

Abstract
Background
Chromosomal and genomic aberrations are common features of human cancers. However, chromosomal numerical and structural aberrations, breakpoints and disrupted genes have yet to be identified in esophageal squamous cell carcinoma (ESCC).
Methods
Using multiplex-fluorescence in situ hybridization (M-FISH) and oligo array-based comparative hybridization (array-CGH), we identified aberrations and breakpoints in six ESCC cell lines. Furthermore, we detected recurrent breakpoints in primary tumors by dual-color FISH.
Results
M-FISH and array-CGH results revealed complex numerical and structural aberrations. Frequent gains occurred at 3q26.33-qter, 5p14.1-p11, 7pter-p12.3, 8q24.13-q24.21, 9q31.1-qter, 11p13-p11, 11q11-q13.4, 17q23.3-qter, 18pter-p11, 19 and 20q13.32-qter. Losses were frequent at 18q21.1-qter. Breakpoints that clustered within 1 or 2 Mb were identified, including 9p21.3, 11q13.3-q13.4, 15q25.3 and 3q28. By dual-color FISH, we observed that several recurrent breakpoint regions in cell lines were also present in ESCC tumors. In particular, breakpoints clustered at 11q13.3-q13.4 were identified in 43.3% (58/134) of ESCC tumors. Both 11q13.3-q13.4 splitting and amplification were significantly correlated with lymph node metastasis (LNM) (P = 0.004 and 0.022) and advanced stages (P = 0.004 and 0.039). Multivariate logistic regression analysis revealed that only 11q13.3-q13.4 splitting was an independent predictor for LNM (P = 0.026).
Conclusions
The combination of M-FISH and array-CGH helps produce more accurate karyotypes. Our data provide significant, detailed information for appropriate uses of these ESCC cell lines for cytogenetic and molecular biological studies. The aberrations and breakpoints detected in both the cell lines and primary tumors will contribute to identify affected genes involved in the development and progression of ESCC.
Background
Chromosomal and genomic rearrangements are significant features of malignant human tumors. Rearrangements are often associated with structural aberrations, such as translocations, insertions and inversions. They could also result in the copy number alterations (CNAs) [1,2]. Characterizing rearrangements and genes affected by the aberrations and breakpoints might help us to understand tumor development and progression better.
The products and implications of chromosomal rearrangements (e.g., fusion genes, truncated genes, and gene dysregulation by ectopic promoters) have been described in leukemia, lymphoma, sarcomas, and epithelial cancers [3,4]. It was initially difficult to detect chromosomal rearrangements and affected genes in the epithelial cancers, mainly due to the technical difficulty of preparing metaphase spreads from primary epithelial tumors and the karyotypic complexity. Until recently, multiple gene rearrangements and even genomic landscapes which reflect the structural aberrations throughout the genomes have been identified in multiple types of epithelial cancers, including prostate cancer [5,6], breast cancer [7,8], lung cancer [9,10], colorectal cancer [11], gastric cancer [12], head and neck cancer [13], hepatocellular carcinoma [14] and so on.
Recently, it has been reported that recurrent rearrangements could affect genes at the boundaries of CNAs [2,15], thus recurrent breakpoints might be important for screening and identifying frequent unbalanced rearrangements and the involved genes. Multiplex-fluorescence in situ hybridization (M-FISH) [16] and spectral karyotyping (SKY) [17] were designed to replace traditional G-banding in chromosomal analyses of tumor cells, but the resolution of these techniques is not sufficient to detect small rearrangements. Array-based comparative genomic hybridization (array-CGH) was developed to analyze the CNAs, including genomic gains, losses, amplifications and deletions [18,19]. It was recently demonstrated that array-CGH could be used to identify unbalanced breakpoints of the rearrangements in many types of cancer cells at a potentially higher resolution [20-24]. Array-CGH has also been used, in combination with cytogenetic information, to determine the breakpoints in reciprocal translocations [25].
Esophageal cancer (EC) is a common malignant epithelial cancer worldwide, causing more than 40,000 deaths each year [26]. The most prevalent type of EC is esophageal squamous cell carcinoma (ESCC), and China is among the highest risk areas [26,27]. Recently, our group reported the karyotype of ESCC cell line KYSE180 [28] and KYSE450 [29] by 12-color M-FISH, as well as the karyotype of KYSE410-4 by 6-color M-FISH [30]. CGH [31-34], SKY and CGH [35], and array-CGH [36-38] experiments from other groups have also been performed on ESCC cell lines and primary tumors. These studies have revealed numerical and structural chromosomal aberrations. However, genomic rearrangements, breakpoints and genes that are involved in ESCC remain to be decoded and clarified.
Our study intended to identify candidate recurrent breakpoints which might affect genes at or near the boundaries. In this study, we describe CNAs and unbalanced genetic rearrangements in six ESCC cell lines through a combination of M-FISH and 44K array-CGH techniques. We found recurrent breakpoint regions in the cell lines and breakage of several regions present in primary ESCC tumors, which may contribute to disruption of critical genes.
Methods
Cell lines and sample collection
ESCC cell lines KYSE30, KYSE150, KYSE180, KYSE450, KYSE510 and YES2 were kindly provided by Yutaka Shimada (Kyoto University, Japan). KYSE150 and KYSE510 were established from female patients, and KYSE30, KYSE180, KYSE450 and YES2 were from male patients. Each cell line was cultured in RPMI-1640 (Invitrogen, USA) supplemented with 10% fetal calf serum (FCS). ESCC tissue samples were procured from Chinese Academy of Medical Sciences Cancer Hospital. All the samples used in this study were residual specimens collected after diagnosis sampling. And all patients received no treatment before surgery, and signed separate informed consent forms for the sampling and molecular analyses. This study has been approved by the Ethics Committee/IRB of Cancer Institute (Hospital), PUMC/CAMS.
Metaphase chromosomes and interphase cell nuclei preparations
Metaphase chromosomes from ESCC cell lines and normal peripheral blood lymphocytes were harvested after incubation with 0.04 μg/ml Colcemid (Invitrogen) at 37°C for 1-2 hours, followed by treatment with a hypotonic solution (0.075 mol/L KCl) for 30 minutes and three successive changes of the fixative solution (methanol/acetic acid, 3:1). ESCC tissue samples were cut into small pieces in phosphate-buffered saline (PBS), and the interphase nuclei were then prepared following the procedures described above. Metaphase chromosomes and interphase cell nuclei in suspensions were stored at 4°C overnight and then stored at -20°C until use. The nuclear suspensions were dropped onto clean slides and aged at room temperature for 2-3 days prior to the FISH experiments.
Fluorescence in situ hybridization (FISH)
M-FISH was performed on the metaphase spreads. The 24-color whole chromosome painting (WCP) and arm-specific probes were directly labeled with diethylaminocoumarin (DEAC)-dUTP (PerkinElmer Inc., USA), Green-dUTP (Abbott Molecular, USA), Cy3-dUTP (GE Healthcare, USA) or Alexa594-dUTP (Invitrogen) by degenerate oligonucleotide primed-polymerase chain reaction (DOP-PCR) [39] or were indirectly labeled with Biotin-11-dCTP (Invitrogen). Biotin was then visualized with streptavidin-conjugated Cy5 (Jackson Immunoresearch Laboratories Inc., USA). The 12-color FISH probes were labeled with Green-dUTP, Cy3-dUTP, Alexa594-dUTP and Cy5-dUTP.
Split regions were detected using dual-color break-apart bacterial artificial chromosome (BAC) DNA clone probes, which were labeled with Green-dUTP and Cy3-dUTP by random priming using BioPrime DNA labeling system (Invitrogen). BAC DNA clones were selected according to their descriptions in Ensembl database ( http://www.ensembl.org). BAC DNA clones used to detect splitting of 11q13.3-q13.4 and associated regions included NONSC16D6 (68,072,319-68,278,585, hg18), Cancer_1D11 (69,162,501-69,323,924, hg18), NONSC2E5 (70,236,623-70,391,405, hg18), NONSC3C5 (71,992,715-72,182,751, hg18) and NONSC15F5 (75,107,934-75,273,492, hg18).
The slides for M-FISH and dual-color break-apart FISH analyses were pretreated with RNase A (100 mg/ml in 2 x saline sodium citrate [SSC]) and pepsin (50 mg/ml in 0.01 mol/l HCl). The slides were subsequently denatured in 70% formamide/2 x SSC at 73°C-75°C for 3 minutes, quickly cooled with two rinses of 2 x SSC at 4°C, dehydrated in a gradient series of ethanol (75%, 85% and 100%), and air dried. The labeled probes were precipitated, and redissolved in the hybridization solution (50% formamide, 10% dextran sulfate, 1% Tween-20, 2 x SSC), denatured at 75°C for 8 minutes, and quick-chilled on ice for 2 minutes. Hybridization was performed in a humid chamber at 37°C for 24-48 hours. Post-hybridization washes were performed in 50% formamide/2 x SSC for 15 minutes at 43°C and were performed twice for 3 minutes each in 2 x SSC. The slides were dehydrated in 75%, 85% and 100% ethanol, air dried, counterstained with 40,6-diamidino-2-phenylindole (DAPI) (1 mg/ml) and covered with coverslips.
For 12-color FISH analysis [28], the slides were hybridized twice on metaphase spreads as previously described, which was named two-round FISH. After digital fluorescence image acquisition, coverslips on the slides were removed by dipping in 100% ethanol for 30 min, and washed twice in 100% ethanol for 3 min each time, then air dried, and then the slides could be denatured as the above procedures.
Microscopy and digital image analysis
FISH images were captured using a Zeiss Axio fluorescence microscope equipped with a cooled charged-coupled device (CCD) camera (Princeton Instruments, USA) or a JAI M4 Plus CCD camera (Metasystems International, Germany). All of the fluorescent images were captured with individual single-band-pass filters specific for visualizing DAPI, DEAC, Green, Cy3, Alexa 594 and Cy5 fluorochromes. Pseudo-color images were constructed and analyzed using MetaMorph (Universal Imaging Corporation, USA) or Metacyte module of Metafer imaging systems (Metasystems International).
Genomic DNA isolation and oligo array-based comparative genomic hybridization (array-CGH)
Genomic DNA from ESCC cell lines was isolated using DNeasy Blood & Tissue Kit (Qiagen, Germany). Genome-wide copy number studies were then performed using an Agilent 44K oligo array platform (Agilent Technologies, USA), with sex-matched normal human DNA (Promega Corporation, USA) used as the reference. Briefly, 1 μg samples of the tested and reference DNA were digested with AluI and RsaI, and differentially labeled with Cy3-dUTP and Cy5-dUTP using Agilent Genomic DNA Enzymatic Labeling Kit Part Number 5190-0449 (Agilent Technologies), respectively. Then Microcon YM-30 (Millipore) was used to clean up the labeled probes. Tested and reference DNA probes were combined and hybridized onto the microarrays enclosed in Agilent SureHyb-enabled hybridization chambers for 40 hours. After hybridization, slides were washed sequentially and scanned with an Agilent DNA Microarray Scanner. Annotations for the probes were based on UCSC hg18 (NCBI Build 36). CNAs and breakpoint data were analyzed via the Agilent Genomic Workbench Software 5.0, set to use the ADM-2 algorithm, an aberration threshold of 5.0 and an absolute average log2 ratio ≥ 0.5.
