Sph2 are justified, as are studies designed to find more potent derivatives.
The immediate implication of the finding that two small molecules inhibit both cell wound repair and cytokinesis is that these who processes likely share in common specific macromolecular players. Single cell wound repair in several different systems entails local activation of Rho GTPases (Benink and Bement, 2005; Kono et al., 2012) and accumulation of F-actin and myosin-2 around wounds (Mandato and Bement, 2001; Abreu-Blanco et al., 2011; Lin et al., 2012), and the similarity of cellular wound repair and cytokinesis has been pointed out before (e.g. Mandato and Bement, 2001; Darenfed and Mandato, 2005). The question at hand is whether or not the known similarities can be used to deduce potential targets of the two agents. With respect to Sph2, the most logical targets are players involved in fusion of compartments with the plasma membrane. This presumption is based on the finding that Sph2 treatment of Xenopus embryos results in tearing of the plasma membrane during cytokinesis, as might be expected if closure of the contractile ring is not balanced by new membrane insertion, as previously described (e.g. Danilchik et al., 1998). Further support for this notion comes from the fact that membrane insertion in echinoderms occurs late during cytokinesis (Shuster and Burgess, 2002), consistent with the late failures of cytokinesis in the presence of Sph2 observed here. In addition, fusion of internal membrane compartments with the plasma membrane can trigger activation of Rho GTPases as a result of compartment mixing (Yu and Bement, 2009), which could explain why suppression of membrane fusion results in attenuation of GTPase signaling during wound healing. Given the fact that recycling endosomes have been implicated in cytokinesis (Neto et al., 2011), these compartments represent potential candidates as Sph2 targets. While there is currently no evidence that recycling endosomes participate in the single cell wound response, the identity of the compartments that undergo damage-induced fusion with the plasma membrane remains an open question (see Sonnemann and Bement, 2011).
The potential target(s) of Sph1 are even more intriguing. That is, analysis of wound healing in the presence of Sp1 reveals a reduction of Rho activity, while the phenotypic analysis of cytokinesis reveals failure of midzone formation, as judged both by imaging of microtubules and accumulation of MgcRacGAP. Two broad hypotheses could explain these results: 1) Sph1 has multiple targets. 2) Sph1 targets a macromolecule or cellular process that directly or indirectly regulates both single cell wound repair and cytokinesis. Based on the specificity considerations presented above, we favor the second hypothesis. While it might seem far-fetched that the relatively small number of known cytokinetic regulators participate in single cell wound repair, recent work indicates that both Ect2 and MgcRacGAP, conserved cytokinetic regulators, also play essential roles in the establishment of cell-cell junctions (Ratheesh et al., 2012). Thus, it may be that proteins previously assigned specific roles in cytokinesis have other, as yet unanticipated functions. Further, suppression of Rho activation in echinoderm embryos (Foe and von Dassow, 2008) and other cell types (Kanada et al., 2009) suppresses midzone formation and/or destabilizes the midzone. Thus, assuming the target of Sph1 directly or indirectly effects Rho activity, both phenotypes could be accounted for.
Based on the observations presented above, further investigation of the targets of Sph1 and Sph2 are warranted, as are studies designed to find more potent derivatives. However, this study also provides a proof of the principle that small molecule screens can be usefully employed in a deliberately “low tech” manner to study otherwise highly complex processes. That is, as noted in the Introduction, the great majority of studies employing small molecule libraries have focused on assays that are either automated, employ readouts based on a limited number of components rather than intact cells, or both. Here, the initial screens were not automated, but rather based on visual screens employing dissecting microscopes, and used intact oocytes or embryos.
The disadvantage of this approach is that it is comparatively slow, or “low throughput” to use the standard terminology. However, this shortcoming is not so dire: we found that 100 or more compounds could be screened in a day by a single person, indicating that screening of larger libraries is entirely feasible.