Statistical analysis
Statistical analyses were carried out by using the SPSS 17.0 software package. The association between splitting of breakpoint regions and clinico-pathological characteristics were assessed by the χ2 test, Fisher’s exact test or Kruskal–Wallis test. Logistic regression analysis was performed to determine the independent predictors of lymph node metastasis. P values < 0.05 were considered significant.
Results
Copy number alterations
M-FISH was performed on the metaphase chromosomes of four ESCC cell lines (KYSE30, KYSE150, KYSE510 and YES2). Modal karyotypes of the cell lines are shown in Figure 1. M-FISH karyotypes of two other cell lines KYSE180 [28] and KYSE450 [29] have been previously reported by our laboratory. We found multiple numerical alterations in the six cell line, which exhibited high level of aneuploidy. An overview of CNAs indicated that imbalances occurred throughout the entire genome of the cell lines. Gains were observed at 1p, 1q, 3p, 3q, 4p, 5q, 7p, 8q, 9q, 11q, 14q, 16p, 16q, 17p, 17q, 18p, 19p, 19q, 20q, and 22q. Losses were primarily detected at 3p, 4p, 4q, 6p, 6q, 9p, and 18q.
Figure 1.
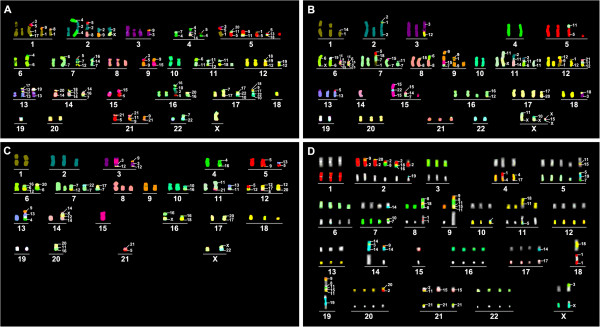
M-FISH profiling of ESCC cell lines. A 24-color analysis technique was used for KYSE30 (A), KYSE150 (B), and YES2 cells (C) and 12-color detection was used for KYSE510 cells (D).
The detail CNAs of these cell lines were detected by array-CGH, and the profiles of gains and losses are shown in Figure 2 and Additional file 1: Table S1. Our results were compared with the data available from Cancer Cell Line Project on the Wellcome Trust Sanger Institute Cosmic website ( http://www.sanger.ac.uk/genetics/CGP/cosmic). Copy number data of KYSE150, KYSE450 and KYSE510 on the website were analyzed using Affymetrix SNP6.0 arrays. Copy number profiles derived from our Agilent 44K platform are very similar to those from the Affymetrix platform. We then compared CNAs among the six cell lines according to the array-CGH data, and frequent gains and losses in at least two cell lines were summarized in Table 1. More gains were found than losses. The results were combined with the data from other 17 ESCC cell lines available on Cosmic website, including KYSE70, KYSE140, KYSE270, KYSE410, KYSE520, Colo-68N, EC-GI-10, HCE-4, TE-1, TE-5, TE-6, TE-8, TE-9, TE-10, TE-11, TE-12 and TE-15. The gains with high frequencies were shown in Additional file 2: Table S2.
Figure 2.

CNAs and unbalanced breakpoints in six ESCC cell lines detected by array-CGH. Gains and amplifications are presented as lines on the right side of the chromosomes, while the lines for losses and deletions are on the left side. Unbalanced breakpoints are at the boundaries of CNAs. Numbers on top of the lines are indicated as the cell lines. 30: KYSE30, 150: KYSE150, 180: KYSE180, 450: KYSE450, 510: KYSE510, 2: YES2.
Table 1.
Frequent gains and losses in six ESCC cell lines analyzed by array-CGH
| CNAs | Positive No. of cell lines | Region |
|---|---|---|
| Gain |
≥5 |
3q26.33-qter, 5p14.1-p11, 7pter-p12.3, 8q24.13-q24.21, 9q31.1-qter, 11p13-p11, 11q11-q13.4, 17q23.3-qter, 18pter-p11, 19, 20q13.32-qter |
| |
3-4 |
1q21.1-q31.1, 2p24.1-p11, 3q13.31-q26.33, 5pter-p11, 5q31.1-qter, 5q32-qter, 6p21.1-p11, 6p21.31-p11, 7pter-p12.3, 7q11-q22.2, 8q21.2-q24.21, 8q23.3-q24.21, 9q22.1-qter, 10q26.3-qter, 11q11-q22.3, 11q13.4-qter, 12pter-p14.4, 16pter-p11, 16q11-qter, 17p13.1-p11, 17q, 20q11-qter, 20q13.12-qter |
| |
2 |
1pter-p36.13, 1p35.2-q31.1, 1p22.2-q31.1, 2pter-p25.1, 2p24.2-p11, 2q11-q13, 3q12.1-qter, 5q15-qter, 6p21.33-p21.1, 7q35-qter, 8q11-q24.21, 9q21.33-qter, 11pter-p14.1, 12q14.4-qter, 13q21.32-q31.3, 13q32.1-qter, 14q22.1-q31.1, 15q11, 15q25.3-qter, 16q11-qter, 17pter-p11, 18q11-q12.2, 22q |
| Loss |
≥5 |
18q21.1-qter |
| |
3-4 |
3p14.1-p11, 4p15.32-p14, 4q22.1-q32.3, 9pter-p24.1, 9p23-p11, 11q23.3-qter, 18q12.2-q21.1, Xpter-p11, Xq21.1-q23 |
| 2 | 2q33.1-q35, 3p14.2-p14.1, 4pter-p14.1, 4q21.1-q32.3, 4q22.1-qter, 6pter-p24.1, 6q11-q13, 7q22.2-qter, 8pter-p22, 9pter-p11, 9q11-q21.13, 10p12.33-p11, 11q24.1-qter, 11q22.3-qter, Xq11-qter, |
Regions with average log2ratio values greater than 1 were defined as amplifications. High-level amplifications (HLAs) and homozygous deletions (HDs) were identified if the absolute average values were no less than 2. According to the positions of HLAs and HDs boundaries, the smallest HLA and HD regions and involved genes among these cell lines were listed in Table 2. HLAs include 7p11 (2/6, 33%), 8q24.21 (2/6, 33%) and 11q13.3-q13.4 (3/6, 50%), harboring several oncogenes, including EGFR, MYC and CTTN (Table 2). Homozygous deletion of 9p21.3, containing tumor suppressor genes CDKN2A, CDKN2B and CDKN2B-AS1, occurred in 67% (4/6) of the cell lines.
Table 2.
High level amplifications and homozygous deletions in different cell lines
| CNAs | Region a | Cell line | Start | Stop | Genes |
|---|---|---|---|---|---|
| HLA |
7p11 |
KYSE30, KYSE450 |
54572103 |
57613746 |
VSTM2A, SEC61G, EGFR, LANCL2, VOPP1, FKBP9L, ZNF713, MRPS17, GBAS, PSPH, CCT6A, SUMF2, PHKG1, CHCHD2, ZNF479, ZNF716 |
| |
8q24.21 |
KYSE450, KYSE510 |
128548643 |
129216964 |
POU5F1B, LOC727677, MYC, PVT1 |
| |
11q13.3-q13.4 |
KYSE30, KYSE180, KYSE510 |
68232360 |
70964483 |
MTL5, CPT1A, MRPL21, IGHMBP2, MRGPRD, MRGPRT, TPCN2, MYEOV, CCND1, ORAOV1, FGF19, FGF4, FGF3, ANO1, FADD, PPFIA1, CTTN, SHANK2, DHCR7, NADSYN1, KRTAP5-7, KRTAP5-8, KRTAP5-9, KRTAP5-10, KRTAP5-11 |
| HD | 9p21.3 | KYSE180, KYSE450, KYSE510, YES2 | 21958099 | 22136626 | CDKN2A, CDKN2B, CDKN2B-AS1 |
HLA: high level amplification; HD: homozygous deletion; the Start and Stop positions are annotated by hg18.
a Regions of amplifications or deletions in at least two cell lines.
Unbalanced breakpoints
Breakpoints were restricted to the boundaries between two adjacent DNA fragments with significantly distinctive log2 ratio values, reflecting different copy numbers. Using this scheme, 261 candidate unbalanced breakpoints were identified ( Additional file 3: Table S3). Among these candidates, 39 occurred in the centromeric regions, and the other 224 were present on chromosome arms. Fifty-seven of arm breakpoints were localized in the vicinity of fragile sites. Breakpoints on chromosome arms and copy number status of the regions at both sides of the breakpoints were listed in Additional file 3: Table S3. Cell lines were ranked according to the number of breakpoints, and the top three were KYSE30, KYSE510 and YES2, respectively. This tendency was similar to that in M-FISH results.
Chromosomal structural aberrations
M-FISH results of four cell lines (KYSE30, KYSE150, KYSE510 and YES2) as well as that of the previously reported two cell lines (KYSE180 and KYSE450) showed that a total of 156 derivative chromosomes resulted from translocations, most of which were unbalanced; only 12.8% (20/156) were reciprocal. Approximately, 35% of the translocation derivative chromosomes were fused at the centromeric regions. Chromosomes 1, 2, 3, 5, 6, 7, 8, 9, 11, 12, 14, 15 and X were frequently rearranged. Combining M-FISH with array-CGH, we further characterized multiple rearrangements present in these cell lines (Table 3). KYSE30 is the cell line with the most complex rearrangements, and array-CGH results have also indicated that much more breakpoints were present in KYSE30 than the other cell lines, which are consistent with M-FISH results.
Table 3.
Chromosomal structural aberrations analyzed by a combination of M-FISH and array-CGH
| Cell line | Chromosomal structural aberrations |
|---|---|
| KYSE30 |
der(1)t(1;8)(p10;q?), der(1;9)(p10;p10), der(1)(3qter → 3q?::5p11 → 5p14.1::1p35.3 → 1q23.2::17p10 → 17p13.1:), der(2;4)(4pter → 4p10::2pter → 2q31.2::4q10 → 4qter), der(2)t(2;9)(p10;q22.3), der(2)t(2;X)(p10;p11), der(2)(5pter → 5p14.1::2p16.2 → 2q31.2::8q10 → 8qter), der(3)t(3;9)(3q10 → qter::9q22.1 → q22.33:), der(4)t(4;5)(q10;p11), der(4)t(4;7)(q13.3;p15.1), der(4)(4pter → 4p10::3q26.33 → 3qter::7p12.1 → 7p11::14q?), der(5)t(1;5)(p36.13;p10), der(5;11)(q10;p10), der(5)(3q? → 3qter::5q10 → 5q15::1q23.2 → 1qter), der(5)(3q26.33 → 3qter::5p14.1 → 5p10::9q22.1 → 9qter), der(5)(5pter → 5p10::11q13.3 → 11q13.4::20q11 → 20q11.23:), der(5)(:5p14.1 → 5p10::13q?::18q11:), der(6)t(4;6)(q?;p24.1), der(7)t(6;7)(p22.3;p14.3), der(5;7)(:5p14.1 → 5p10::7p14.3 → 7q22.1::12p10 → 12pter), der(7)(16pter → 16p11;14q?;7p12.1 → 7q22.1:), der(15)(9q22.3 → 9qter::15q10 → 15qter), der(11)t(6;11)(p10;p12), der(11;18)(p10;p10), der(7;11)(:11q10 → 11q13.3::7q10 → 7q22.1::12q11 → 12q14.1:), der(12;19)(p10;p10), der(12)(12pter → 12q14.1::19p?::18p?), der(13;19)(q10;q10), der(17)(:13q21.32 → 13q31.3::17p13.1 → 17p10::13q21.32 → 13q31.3::17p13.1 → 17p10:), der(14;20)(q10;p10), der(?)(14q?::16p?::7p11 → 7p12.1:), der(14)(18pter → 18p10::13q?::14q10 → 14q31.1:), der(15)t(5;15)(q15;q10), der(16;20)(q10;p10), der(16)(16pter → 16p10::10pter → 10p11.21::2q11 → 2q33.1::?), der(17)(:7p12.1 → 7p11::17q10 → 17qter::17q10 → 17qter), der(17;20)(:17p13.1 → 17p10::22q10 → 22q11.21::10q26.3 → 10qter), der(17)(:18q11::9q22.1 → 9q22.33::17p? → 17p10::22q12.1 → 22q12.3::10p11.21 → 10pter), der(21)t(5;21)(q15;q10), der(21)t(9;21)(q22;q10), der(21)(:9q22.1 → 9q22.33::11q13.3 → 11q13.4::21q10 → 21qter), der(22)t(7;22)(p12.1;q10) |
| KYSE150 |
der(1)t(1;14)(q31.1;q1?), der(2)(2pter → 2p24.3::1q32.2 → 1q41::2p24.1 → 2q35:), der(3)t(3;12)(p14.2;q14.1), der(5;11)(q10;p10), der(6)t(6;X)(q?;q27.1), t(15;6;15;6;15)(:15q11::6p22.2 → 6p21.33::15q10 → 15q11::6p12.2 → 6p12.1::15q11:), der(7)t(7;11)(p10;q?), der(7)t(7;15)(q22.1;q11), der(5;7)(1pter → 1p22.2::7q10 → 7q22.1::18q21.1 → 18q21.1::5p10 → 5pter), der(6;8)(p10;q10), der(8)t(8;18)(p22;p11), der(9)(:9p24.1 → 9p23::1q32.3 → 1q41::9p21.1 → 9qter), der(9)(9pter → 9q?::15q11 → 15qter::7q? → 7qter), der(11;19)(q10;p10), der(11)t(11;X)(p14.1;q10), der(11)(:2q35::11p14.1 → 11p10::6p22.2 → 6p21.33:), der(12)t(1;12)(?;q14.1), der(12)(1pter → 1q31.1::9p2? → 9p2?::12p10 → 12pter), t(21;11;12)(?::q?::q? → qter), der(13)t(5;13)(?;q10), der(?)(7q?::14q23.2 → 14qter::15q10 → 15q11:), der(14)(2qter → 2q35::11p1? → 11p1?::14q10 → 14qter), der(15;22)(15q10 → 15qter::22q10 → 22qter::15q10 → 15qter), der(16)t(12;16)(?;pter), der(17;20)(p10;q10), der(18)t(3;18)(p14.2;q10), der(6;19)(q10;p10), der(X)t(11;X)(p14.1;p10), t(X;15;X)(?::15q?::?) |
| KYSE510 |
der(2)t(2;8)(q10;q23.3), der(2;9)(q10;q10), der(2)t(2;19)(p10;q1?), der(2)t(2;20)(q33.1;q?), der(2)(2pter → 2p10::18q12.1 → 18q12.2::16p?), der(3;6)(q10;p10), der(4)t(1;4)(p21.1;q10), der(4)t(4;17)(q34.3;?), der(5)(5pter → 5p10::19q13.11 → 19q13.42::11q22.2 → 11q22.3::15q25.3 → 15qter::7pter → 7p12.3), der(7;10)(10q10 → 10qter::7q10 → 7q21.2::14q22.1 → 14q31.1:), der(8)(8pter → 8q12.1::1q21.2 → 1q23.3::18q12.1 → 18q12.2::1q21.2 → 1q23.3:: 8pter → 8q12.1), der(9)(9q10 → 9qter::8q23.3 → 8q24.21::11q13.2 → 11q13.4::15q25.3 → 15qter::11q11 → 11q13.2:), der(10)t(10;19)(q10;q12), der(11)t(11;18)(p10;q11), der(11)(5pter → 5p11::11p13 → 11q14.3::18pter → 18p11.31), der(14)t(9;14)(p11;q10), der(14)(14q?::3q28 → 3qter::14q22.1 → 14q31.1::14q?), der(15)t(15;21)(q10;q?), der(17)t(14;17)(?;q10), der(18)(:1q21.1 → 1q23.3::18pter → 18p10::1q23.3 → 1qter), der(11)(9pter → 9p21.3::19q13.11 → 19q13.42::8pter → 8p21.3::11q13.2 → 11q13.4::15q25.3 → 15qter::11q10 → q?), der(20)t(2;20)(q33.1;q10), der(21)t(15;21)(q25.3;q10), der(21)(:11p13 → 11p11::2p11 → 2p11::21q10 → 21q?), der(X)t(3;X)(p14.1;p22.31) |
| YES2 | der(3)t(3;12)(q29;?), der(3)(9pter → 9p21.3::3q10 → 3q13.31::?), der(4)t(4;16)(q?;p11), der(5)t(5;9)(q?;?), der(5)t(5;13)(p10;q32.1), der(6)t(6;20)(p12.3;q10), der(12)(16pter → 16p11::6p21.1 → 6p12.3::12q10 → 12q21.1:), der(7)t(7;12)(q11.21;q24.11), der(17)t(7;17)(q31.1;p10), der(22)t(7;22)(q11.21;q10), der(10)t(10;16)(p10;p11), der(11)t(11;13)(p10;q32.1), der(11)t(11;21)(pter;?), der(12)t(5;12)(q23.3;p10), der(12)t(12;20)(q21.1;q?), der(12)(12pter → 12p10::17q2?::8q23.3 → 8q24.1:), der(13)(9q21.32 → 9qter::13q10 → 13q32.1::?::4q?), der(14)(14pter → 14q12::X?::12q?::14q22.1 → 14qter), der(15)(15qter → q10::15q10 → 15qter), der(16)t(16;18)(p10;q11), der(16)t(16;X)(q11;?), der(17)(:17q10 → 17q24.3::20q11.21 → 20q?), der(20)(:16p11 → 16p11::11p13 → p11::20p10 → 20pter), der(21)t(5;21)(q10;q21.1), der(X)t(22;X)(q11;p10) |
Recurrent breakpoint regions in ESCC cell lines and primary tumor tissues
Positions of the breakpoints were compared among different cell lines, and further the distances of near breakpoint regions were calculated. Five regions (11q13.4, 9p21.3, 15q25.3, 3q28 and 10q26.3) had breakpoints less than 1 Mb apart, and twelve (11q13.3, 4p13, 11p13, 8q24.21, 2q35, 1q31.1, 21q21.1, 9p21.3, 18q12.2, 3p14.2, 3q12.1-q12.2 and 6p12.3-p12.2) had breakpoints less than 2 Mb apart in different cell lines (Table 4). For example, breakpoints at 11q13.4 were detected in KYSE30 and KYSE510, while breakpoints at 11q13.3 were detected in KYSE30 and KYSE180 (Figure 3A). The three cell lines presented gain of 11q and amplification of 11q13, in which copy numbers of the regions flanking centromere to 11q13 amplicon was higher than the region distal to the amplicon. Losses of the region distal to 11q13 were also found as del(11q13.4-qter) in KYSE30, del(11q14.3-q21) and del(11q22.3-qter) in KYSE510. Translocations of highly amplified regions were also observed (Table 3).
Table 4.
Recurrent breakpoint regions analyzed by array-CGH in ESCC cell lines
| Region | Cell line | CN status a | BP Intervals (hg 18) in array-CGH | Distance between cell lines b | Genes c | CFSs |
|---|---|---|---|---|---|---|
| 9p21.3 a |
KYSE180 |
Neutral/Del |
21958099-21968346 |
115.1 kb |
C9orf53§, CDKN2A
*
, CDKN2B†, CDKN2B-AS1† |
FRA9C |
| |
KYSE450 |
Neutral/Del |
21853263-21968346 |
|
MTAP§, C9orf53§, CDKN2A
*
|
|
| |
KYSE510 |
Loss/Del |
21958099-21968346 |
|
C9orf53§, CDKN2A
*
, CDKN2B†, CDKN2B-AS1† |
|
| |
YES2 |
Neutral/Del |
21853263-21957548 |
|
MTAP§, C9orf53†, CDKN2A† |
|
| 15q25.3 |
KYSE450 |
Neutral/Gain |
86361096-86429254 |
154.2 kb |
LINC00052§, NTRK3
*
, MRPL46†, MRPS11† |
|
| |
KYSE510 |
Neutral/Gain |
86275066-86429254 |
|
LINC00052§, NTRK3
*
, MRPL46†, MRPS11† |
|
| 11q13.4 |
KYSE30 |
Amp/Loss |
70964483-71305189 |
340.1 kb |
KRTAP5-11§, FAM86C1, DEFB108B, RNF121† |
FRA11H |
| |
KYSE510 |
Amp/Neutral |
70964483-71305189 |
|
KRTAP5-11§, FAM86C1, DEFB108B, RNF121† |
|
| 3q28 |
KYSE180 |
Gain/Gain |
191171376-191222891 |
517.0 kb |
TP63§, LEPREL1
*
, CLDN1† |
|
| |
KYSE510 |
Neutral/Gain |
191610761-191688399 |
|
CLDN1§, CLDN16, TMEM207†, IL1RAP† |
|
| 10q26.3 |
KYSE30 |
Neutral/Gain |
133045086-133476780 |
801.8 kb |
TCERG1L§, PPP2R2D† |
|
| |
YES2 |
Neutral/Gain |
133795639-133846905 |
|
BNIP3§, JAKMIP3
*
, DPYSL4† |
|
| 11q13.3 |
KYSE30 |
Amp/Amp |
69339391-69569221 |
1.05 Mb |
FGF3§, ANO1†, FADD† |
FRA11H |
| |
KYSE180 |
Amp/Amp |
70182767-70386856 |
|
CTTN§, SHANK2
*
, DHCR7† |
|
| 4p13 |
KYSE150 |
Amp/Loss |
41832777-42109513 |
1.15 Mb |
SLC30A9§, BEND4, SHISA3†, ATP8A1† |
|
| |
KYSE510 |
Neutral/Loss |
40955943-41226036 |
|
UCHL1§, LIMCH1
*
, PHOX2B† |
|
| 11p13 |
KYSE510 |
Neutral/Gain |
33107818-33136537 |
1.2 Mb |
TCP11L1§, PIGCP1§, CSTF3
*
, HIPK3† |
|
| |
YES2 |
Neutral/Gain |
34278741-34307224 |
|
NAT10§, ABTB2
*
, CAT† |
|
| 8q24.21 |
KYSE450 |
Amp/Neutral |
129216964-129574570 |
1.23 Mb |
FAM84B§, POU5F1B, MYC†, PVT1† |
|
| |
KYSE510 |
Amp/Loss |
129972316-130159085 |
|
FAM84B§, POU5F1B, MYC†, PVT1† |
|
| |
YES2 |
Amp/Neutral |
130159144-130451718 |
|
far from genes, PVT1§, GSDMC† |
|
| 2q35 |
KYSE30 |
Loss/Neutral |
217432472-218386863 |
1.37 Mb |
DIRC3§, TNS1, CXCR2P1† |
|
| |
KYSE150 |
Loss/Gain |
218517852-218801703 |
|
TNS1§, CXCR2P1, CXCR2, CXCR1, HMGB1P9, ARPC2† |
|
| 1q31.1 |
KYSE150 |
Gain/Loss |
186080345-186315797 |
1.40 Mb |
PLA2G4A§, FAM5C† |
FRA1K |
| |
KYSE450 |
Gain/Neutral |
184912220-185051701 |
|
PTGS2§, PLA2G4A† |
|
| 21q21.1 |
KYSE30 |
Gain/Neutral |
17060792-17145790 |
1.48 Mb |
USP25§, C21orf34§, CXADR† |
|
| |
YES2 |
Gain/Neutral |
18435266-18540695 |
|
CHODL-AS1§, CHODL†, TMPRSS15† |
|
| 9p21.3 b |
KYSE180 |
Del/Neutral |
21999029-22136626 |
1.45 Mb |
CDKN2A
*
, CDKN2B, CDKN2B-AS1, DMRTA1† |
FRA9C |
| |
KYSE450 |
Del/Neutral |
21980581-21993651 |
|
CDKN2A
*
, CDKN2B, CDKN2B-AS1
*
, DMRTA1† |
|
| |
KYSE510 |
Del/Loss |
22992377-23425976 |
|
DMRTA1
*
, ELAVL2† |
|
| |
YES2 |
Del/Neutral |
21999029-22136626 |
|
CDKN2B, CDKN2B-AS1, DMRTA1† |
|
| 18q12.2 |
KYSE450 |
Gain/Loss |
33583906-33747373 |
1.64 Mb |
CELF4§, LOC647946† |
FRA18A |
| |
KYSE510 |
Amp/Loss |
32107441-32200063 |
|
MOCOS§, FHOD3, C18orf10† |
|
| 3p14.2 |
KYSE150 |
Gain/Loss |
58573676-58887412 |
1.69 Mb |
FAM107A§, FAM3D, C3orf67, FHIT† |
FRA3B |
| |
KYSE450 |
Neutral/Del |
59933661-60267262 |
|
FHIT
#
|
|
| 3q12.1-q12.2 |
KYSE180 |
Neutral/Amp |
100190484-100877203 |
1.86 Mb |
TFG§, ABI3BP
*
, IMPG2† |
|
| |
KYSE450 |
Neutral/Gain |
102009730-102076392 |
|
DCBLD2§, COL8A1† |
|
| 6p12.3-p12.2 |
KYSE150 |
Loss/Amp |
51932658-52161439 |
1.9 Mb |
PKHD1§, IL17A†, MCM3† |
|
| YES2 | Gain/Gain | 50261630-50627364 | DEFB112§, TFAP2D† |
a Copy number status on the left and right side of the breakpoint regions. CN: copy number, Amp: amplification, Del: deletion.