The advantages of this approach are several: First, like classic forward genetics, it is truly blind and therefore has the potential to reveal completely novel players and subprocesses important for complex biological events. Second, when coupled with appropriate secondary screens, not only are hits identified in the primary screen that are of less interest easy to eliminate, their likely mode of action can be rapidly narrowed. This is true not only for relatively sophisticated secondary screens such as imaging of specific molecules, but also for DIC-based imaging of cell division itself. For example, loss or depletion of microtubules is readily manifest as smaller asters and displaced spindles. Third, in contrast to screens that are based on automated screens or on highly focused readouts involving purified or partially purified components, the approach used here has the potential to identify agents that have important biological effects independent of the original goal of the screen. For example, simply letting the embryos sit in the 96 well plates overnight could permit the identification of, for example, inhibitors of gastrulation or ciliogenesis. Finally, the low tech nature of the primary screen makes it accessible to situations where resources are relatively limited such as small laboratories or laboratories that are focused on education. The potential for this approach in the latter case is particularly striking. That is, given the growing number of small molecules in search of an application, and the wealth of undergraduate biology students who need lab experience, it would seem to be logical to institute lab classes designed around small molecule screens, with corresponding efforts to disseminate the findings from different screens.
Acknowledgments
This work was supported by grants from the NIH to WMB (GM52932) and GF (GM077622) and from the NSF to GvD (MCB-0917887)
References
- Abreu-Blanco MT, Verboon JM, Parkhurst SM. Cell wound repair in Drosophila occurs through three distinct phases of membrane and cytoskeletal remodeling. J Cell Biol. 2011 May 2;193(3):455–64. doi: 10.1083/jcb.201011018. Epub 2011 Apr 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkel BM, von Dassow G, Bement WM. Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil Cytoskeleton. 2007 Nov;64(11):822–32. doi: 10.1002/cm.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkel BM, Benink HA, Vaughan EM, von Dassow G, Bement WM. A Rho GTPase Signal Treadmill Backs a Contractile Array. Dev Cell. 2012 Aug 14;23(2):384–96. doi: 10.1016/j.devcel.2012.05.025. Epub 2012 Jul 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilchik MV, Funk WC, Brown EE, Larkin K. Requirement for microtubules in new membrane formation during cytokinesis of Xenopus embryos. Dev Biol. 1998 Feb 1;194(1):47–60. doi: 10.1006/dbio.1997.8815. [DOI] [PubMed] [Google Scholar]
- Darenfed H, Mandato CA. Wound-induced contractile ring: a model for cytokinesis. Biochem Cell Biol. 2005 Dec;83(6):711–20. doi: 10.1139/o05-164. Review. [DOI] [PubMed] [Google Scholar]
- Foe VE, von Dassow G. Stable and dynamic microtubules coordinately shape the myosin activation zone during cytokinetic furrow formation. J Cell Biol. 2008 Nov 3;183(3):457–70. doi: 10.1083/jcb.200807128. Epub 2008 Oct 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NS. Combinatorial libraries and biological discovery. Curr Opin Neurobiol. 2001 Oct;11(5):608–14. doi: 10.1016/s0959-4388(00)00257-9. Review. [DOI] [PubMed] [Google Scholar]
- Green RA, Paluch E, Oegema K. Cytokinesis in Animal Cells. Annu Rev Cell Dev Biol. 2012 Jul 9; doi: 10.1146/annurev-cellbio-101011-155718. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Kanada M, Nagasaki A, Uyeda TQ. Stabilization of anaphase midzone microtubules is regulated by Rho during cytokinesis in human fibrosarcoma cells. Exp Cell Res 2009. 2009 Oct 1;315(16):2705–14. doi: 10.1016/j.yexcr.2009.06.027. Epub 2009 Jul 1. [DOI] [PubMed] [Google Scholar]
- Kono K, Saeki Y, Yoshida S, Tanaka K, Pellman D. Proteasomal degradation resolves competition between cell polarization and cellular wound healing. Cell. 2012 Jul 6;150(1):151–64. doi: 10.1016/j.cell.2012.05.030. Epub 2012 Jun 21. [DOI] [PubMed] [Google Scholar]