b The distance between two outermost breakpoints of all the different cell lines.
c These genes are located at or close to breakpoints in each cell line. “*”: Obvious breakpoints were detected inside of genes. “§” and “†”: Genes at the left and right side of the breakpoint regions, respectively. Genes that are not labeled are located in the breakpoint regions, but positions of the exact breakpoints are not determined. “#”: Genes with an inside homozygous deletion (HD), and thus might also be disrupted.
Figure 3.

11q13.3 and 11q13.4 are recurrent breakpoint regions in both the ESCC cell lines and primary tumors. (A) Breakpoints in 11q13.3 and 11q13.4 in the cell lines detected by array-CGH. Breakpoints fall between the probes that displayed significant copy number discrepancies (blue arrows). Amplifications are indicated with red signals. (B) The 1-Mb BAC clones that are used in dual-color FISH experiments are shown in the ideogram. Also, 11q13.3 and 11q13.4 are divided into smaller regions according to the positions of BAC DNA clones: 11q13.3(1) (from NONSC16D6 to Cancer_1D11), 11q13.3(2) (from Cancer_1D11 to NONSC2E5) and 11q13.4(1) (from NONSC2E5 to NONSC3C5). (C) Detection of 11q13.3-q13.4 breakpoint regions by dual-color FISH in ESCC tumors. BAC DNA probes at two sides of the breakpoint regions are labeled with Cy3-dUTP (red) and Green-dUTP (green). The BAC clones used for each region are listed beside the panel. Two examples of positive tumors are shown for each region. “Tumor 1” and “Tumor 2” samples shown for different regions may not be from the same cases. Most splitting-positive nuclei exhibited amplifications on one side, even high-level amplifications, revealing the breakages between red and green signals. Normal: peripheral blood from normal persons.
Genes which might be interrupted by the recurrent breakpoints in each cell line were listed in Table 4. Ten of these common breakpoint regions were localized in the vicinity of fragile sites. Genes in these cell lines with inner breakpoints included CDKN2A, LEPREL1, JAKMIP3, LIMCH1, CSTF3, ABTB2, CDKN2B-AS1, FHIT and ABI3BP. For these genes, one breakpoint could be detected. Small HDs were also observed inside some genes, resulting in two breakpoints, such as FHIT gene in KYSE450. Other genes flanking or close to the boundaries might also be influenced by the breakpoints.
To determine whether genomic aberrations found in these cell lines are also present in primary tumors, we first tested a small sample of 15 ESCC tumors by dual-color FISH. This analysis revealed splitting of regions 11q13.3-q13.4, 9p21, 15q25.3 and 3q28, which presented the highest frequency of disruption in the cell lines. Splitting of these regions had occurred in 5, 1, 2 and 3 out of 15 tumors, respectively. We also examined online data of ESCC cell lines. The results showed that both high level amplifications and breakages existed at 67-72 Mb positions in 11q13 (Figure 3). Multiple breakpoints are present in most of the cell lines, revealing these positions may be highly rearranged.
Due to the highest splitting frequency of 11q13.3-q13.4 in the initial 15 cases, we further expanded the sample pool to further characterize splitting of this region in primary ESCC cases (Figures 3B and 3C). Splitting frequencies of 11q13.4 and 11q13.3 were 36.6% (49/134) and 23.4% (32/137), respectively. Overall, breakage of 11q13.3-q13.4 was observed in 58 out of 134 cases (43.3%). Next, we divided the whole 11q13.3-q13.4 region into several parts, including 11q13.3(1) (CPT1A, MRPL21, IGHMBP2, MRGPRD, MRGPRF, TPCN2, MYEOV), 11q13.3(2) (CCND1, ORAOV1, FGF19, FGF4, FGF3, ANO1, FADD, PPFIA1, CTTN and SHANK2), 11q13.4(1) (DHCR7, NADSYN1, KRTAP5-7, KRTAP5-8, KRTAP5-9, KRTAP5-10, KRTAP5-11, FAM86C1, DEFB108B, RNF121, IL18BP, NUMA1, LRTOMT, FOLR3, FOLR1, FOLR2, INPPL1, PHOX2A and CLPB). Regions of 11q13.3(1), 11q13.3(2), 11q13.4(1), 11q13.3(2)-q13.4 and 11q13.3(2)-q13.4(1) were split in 16.3% (22/135), 8.4% (11/131), 24.1% (32/133), 41.0% (55/134) and 30.0% (39/130) of the primary ESCC tumors, respectively. Almost all of the array-CGH images of the cell lines in Figure 3A and Additional file 4: Figure S1 showed amplification of the region proximal or distal to the breakpoints. Similarly, most of the splitting-positive ESCC tumors examined by FISH presented focal high-level amplification of the region. The majority of breakpoints between NONSC16D6 and Cancer_1D11 were proximal to the amplicon, while most of the breakpoints between Cancer_1D11 and NONSC2E5 as well as those between NONS2E5 and NONSC15F5 were distal to the amplicon (Figure 3C and Additional file 5: Table S4).
Correlations between split and amplified regions and clinicopathological characteristics
Clinicopathological parameters of each patient were listed in Additional file 6: Table S5, and the relationships between regional splitting events and clinicopathological characteristics were summarized in Table 5. Splitting of 11q13.3-q13.4 was significantly correlated with lymph node metastasis (LNM) (P = 0.004) and advanced stages (P = 0.004). In LNM-positive group, 54.9% (39/71) of the tumors exhibited splitting compared with 30.2% (19/63) in LNM-negative group. Tumors at stages IIb/III/IV (53.9%, 41/76) showed higher frequencies of splitting than those at stages I/IIa (29.3%, 17/58). Breakpoint regions of 11q13.4, 11q13.4(1), 11q13.3(2)-q13.4(1) and 11q13.3(2)-q13.4 were also associated with LNM and advanced stages (P < 0.05, Additional file 7: Table S6). No correlations were observed between splitting in these regions and other clinico-pathological parameters, including gender, age, tumor size, and differentiation (Table 5 and Additional file 7: Table S6). We also tested the relationship between amplification of this region and clinical features. 11q13.3-q13.4 amplifications were defined as the number of FISH signals was at least 3 ( Additional file 5: Table S4). A positive correlation was observed between 11q13.3-q13.4 amplification and LNM (P = 0.022) or advanced stages (P = 0.039) (Table 5). Amplification of 11q13.3 or 11q13.4 alone was not associated with the parameters ( Additional file 8: Table S7).
Table 5.
Relationship between splitting and amplification of 11q13.3-q13.4 and clinical features of primary ESCC tumors
|
Clinical features |
Splitting |
Amplification |
||
|---|---|---|---|---|
| Frequency | P value | Frequency | P value | |
| Gender | ||||
| Male |
47.2% (50/106) |
0.089 a |
86.2% (94/109) |
1.000 a |
| Female |
28.6% (8/28) |
|
89.3% (25/28) |
|
| Age | ||||
| < 60 |
47.8% (33/69) |
0.274 |
87.1% (61/70) |
1.000 a |
| ≥ 60 |
38.5% (25/65) |
|
86.6% (58/67) |
|
| Tumor size | ||||
| T1, T2 |
50.0% (10/20) |
0.511 |
85.0% (17/20) |
0.728 a |
| T3, T4 |
42.1% (48/114) |
|
87.2% (102/117) |
|
| Lymph node metastasis | ||||
| N0 |
30.2% (19/63) |
0.004 |
79.4% (50/63) |
0.022 a |
| N1 |
54.9% (39/71) |
|
93.2% (69/74) |
|
| Stage | ||||
| I, IIa |
29.3% (17/58) |
0.004 |
79.3% (46/58) |
0.039 a |
| IIb, III, IV |
53.9% (41/76) |
|
92.4% (73/79) |
|
| Differentiation | ||||
| G1 |
40.7% (11/27) |
0.762 b |
85.2% (23/27) |
0.118 b |
| G2 |
41.7% (30/72) |
|
91.9% (68/74) |
|
| G3 | 48.6% (17/35) | 77.8% (28/36) | ||
a Fisher’s test.
b Kruskal–Wallis test.
The P value which is not labeled with “a” or “b” is assessed by χ2 test.
In order to create a multivariate model describing the risk for LNM, univariate and multivariate logistic regression analyses were performed with respect to gender, age, tumor size, differentiation status, as well as 11q13.3-q13.4 splitting and amplification. Multivariate analysis indicated that only splitting of 11q13.3-q13.4 was an independent predictor for LNM in ESCC (P = 0.026, RR = 2.357, Table 6).
Table 6.
Logistic regression analyses of the impact of clinico-pathological factors and 11q13.3-q13.4 splitting and amplification on LNM
|
Clinical features |
LNM |
Univariate analysis |
Multivariate analysis |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N0 | N1 | P value | RR (95% CI) | P value a | RR (95% CI) | |||||||
| Gender |
|
|
|
|
|
|
||||||
| Male |
48 |
61 |
0.368 |
1.466 (0.637-3.374) |
- |
|
||||||
| Female |
15 |
13 |
|
|
|
|
||||||
| Age |
|
|
|
|
|
|
||||||
| < 60 |
32 |
38 |
0.948 |
0.978 (0.499-1.915) |
- |
|
||||||
| ≥ 60 |
31 |
36 |
|
|
|
|
||||||
| Tumor size |
|
|
|
|
|
|
||||||
| T1, T2 |
11 |
9 |
0.384 |
1.528 (0.589-3.964) |
- |
|
||||||
| T3, T4 |
52 |
65 |
|
|
|
|
||||||
| Differentiation |
|
|
|
|
|
|
||||||
| G1 |
16 |
11 |
0.295 |
1.308 (0.792-2.161) |
- |
|
||||||
| G2 |
31 |
43 |
|
|
|
|
||||||
| G3 |
16 |
20 |
|
|
|
|
||||||
| Splitting status |
|
|
|
|
|
|
||||||
| Non-splitting |
44 |
32 |
0.004 |
2.822 (1.384-5.757) |
0.026 |
2.357 (1.110-5.004) |
||||||
| Splitting |
19 |
39 |
|
|
|
|
||||||
| Amplification status |
|
|
|
|
|
|
||||||
| Non-amp |
13 |
5 |
0.022 |
3.588 (1.202-10.712) |
0.165 |
2.265 (0.715-7.175) |
||||||
| Amp | 50 | 69 | ||||||||||
a The P value of each variable which is not significantly correlated with LNM in univariate analysis is indicated with “-” in the multivariate analysis. Multivariate logistic regression analysis is performed using forward procedures.
LNM: lymph node metastasis; RR, relative risk; CI, confidence interval; amp: amplification.
Discussion
Genomic numerical and structural alterations are common features in ESCC. Our study characterized CNAs, structural aberrations, and recurrent breakpoints in six ESCC cell lines by a combination of M-FISH and array-CGH analyses, which helps provide accurate karyotypes of these cell lines. We further found the correlation between splitting of an amplified region 11q13.3-q13.4 and lymph node metastasis.
Genomic CNAs may influence gene expression through the following mechanisms. A well known mechanism is that gains or losses may result in gene amplifications or deletions, and thus upregulate or downregulate the protein expression [40]. Different situations may occur on genes at the boundaries of gain or loss regions. CNA boundaries inside of the genes usually indicate gene breakage. Gene rearrangements may result from such breakages, leading to the formation of an aberrant gene product [41]. If the CNA boundaries occur in non-coding regions flanking genes, expression may be controlled by proximity to regulatory sequences from other genes. Alternatively, the recurrent breakpoint may indicate loss of a tumor suppressor gene distal to the CNA boundary [42]. Small deletions inside of the genes may result in structural aberrant proteins, truncated proteins, or even loss-of-function proteins. Small amplifications and deletions inside of genes may also indicate gene breakage, and the gene products may also be affected by rearrangements with the partner gene. On the other hand, many recurrent rearrangements occurred at boundaries of the breakpoints, resulting in fusion genes, truncated genes, as well as other structural variants [2]. Therefore, we focused on the breakpoints with CNAs involved in genomic rearrangements and breakpoints mapped to specific sites.
Copy number profiling of ESCC have been analyzed from different studies. Gains involved regions 5p, 7p, 7q, 8q, 11q, 12p, 14q, 16p, 19q, 20q and have been reported in ESCC cell lines by SKY and CGH [35], as well as at least 60% primary tumors by 32K array-CGH [36]. Recently, gains of 19p13.3-q13.43, 11q13.1-q13.4, 20p13-q13.33, 3q24-q29, 22q11.21-q12.1 have been reported [38]. We detected six cell lines, and compared with online database, the high frequency of gains mainly include 3q26.33-qter, 5p14.1-p11, 7pter-p12.3, 8q24.13-q24.21, 9q31.1-qter, 11p13-p11, 11q11-q13.4, 17q23.3-qter, 18pter-p11, 19p, 19q and 20q13.32-qter. Gain of 3q is prevalent in ESCC, and 3q26-qter was found in 76.5% (39/51) primary tumors [43] and 66.7% (4/6) cell lines [44], suggesting cancer-related genes may be present in 3q26-qter. Evidence has been found that PIK3CA[35,45], PRKCI[46], and ZNF639[47] are amplified and overexpressed in ESCC. PIK3CA[45] and PRKCI[46] are associated with LNM and overexpression of PIK3CA and TFRC are associated with poor prognosis [48]. We found gain of 5p14.1-p11 existed in 83.3% (5/6) cell lines. In the previous study, gain of 5p13 was detected in 10% (3/29) ESCC cell lines. SKP2 on 5p13 was amplified and overexpressed in 50% (23/46) ESCC tumors, and was associated with LNM and stage. SKP2 overexpression could protect cancer cells from anoikis, which was mediated in part by the phosphoinositidyl 3-kinase-Akt pathway [49]. Gains of 18p11.2-p11.3 and 18p11.3 were also found in 20.7% and 17.2% cell lines by CGH [31] and FISH [50]. AURKA at 20q13 encodes a cell cycle-regulated kinase. Yang et al. found that overexpression of AURKA was existed in 85.7% (6/7) cell lines and 93.1% (27/29) tumors [51]. Recent studies have reported that AURKA is a direct target of the MAPK pathway [52]. Overexpression of AURKA is independently associated with chromosomal instability in colorectal cancer [53], and AURKA expression has a prognostic value in ovarian carcinoma [54]. High-level amplifications of 11q13.3-q13.4, 7p11.2, and 8q24.21 have been observed in this study. Amplification of 11q13.3-q13.4 will be discussed later. Amplification of 7p11.2, which harbors an important oncogene EGFR, was also found in ESCC from other studies [36]. Amplification and overexpression of EGFR gene may play roles in the invasion and progression in cancer [55], and the elevated expression may be an indicator for tumor recurrence and lower survival in head and neck squamous cell carcinoma (HNSCC) [56]. Amplification of MYC in 8q24.21 may contribute to the progression of breast cancer [57].
Losses were previously found on 3p, 5q, 8p, 9p and 11q [36], as well as 4p16.3-q35.2, 13q12.11-q34, 18p11.32-q23, and chromosome Y [38]. In our study, losses were observed on 18q12.2-qter, 3p14.1-p11, 4p15.32-p14, 4q22.1-q32.3, 9pter-p24.1, 9p23-p11, 11q23.3-qter, 18q12.2-q21.1, Xpter-p11, Xq21.1-q23 in more than 50% (3/6) of the cell lines, indicating several tumor suppressor genes may be located in these regions. 9p21.3 (CDKN2A and CDKN2B) is homozygously deleted in some of ESCC [58,59]. It has been found that CDKN2A was fused to IGH through the translocation t(9;14)(p21;q32) in a pre-B acute lymphoblastic leukemia cell line [60]. The functional study demonstrated that restoring wild-type CDKN2A into the gene deleted ESCC cells significantly inhibited cell invasion, suggesting that inactivation of CDKN2A may be involved in ESCC invasion [59]. Our array-CGH results confirmed that HD frequency of 9p21.3 was 66.7% (4/6). Interestingly, CDKN2A was deleted in only one cell line, and the other three harbored at least one breakpoint inside of CDKN2A. For CDKN2B, the inside breakpoint was detected in one cell line, while the other three were homozygously deleted.
Recurrent breakpoint regions were detected in at least two cell lines, including 1q31.1, 2q35, 3p14.2, 3q12.1-q12.2, 3q28, 4p13, 4q22.1, 6p12.3-p12.2, 6p22.2-p22.1, 7q22.2-q22.3, 8q24.21, 9p21.3, 10q26.3, 11p13, 11q13.3, 11q13.4, 13q21.32, 15q25.3, 18q12.2 and 21q21.1. Many of these breakpoints were different from those detected by SKY in other ESCC cell lines [35]. The correlation between breakpoints of fusion genes and fragile sites has been emphasized in previous studies. Burrow et al. analyzed 444 pairs of genes involved in cancer-specific recurrent translocations, and found that 52% of the breakpoints in at least one gene of the fusion-gene pairs were localized within the vicinity of a fragile site [61]. Thus, understanding breakpoints near fragile sites may be helpful for further discovering cancer-related gene rearrangements.
11q13 is an important region that presents various aberrations in many malignancies. Gain of 11q13 has previously been described in ESCC [34,36-38,62,63] and other solid tumors, including oral [42], gastric [64], breast [65-67], ovarian [68], prostate [69], bladder [70], laryngeal [71], nasopharyngeal [72], and liver tumors [73] as well as head and neck [74,75] cancer. Gain frequencies of 11q13 varied between studies. The probable target genes that are amplified and/or overexpressed in different cancers have been reported to include CCND1[62,76,77], FGF4[62,78], PPFIA1[67], CTTN[62,76] and ORAOV1[79]. We observed that 11q13.3-q13.4 was a region with high-level amplification adjacent to the breakpoint boundaries. Breakpoints in 11q13.4 and 11q13.3 were both found in two cell lines, and breakpoints that were identified in 11q13.4 between KYSE30 and KYSE510 were closer to each other. Furthermore, breakpoints in the entire region of 11q13.3-q13.4 were present in more than 40% of ESCC tumors, suggesting that 11q13.3-q13.4 may be a frequently split region in ESCCs.
11q13 is also involved in various chromosomal rearrangements in both hematological malignancies and epithelial carcinomas. The t(11;14)(q13;q32) translocation is associated with 70%-90% of mantle cell lymphomas (MCL) [80,81], a small portion of multiple myeloma (MM) [81,82], acute myeloid leukemia (AML) [83] and other lymphoproliferative disorders [84]. As a result of this translocation, CCND1 is fused to the enhancer of immunoglobulin heavy chain gene (IGH), and thus overexpressed in MCL and MM [81]. MYEOV, proximal to CCND1, was also overexpressed in a subset of t(11;14)-positive MM cell lines, in which both MYEOV and CCND1 were under the control of IGH enhancers due to translocations [85]. There are also other partner genes fused to CCND1, including IGK involved in t(2;11)(p11;q13) in leukemic small-cell B-non-Hodgkin lymphoma (NHL) [86] and an unknown partner gene in AML with t(5;11)(q35;q13) [87]. 11q13 rearrangements with 6p21, 7q11.2, 1p13 and 5q35 were observed in renal oncocytoma. Jhang et al. demonstrated that CCND1 was overexpressed only in the cases with 11q13 translocation. However, not all of the cases with 11q13 translocations could lead to CCND1 overexpression [88]. Evidence of fusions involving other genes in 11q13 has been reported. NUMA-RARA and RUNX1-MACROD1 were present in the monocytic leukemia with t(11;17)(q13;q21) [89-91] and APL with t(11;21)(q13;q22) [92], respectively. Rearrangement of LRP5 was found in AML, although the partner gene has not been identified [93]. Most of the above translocations in lymphomas and leukemia are balanced and not complicated, while more complex rearrangements of 11q13 were detected in epithelial carcinomas, including cervical carcinoma cell lines [94], serous ovarian adenocarcinoma [95], hepatocellular carcinoma [96], gastric cancer [97] and oral squamous cell carcinoma (OSCC) [98,99]. Our M-FISH results were much similar to the observations in these carcinomas. We found that chromosome 11 was frequently rearranged, especially in KYSE30, KYSE150 and KYSE510. In each cell line, five to six derivative chromosomes associated with chromosome 11 were easily found, and some complex derivative chromosomes involved q13 band of the chromosome. Genes involved in breakpoints of these rearrangements remain to be clarified.