- Lin P, Zhu H, Cai C, Wang X, Cao C, Xiao R, Pan Z, Weisleder N, Takeshima H, Ma J. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. 2012 May;26(5):1875–83. doi: 10.1096/fj.11-188599. Epub 2012 Jan 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomenick B, Olsen RW, Huang J. Identification of direct protein targets of small molecules. ACS Chem Biol. 2011 Jan 21;6(1):34–46. doi: 10.1021/cb100294v. Epub 2010 Nov 30. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandato CA, Bement WM. Contraction and polymerization cooperate to assemble and close actomyosin rings around Xenopus oocyte wounds. J Cell Biol. 2001 Aug 20;154(4):785–97. doi: 10.1083/jcb.200103105. Epub 2001 Aug 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandato CA, Bement WM. Actomyosin transports microtubules and microtubules control actomyosin recruitment during Xenopus oocyte wound healing. Curr Biol. 2003 Jul 1;13(13):1096–105. doi: 10.1016/s0960-9822(03)00420-2. [DOI] [PubMed] [Google Scholar]
- Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999 Oct 29;286(5441):971–4. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- Miller AL, Bement WM. Regulation of cytokinesis by Rho GTPase flux. Nat Cell Biol. 2009 Jan;11(1):71–7. doi: 10.1038/ncb1814. Epub 2008 Dec 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neto H, Collins LL, Gould GW. Vesicle trafficking and membrane remodelling in cytokinesis. Biochem J. 2011 Jul 1;437(1):13–24. doi: 10.1042/BJ20110153. Review. [DOI] [PubMed] [Google Scholar]
- Ratheesh A, Gomez GA, Priya R, Verma S, Kovacs EM, Jiang K, Brown NH, Akhmanova A, Stehbens SJ, Yap AS. Centralspindlin and α-catenin regulate Rho signalling at the epithelial zonula adherens. Nat Cell Biol. 2012 Aug;14(8):818–28. doi: 10.1038/ncb2532. Epub 2012 Jul 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuster CB, Burgess DR. Targeted new membrane addition in the cleavage furrow is a late, separate event in cytokinesis. Proc Natl Acad Sci U S A. 2002 Mar 19;99(6):3633–8. doi: 10.1073/pnas.052342699. Epub 2002 Mar 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnemann KJ, Bement WM. Wound repair: toward understanding and integration of single-cell and multicellular wound responses. Annu Rev Cell Dev Biol. 2011 Nov;10(27):237–63. doi: 10.1146/annurev-cellbio-092910-154251. Epub 2011 Jun 20. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Haggarty SJ, Schreiber SL. High-throughput screening of small molecules in miniaturized mammalian cell-based assays involving post-translational modifications. Chem Biol. 1999 Feb;6(2):71–83. doi: 10.1016/S1074-5521(99)80004-0. [DOI] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science. 2003 Mar 14;299(5613):1743–7. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Titov DV, Liu JO. Identification and validation of protein targets of bioactive small molecules. Bioorg Med Chem. 2012 Mar 15;20(6):1902–9. doi: 10.1016/j.bmc.2011.11.070. Epub 2011 Dec 20. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Dassow G, Verbrugghe KJ, Miller AL, Sider JR, Bement WM. Action at a distance during cytokinesis. J Cell Biol. 2009 Dec 14;187(6):831–45. doi: 10.1083/jcb.200907090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EA, Glotzer M. Centralspindlin: At the heart of cytokinesis. Cytoskeleton (Hoboken) 2012 Aug 24; doi: 10.1002/cm.21065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TY, Wagner KW, Bursulaya B, Schultz PG, Deveraux QL. Development and characterization of nonpeptidic small molecule inhibitors of the XIAP/caspase-3 interaction. Chem Biol. 2003 Aug;10(8):759–67. doi: 10.1016/s1074-5521(03)00157-1. [DOI] [PubMed] [Google Scholar]
- Yu HY, Bement WM. Control of local actin assembly by membrane fusion-dependent compartment mixing. Nat Cell Biol. 2007 Feb;9(2):149–59. doi: 10.1038/ncb1527. Epub 2007 Jan 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Mc Henry KT, Lane WS, Fenteany G. A chemical inhibitor reveals the role of Raf kinase inhibitor protein in cell migration. Chem Biol. 2005 Sep;12(9):981–91. doi: 10.1016/j.chembiol.2005.07.007. [DOI] [PubMed] [Google Scholar]