The current array-CGH profiling enabled us to set the boundaries of 11q13 amplicons in ESCC cell lines. We observed that multiple breakpoints existed in high level amplification regions involving 11q13.3 were located in 67-72 Mb position in three ESCC cell lines we detected and ten online cell lines, which is similar to the amplification peak in HNSCC [75]. The mechanisms for formation of several amplicons have been well described by a model of breakage-fusion-bridge (BFB) cycle. According to this model, the formation of amplicons is initiated by distal DNA breakages at fragile sites. During DNA replication, a dicentric chromosome with an inverted duplication may be resulted from the sister chromatid fusion (SCF). Breakage-fusion-bridge cycle may continue when another break between two centromeres occurs. The cycle may be then stabilized by a telomere or by translocation [42,98,100-102]. Albertson suggested that amplicon boundaries might also be set by selection for overexpressed genes in the amplicons, or by selection against expression changes of genes outside of amplified regions induced by CNAs [101,103]. 11q13 harbors three fragile sites, FRA11A, FRA11H and FRA11F[104]. FRA11A is a rare fragile site, while FRA11H and FRA11F are common fragile sites. FRA11A is located between RIN1 (11q13.2) and CCND1 (11q13.3) [98]. FRA11H is positioned at 11q13, but the exact location still needs to be characterized. FRA11F is located between the BAC clones of RP11-281H14 and RP11-841F15 in 11q14.2 [42]. Reshmi et al. found that OSCC cell lines with complex 11q rearrangements were affected by FRA11F, and gene amplifications in 11q13 region in OSCC cell lines may be initiated by breakage at FRA11F[42]. Shuster et al. demonstrated that breakages at FRA11A between RIN1 and CCND1 may promote the BFB cycles [98]. They also found the involvement of FRA11H in some OSCC cases with amplifications of genes in 11q13 [42,105]. In the present study, distal boundaries of amplicons in the majority of ESCC cell lines and primary tumors with 11q13 amplification were clustered within 67-72 Mb region of 11q13.3, which may involve FRA11H breakages for these cases. Another breakpoint was observed at NAALAD2 gene in KYSE510, and it was located within FRA11F. In addition, breakpoints proximal of 11q13 amplicons in KYSE180, KYSE510 and five online cell lines were located in FRA11A region in 11q13.3, while the proximal breakpoints in KYSE30 and other online cell lines were distal to FRA11A or in FRA11H. In the tested ESCC tumors, the majority of breakpoints in 11q13.3(1) were proximal to the amplicons, and most of those in 11q13.3(2) and 11q13.4 were distal to the amplicons. Thus, we speculate that initial distal breakages may primarily occur at FRA11H, and the process may involve FRA11F in some cases. FRA11A or FRA11H may contribute to setting amplicon boundaries by promoting subsequent steps of BFB cycle. Concerning the presentation of multiple breakpoint boundaries in some of ESCC cell lines and primary tumors with high-level amplification of 11q13, several cycles of random breakages may be undergone.
In the current study, we have noticed that copy numbers of the regions from centromere to boundaries at initial breaks were higher than those of the regions distal to breakpoints in most of ESCC cell lines with 11q13 amplifications. Gains of proximal regions, losses of distal regions, intrachromosomal or interchromosomal rearrangements of 11q13 have been found in the cell lines or primary tumors of human cancer and demonstrated to be indicators of BFB cycle [98,101]. At the end of BFB cycles, distal breakpoints of 11q13.3-q13.4 amplicon may undergo intrachromosomal rearrangements or translocating to other chromosomes, which may affect genes at distal boundaries through forming intragenic rearrangements or fusing to other genes. Notably, most of 11q13.3-q13.4 splitting cell lines according to our and online array-CGH data showed high-level amplification of 11q13 proximal or distal to the breakpoints in ESCC cell lines and primary tumors. Moreover, amplicons involving intrachromosomal or interchromosomal rearrangements have also been detected. Thus, recurrent breakage at 11q13.3-q13.4 may reflect the following aspects. On one hand, genes between two BACs flanking the regions may be amplified, with proximal gain and gene overexpression. On the other hand, breakages between two BACs and thus rearrangements of genes at the amplicon boundaries may also dysregulate expression of these genes.
The relationship between gain of 11q13 and LNM or prognosis have been analyzed and discussed in several studies. However, contrary opinions still exist. Tada et al. conducted CGH on 36 ESCC specimens, and demonstrated that gain of 11q13 did not occur at a significantly different rate between LNM and non-LNM groups [106]. Genes located in 11q13 were analyzed. Amplification of CTTN was correlated with LNM, while no significant association was found between CCND1 amplification and LNM [76], however, predicting CCND1 amplification using plasma DNA may be an independent prognostic factor in ESCCs [107]. Komatsu et al. found that overexpression of ORAOV1 showed a significant association with LNM and stages. Gain of 11q13.2 was determined to be an independent prognostic factor for predicting poor outcome, and amplification of CPT1A in 11q13.2 was correlated with shorter overall survival in ESCC. Here, we found the correlation between 11q13.3-q13.4 amplification and LNM as well as advanced stages. The relationship between gene status in 11q13 and LNM has also been evaluated in other cancers. Amplification of 11q13 DNA is associated with lymph node involvement in HNSCC [108]. CCND1 amplification and overexpression are significantly associated with LNM and survival in OSCC [109]. Another study confirmed amplifications of 11 genes in 11q13, and found two amplification cores, including core 1 (TPCN2 and MYEOV), and core 2 (from CCND1 to CTTN). Amplification of CTTN (core 2) and/or TPCN2/MYEOV (core 1) was further demonstrated to be associated with LNM in OSCC [110]. However, Huang et al. reported that there was no correlation between LNM and amplification or expression of the tested genes in 11q13 in OSCC [111]. Fortin et al. also found 11q13 amplifications not appear to be a reliable marker for subclinical LNM prediction in oral and oropharyngeal carcinomas [112]. A study by Xia et al. indicated that amplifications of ORAOV1 and CTTN are indicated to be associated with LNM [113]. In the breast cancer, PPFIA1 is coamplified with CCND1, which is significantly associated with high-grade phenotype but not tumor stage or nodal stage [67].
In this report, we demonstrated that both of 11q13.3-q13.4 splitting and amplification are significantly correlated with LNM and advanced stages, indicating that breakage and amplification of this region may play important roles in the tumor progression. According to multivariate logistic regression analysis, however, it was splitting rather than amplification that could be an independent predictor for the higher tendency of metastasis. In addition, two smaller regions 11q13.4(1) (SHANK2, DHCR7, NADSYN1, KRTAP5-7, KRTAP5-8, KRTAP5-9, KRTAP5-10, KRTAP5-11, FAM86C1, RNF121, IL18BP, NUMA1, LRTOMT, FOLR3, FOLR1, FOLR2, INPPL1, PHOX2A and CLPB) and 11q13.3(2)-q13.4(1) (CCND1, ORAOV1, FGF19, FGF4, FGF3, ANO1, FADD, PPFIA1, CTTN, SHANK2, DHCR7, NADSYN1, KRTAP5-7, KRTAP5-8, KRTAP5-9, KRTAP5-10, KRTAP5-11, FAM86C1, RNF121, IL18BP, NUMA1, LRTOMT, FOLR3, FOLR1, FOLR2, INPPL1, PHOX2A and CLPB) and were also associated with these parameters. However, no significant difference was found for 11q13.3(1) (CPT1A, MRPL21, IGHMBP2, MRGPRD, MRGPRT, TPCN2, MYEOV). These results suggested that genes located in 11q13.3(2) and 11q13.4(1) may play more important roles in LNM than 11q13.3(1). It will be interesting to further investigate gene and protein alterations caused by genomic breakages. Since the exact breakpoint locations may be distinctive in different cases, further studies will be focused on determining which of these genes were rearranged or disrupted in specific cases, identifying possible rearranged forms and roles of these alterations may play in ESCC development.
Conclusions
Our data provide detailed information on chromosomal and genomic aberrations present in six ESCC cell lines. Using a combination of M-FISH and array-CGH enabled us to produce more accurate karyotypes, which will help to determine appropriate applications of these cell lines for cytogenetic and molecular biological studies. The recurrent genomic breakpoints present in both the cell lines and primary tumors may help to identify aberrant genes associated with the development and progression of ESCC.
Competing interests
There was no conflict of interest in this study.
Authors' contributions
HJJ carried out M-FISH experiments, participated in the data analyses, and draft the manuscript. SZZ carried out array-CGH experiments. ZZX organized clinico-pathological information. ZY participated in the design of the study. GT carried out part of the FISH experiments. LCX carried out part of the FISH experiments. ZT performed some statistical analysis. CY performed some statistical analysis. DJT provided FISH probe templates (BAC-DNA) and gave experimental suggestions. FSB provided the statistical analysis suggestion. ZQM gave experimental design suggestions. WMR conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors have read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
Table S1. Genomic copy number alterations in ESCC cell lines detected by array-CGH.
Table S2. Regions of gain and the frequencies in the ESCC cell lines.
Table S3. Unbalanced breakpoints analyzed according to the copy number alterations.
Figure S1. High level amplifications and breakages of 11q13 in ten ESCC cell lines. (A) KYSE140, (B) KYSE410, (C) EC-GI-10, (D) HCE-4, (E) TE-6, (F) TE-8, (G) TE-9, (H) TE-10, (I) TE-11, (J) TE-15. Amplifications are mainly located within 67-72 Mb position. The smallest amplification region is similar to previously reported. More than two breakpoints are present in most of the cell lines, revealing these positions may be highly rearranged. (EPS 3433 kb)
Table S4. Splitting and amplification of 11q13.3-q13.4 in the primary ESCC tumors by FISH analyses.
Table S5. Clinico-pathological parameters of the examined ESCC patients.
Table S6. Relationship between region splittings and clinico-pathological features of ESCC.
Table S8. Relationship between region amplifications and clinico-pathological features of ESCC.
Contributor Information
Jia-Jie Hao, Email: haojj2011@gmail.com.
Zhi-Zhou Shi, Email: zhizhoushi@gmai.com.
Zhi-Xin Zhao, Email: zhaozx2006@gmail.com.
Yu Zhang, Email: zhangyu909@gmail.com.
Ting Gong, Email: gongting8@gmail.com.
Chun-Xiang Li, Email: chunxiangl@gmail.com.
Ting Zhan, Email: zhanting407@163.com.
Yan Cai, Email: caiyan0818@yahoo.com.cn.
Jin-Tang Dong, Email: jin-tang.dong@emory.edu.
Song-Bin Fu, Email: fusb@ems.hrbmu.edu.cn.
Qi-Min Zhan, Email: zhanqimin@pumc.edu.cn.
Ming-Rong Wang, Email: wangmr2015@cicams.ac.cn.
Acknowledgements
We thank Professor Hong Chen (Basic Medical Institute, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China) for the kind gift of Whole-chromosome painting (WCP) probes. This work was supported by Chinese Hi-Tech R&D Program Grant (2012AA02A503), National Science Fund (81021061, 30872936) and Specialized Research Fund for the Doctoral Program of Higher Education of China (20101106110015).
References
- Frohling S, Dohner H. Chromosomal abnormalities in cancer. N Engl J Med. 2008;359(7):722–734. doi: 10.1056/NEJMra0803109. [DOI] [PubMed] [Google Scholar]
- Ritz A, Paris PL, Ittmann MM, Collins C, Raphael BJ. Detection of recurrent rearrangement breakpoints from copy number data. BMC Bioinformatics. 2011;12:114. doi: 10.1186/1471-2105-12-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7(4):233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- Edwards PA. Fusion genes and chromosome translocations in the common epithelial cancers. J Pathol. 2010;220(2):244–254. doi: 10.1002/path.2632. [DOI] [PubMed] [Google Scholar]
- Clark JP, Cooper CS. ETS gene fusions in prostate cancer. Nat Rev Urol. 2009;6(8):429–439. doi: 10.1038/nrurol.2009.127. [DOI] [PubMed] [Google Scholar]
- Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C. et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470(7333):214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ. et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462(7276):1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaki K, Hillmer AM, Ukil L, Yao F, Woo XY, Vardy LA, Zawack KF, Lee CW, Ariyaratne PN, Chan YS. et al. Transcriptional consequences of genomic structural aberrations in breast cancer. Genome Res. 2011;21(5):676–687. doi: 10.1101/gr.113225.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y. et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18(3):375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK, Kim YT, Kim JI, Kang JH, Seo JS. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012;22(3):436–445. doi: 10.1101/gr.133645.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass AJ, Lawrence MS, Brace LE, Ramos AH, Drier Y, Cibulskis K, Sougnez C, Voet D, Saksena G, Sivachenko A. et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat Genet. 2011;43(10):964–968. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, Han B, Cao Q, Cao X, Suleman K. et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med. 2010;16(7):793–798. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A. et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totoki Y, Tatsuno K, Yamamoto S, Arai Y, Hosoda F, Ishikawa S, Tsutsumi S, Sonoda K, Totsuka H, Shirakihara T. et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43(5):464–469. doi: 10.1038/ng.804. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R. et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Speicher MR. Gwyn Ballard S, Ward DC: Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat Genet. 1996;12(4):368–375. doi: 10.1038/ng0496-368. [DOI] [PubMed] [Google Scholar]
- Schrock E, du Manoir S, Veldman T, Schoell B, Wienberg J, Ferguson-Smith MA, Ning Y, Ledbetter DH, Bar-Am I, Soenksen D. et al. Multicolor spectral karyotyping of human chromosomes. Science. 1996;273(5274):494–497. doi: 10.1126/science.273.5274.494. [DOI] [PubMed] [Google Scholar]
- Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y. et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20(2):207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- Speicher MR, Carter NP. The new cytogenetics: blurring the boundaries with molecular biology. Nat Rev Genet. 2005;6(10):782–792. doi: 10.1038/nrg1692. [DOI] [PubMed] [Google Scholar]
- Camps J, Grade M, Nguyen QT, Hormann P, Becker S, Hummon AB, Rodriguez V, Chandrasekharappa S, Chen Y, Difilippantonio MJ. et al. Chromosomal breakpoints in primary colon cancer cluster at sites of structural variants in the genome. Cancer Res. 2008;68(5):1284–1295. doi: 10.1158/0008-5472.CAN-07-2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, James SY, Yanez-Munoz RJ, Chaplin T, Molloy G, Oliver RT, Young BD, Lu YJ. Rapid high-resolution karyotyping with precise identification of chromosome breakpoints. Genes Chromosomes Cancer. 2007;46(7):675–683. doi: 10.1002/gcc.20452. [DOI] [PubMed] [Google Scholar]
- Watson SK, DeLeeuw RJ, Horsman DE, Squire JA, Lam WL. Cytogenetically balanced translocations are associated with focal copy number alterations. Hum Genet. 2007;120(6):795–805. doi: 10.1007/s00439-006-0251-9. [DOI] [PubMed] [Google Scholar]
- Kawamata N, Ogawa S, Zimmermann M, Niebuhr B, Stocking C, Sanada M, Hemminki K, Yamatomo G, Nannya Y, Koehler R. et al. Cloning of genes involved in chromosomal translocations by high-resolution single nucleotide polymorphism genomic microarray. Proc Natl Acad Sci U S A. 2008;105(33):11921–11926. doi: 10.1073/pnas.0711039105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson F, Winnes M, Andren Y, Wedell B, Dahlenfors R, Asp J, Mark J, Enlund F, Stenman G. High-resolution array CGH analysis of salivary gland tumors reveals fusion and amplification of the FGFR1 and PLAG1 genes in ring chromosomes. Oncogene. 2008;27(21):3072–3080. doi: 10.1038/sj.onc.1210961. [DOI] [PubMed] [Google Scholar]
- Howarth KD, Blood KA, Ng BL, Beavis JC, Chua Y, Cooke SL, Raby S, Ichimura K, Collins VP, Carter NP. et al. Array painting reveals a high frequency of balanced translocations in breast cancer cell lines that break in cancer-relevant genes. Oncogene. 2008;27(23):3345–3359. doi: 10.1038/sj.onc.1210993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Parkin DM, Bray FI, Devesa SS. Cancer burden in the year 2000. The global picture. Eur J Cancer. 2001;37(Suppl 8):S4–66. doi: 10.1016/s0959-8049(01)00267-2. [DOI] [PubMed] [Google Scholar]
- Wu YP, Yang YL, Yang GZ, Wang XY, Luo ML, Zhang Y, Feng YB, Xu X, Han YL, Cai Y. et al. Identification of chromosome aberrations in esophageal cancer cell line KYSE180 by multicolor fluorescence in situ hybridization. Cancer Genet Cytogenet. 2006;170(2):102–107. doi: 10.1016/j.cancergencyto.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Wu YP, Yang YL, Han YL, Xu X, Cai Y, Yang GZ, Wang XY, Zhan QM, Wu M, Wang MR. Identification of complex chromosome abnormalities in esophageal carcinoma cells KYSE450 by multicolor fluorescence in situ hybridization. Shijie Huaren Xiaohua Zazhi. 2006;14(8):747–751. [Google Scholar]
- Yang Y, Chu J, Wu Y, Luo M, Xu X, Han Y, Cai Y, Zhan Q, Wang M. Chromosome analysis of esophageal squamous cell carcinoma cell line KYSE 410-4 by repetitive multicolor fluorescence in situ hybridization. J Genet Genomics. 2008;35(1):11–16. doi: 10.1016/S1673-8527(08)60002-8. [DOI] [PubMed] [Google Scholar]
- Pimkhaokham A, Shimada Y, Fukuda Y, Kurihara N, Imoto I, Yang ZQ, Imamura M, Nakamura Y, Amagasa T, Inazawa J. Nonrandom chromosomal imbalances in esophageal squamous cell carcinoma cell lines: possible involvement of the ATF3 and CENPF genes in the 1q32 amplicon. Jpn J Cancer Res. 2000;91(11):1126–1133. doi: 10.1111/j.1349-7006.2000.tb00895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Ni J, Wu SS, Liu H, Xu X, Han YL, Cai Y, Zhang JW, Chen XJ, Pang H. et al. Cytogenetic studies of esophageal squamous cell carcinomas in the northern Chinese population by comparative genomic hybridization. Cancer Genet Cytogenet. 2002;138(1):38–43. doi: 10.1016/S0165-4608(02)00586-1. [DOI] [PubMed] [Google Scholar]
- Qin YR, Wang LD, Fan ZM, Kwong D, Guan XY. Comparative genomic hybridization analysis of genetic aberrations associated with development of esophageal squamous cell carcinoma in Henan, China. World J Gastroenterol. 2008;14(12):1828–1835. doi: 10.3748/wjg.14.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai N, Kajiyama Y, Iwanuma Y, Tomita N, Amano T, Isayama F, Ouchi K, Tsurumaru M. Study of abnormal chromosome regions in esophageal squamous cell carcinoma by comparative genomic hybridization: relationship of lymph node metastasis and distant metastasis to selected abnormal regions. Dis Esophagus. 2010;23(5):415–421. doi: 10.1111/j.1442-2050.2009.01026.x. [DOI] [PubMed] [Google Scholar]
- Yen CC, Chen YJ, Lu KH, Hsia JY, Chen JT, Hu CP, Chen PM, Liu JH, Chiou TJ, Wang WS. et al. Genotypic analysis of esophageal squamous cell carcinoma by molecular cytogenetics and real-time quantitative polymerase chain reaction. Int J Oncol. 2003;23(4):871–881. [PubMed] [Google Scholar]
- Carneiro A, Isinger A, Karlsson A, Johansson J, Jonsson G, Bendahl PO, Falkenback D, Halvarsson B, Nilbert M. Prognostic impact of array-based genomic profiles in esophageal squamous cell cancer. BMC Cancer. 2008;8:98. doi: 10.1186/1471-2407-8-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu N, Wang C, Ng D, Clifford R, Yang HH, Tang ZZ, Wang QH, Han XY, Giffen C, Goldstein AM. et al. Genomic characterization of esophageal squamous cell carcinoma from a high-risk population in China. Cancer Res. 2009;69(14):5908–5917. doi: 10.1158/0008-5472.CAN-08-4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi ZZ, Liang JW, Zhan T, Wang BS, Lin DC, Liu SG, Hao JJ, Yang H, Zhang Y, Zhan QM. et al. Genomic alterations with impact on survival in esophageal squamous cell carcinoma identified by array comparative genomic hybridization. Genes Chromosomes Cancer. 2011;50(7):518–526. doi: 10.1002/gcc.20875. [DOI] [PubMed] [Google Scholar]
- Telenius H, Carter NP, Bebb CE, Nordenskjold M, Ponder BA, Tunnacliffe A. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 1992;13(3):718–725. doi: 10.1016/0888-7543(92)90147-K. [DOI] [PubMed] [Google Scholar]
- Albertson DG, Collins C, McCormick F, Gray JW. Chromosome aberrations in solid tumors. Nat Genet. 2003;34(4):369–376. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- Nambiar M, Kari V, Raghavan SC. Chromosomal translocations in cancer. Biochim Biophys Acta. 2008;1786(2):139–152. doi: 10.1016/j.bbcan.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Reshmi SC, Huang X, Schoppy DW, Black RC, Saunders WS, Smith DI, Gollin SM. Relationship between FRA11F and 11q13 gene amplification in oral cancer. Genes Chromosomes Cancer. 2007;46(2):143–154. doi: 10.1002/gcc.20394. [DOI] [PubMed] [Google Scholar]
- Ueno T, Tangoku A, Yoshino S, Abe T, Toshimitsu H, Furuya T, Kawauchi S, Oga A, Oka M, Sasaki K. Gain of 5p15 detected by comparative genomic hybridization as an independent marker of poor prognosis in patients with esophageal squamous cell carcinoma. Clin Cancer Res. 2002;8(2):526–533. [PubMed] [Google Scholar]
- Tada K, Oka M, Hayashi H, Tangoku A, Oga A, Sasaki K. Cytogenetic analysis of esophageal squamous cell carcinoma cell lines by comparative genomic hybridization: relationship of cytogenetic aberrations to in vitro cell growth. Cancer Genet Cytogenet. 2000;117(2):108–112. doi: 10.1016/S0165-4608(99)00160-0. [DOI] [PubMed] [Google Scholar]
- Akagi I, Miyashita M, Makino H, Nomura T, Hagiwara N, Takahashi K, Cho K, Mishima T, Ishibashi O, Ushijima T. et al. Overexpression of PIK3CA is associated with lymph node metastasis in esophageal squamous cell carcinoma. Int J Oncol. 2009;34(3):767–775. doi: 10.3892/ijo_00000202. [DOI] [PubMed] [Google Scholar]
- Yang YL, Chu JY, Luo ML, Wu YP, Zhang Y, Feng YB, Shi ZZ, Xu X, Han YL, Cai Y. et al. Amplification of PRKCI, located in 3q26, is associated with lymph node metastasis in esophageal squamous cell carcinoma. Genes Chromosomes Cancer. 2008;47(2):127–136. doi: 10.1002/gcc.20514. [DOI] [PubMed] [Google Scholar]
- Imoto I, Yuki Y, Sonoda I, Ito T, Shimada Y, Imamura M, Inazawa J. Identification of ZASC1 encoding a Kruppel-like zinc finger protein as a novel target for 3q26 amplification in esophageal squamous cell carcinomas. Cancer Res. 2003;63(18):5691–5696. [PubMed] [Google Scholar]
- Wada S, Noguchi T, Takeno S, Kawahara K. PIK3CA and TFRC located in 3q are new prognostic factors in esophageal squamous cell carcinoma. Ann Surg Oncol. 2006;13(7):961–966. doi: 10.1245/ASO.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Wang XC, Wu YP, Ye B, Lin DC, Feng YB, Zhang ZQ, Xu X, Han YL, Cai Y, Dong JT. et al. Suppression of anoikis by SKP2 amplification and overexpression promotes metastasis of esophageal squamous cell carcinoma. Mol Cancer Res. 2009;7(1):12–22. doi: 10.1158/1541-7786.MCR-08-0092. [DOI] [PubMed] [Google Scholar]
- Nakakuki K, Imoto I, Pimkhaokham A, Fukuda Y, Shimada Y, Imamura M, Amagasa T, Inazawa J. Novel targets for the 18p11.3 amplification frequently observed in esophageal squamous cell carcinomas. Carcinogenesis. 2002;23(1):19–24. doi: 10.1093/carcin/23.1.19. [DOI] [PubMed] [Google Scholar]
- Yang SB, Zhou XB, Zhu HX, Quan LP, Bai JF, He J, Gao YN, Cheng SJ, Xu NZ. Amplification and overexpression of Aurora-A in esophageal squamous cell carcinoma. Oncol Rep. 2007;17(5):1083–1088. [PubMed] [Google Scholar]
- Furukawa T, Kanai N, Shiwaku HO, Soga N, Uehara A, Horii A. AURKA is one of the downstream targets of MAPK1/ERK2 in pancreatic cancer. Oncogene. 2006;25(35):4831–4839. doi: 10.1038/sj.onc.1209494. [DOI] [PubMed] [Google Scholar]
- Baba Y, Nosho K, Shima K, Irahara N, Kure S, Toyoda S, Kirkner GJ, Goel A, Fuchs CS, Ogino S. Aurora-A expression is independently associated with chromosomal instability in colorectal cancer. Neoplasia. 2009;11(5):418–425. doi: 10.1593/neo.09154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendiola M, Barriuso J, Marino-Enriquez A, Redondo A, Dominguez-Caceres A, Hernandez-Cortes G, Perez-Fernandez E, Sanchez-Navarro I, Vara JA, Suarez A. et al. Aurora kinases as prognostic biomarkers in ovarian carcinoma. Hum Pathol. 2009;40(5):631–638. doi: 10.1016/j.humpath.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Miyai K, Yamamoto S, Asano T, Tamai S, Matsubara O, Tsuda H. Protein overexpression and gene amplification of epidermal growth factor receptor in adult testicular germ cell tumors: potential role in tumor progression. Cancer Sci. 2010;101(9):1970–1976. doi: 10.1111/j.1349-7006.2010.01638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin Grandis J, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, Drenning SD, Tweardy DJ. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90(11):824–832. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- Singhi AD, Cimino-Mathews A, Jenkins RB, Lan F, Fink SR, Nassar H, Vang R, Fetting JH, Hicks J, Sukumar S. et al. MYC gene amplification is often acquired in lethal distant breast cancer metastases of unamplified primary tumors. Mod Pathol. 2012;25(3):378–387. doi: 10.1038/modpathol.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maesawa C, Tamura G, Nishizuka S, Ogasawara S, Ishida K, Terashima M, Sakata K, Sato N, Saito K, Satodate R. Inactivation of the CDKN2 gene by homozygous deletion and de novo methylation is associated with advanced stage esophageal squamous cell carcinoma. Cancer Res. 1996;56(17):3875–3878. [PubMed] [Google Scholar]
- Kim DH, Muto M, Kuwahara Y, Nakanishi Y, Watanabe H, Aoyagi K, Ogawa K, Yoshida T, Sasaki H. Array-based comparative genomic hybridization of circulating esophageal tumor cells. Oncol Rep. 2006;16(5):1053–1059. [PubMed] [Google Scholar]
- Urashima M, Hoshi Y, Sugimoto Y, Kaihara C, Matsuzaki M, Chauhan D, Ogata A, Teoh G, DeCaprio JA, Anderson KC. A novel pre-B acute lymphoblastic leukemia cell line with chromosomal translocation between p16(INK4A)/p15(INK4B) tumor suppressor and immunoglobulin heavy chain genes: TGFbeta/IL-7 inhibitory signaling mechanism. Leukemia. 1996;10(10):1576–1583. [PubMed] [Google Scholar]
- Burrow AA, Williams LE, Pierce LC, Wang YH. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics. 2009;10:59. doi: 10.1186/1471-2164-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai H, Ueno T, Tangoku A, Yoshino S, Abe T, Kawauchi S, Oga A, Furuya T, Oka M, Sasaki K. Detection of amplified oncogenes by genome DNA microarrays in human primary esophageal squamous cell carcinoma: comparison with conventional comparative genomic hybridization analysis. Cancer Genet Cytogenet. 2003;146(1):16–21. doi: 10.1016/S0165-4608(03)00106-7. [DOI] [PubMed] [Google Scholar]
- Hirasaki S, Noguchi T, Mimori K, Onuki J, Morita K, Inoue H, Sugihara K, Mori M, Hirano T. BAC clones related to prognosis in patients with esophageal squamous carcinoma: an array comparative genomic hybridization study. Oncologist. 2007;12(4):406–417. doi: 10.1634/theoncologist.12-4-406. [DOI] [PubMed] [Google Scholar]
- Kuroda A, Tsukamoto Y, Nguyen LT, Noguchi T, Takeuchi I, Uchida M, Uchida T, Hijiya N, Nakada C, Okimoto T. et al. Genomic profiling of submucosal-invasive gastric cancer by array-based comparative genomic hybridization. PLoS One. 2011;6(7):e22313. doi: 10.1371/journal.pone.0022313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letessier A, Sircoulomb F, Ginestier C, Cervera N, Monville F, Gelsi-Boyer V, Esterni B, Geneix J, Finetti P, Zemmour C. et al. Frequency, prognostic impact, and subtype association of 8p12, 8q24, 11q13, 12p13, 17q12, and 20q13 amplifications in breast cancers. BMC Cancer. 2006;6:245. doi: 10.1186/1471-2407-6-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwek SS, Roy R, Zhou H, Climent J, Martinez-Climent JA, Fridlyand J, Albertson DG. Co-amplified genes at 8p12 and 11q13 in breast tumors cooperate with two major pathways in oncogenesis. Oncogene. 2009;28(17):1892–1903. doi: 10.1038/onc.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancau AM, Wuth L, Waschow M, Holst F, Krohn A, Choschzick M, Terracciano L, Politis S, Kurtz S, Lebeau A. et al. PPFIA1 and CCND1 are frequently coamplified in breast cancer. Genes Chromosomes Cancer. 2010;49(1):1–8. doi: 10.1002/gcc.20713. [DOI] [PubMed] [Google Scholar]
- Tan DS, Lambros MB, Rayter S, Natrajan R, Vatcheva R, Gao Q, Marchio C, Geyer FC, Savage K, Parry S. et al. PPM1D is a potential therapeutic target in ovarian clear cell carcinomas. Clin Cancer Res. 2009;15(7):2269–2280. doi: 10.1158/1078-0432.CCR-08-2403. [DOI] [PubMed] [Google Scholar]
- Das K, Lau W, Sivaswaren C, Ph T, Fook-Chong S, Sl T, Cheng C. Chromosomal changes in prostate cancer: a fluorescence in situ hybridization study. Clin Genet. 2005;68(1):40–47. doi: 10.1111/j.1399-0004.2005.00452.x. [DOI] [PubMed] [Google Scholar]
- Zaharieva BM, Simon R, Diener PA, Ackermann D, Maurer R, Alund G, Knonagel H, Rist M, Wilber K, Hering F. et al. High-throughput tissue microarray analysis of 11q13 gene amplification (CCND1, FGF3, FGF4, EMS1) in urinary bladder cancer. J Pathol. 2003;201(4):603–608. doi: 10.1002/path.1481. [DOI] [PubMed] [Google Scholar]
- Jarmuz M, Grenman R, Golusinski W, Szyfter K. Aberrations of 11q13 in laryngeal squamous cell lines and their prognostic significance. Cancer Genet Cytogenet. 2005;160(1):82–88. doi: 10.1016/j.cancergencyto.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Hui AB, Or YY, Takano H, Tsang RK, To KF, Guan XY, Sham JS, Hung KW, Lam CN, van Hasselt CA. et al. Array-based comparative genomic hybridization analysis identified cyclin D1 as a target oncogene at 11q13.3 in nasopharyngeal carcinoma. Cancer Res. 2005;65(18):8125–8133. doi: 10.1158/0008-5472.CAN-05-0648. [DOI] [PubMed] [Google Scholar]
- Wen JM, Huang JF, Hu L, Wang WS, Zhang M, Sham JS, Xu JM, Zeng WF, Xie D, Liang LJ. et al. Establishment and characterization of human metastatic hepatocellular carcinoma cell line. Cancer Genet Cytogenet. 2002;135(1):91–95. doi: 10.1016/S0165-4608(01)00636-7. [DOI] [PubMed] [Google Scholar]
- Jin C, Jin Y, Gisselsson D, Wennerberg J, Wah TS, Stromback B, Kwong YL, Mertens F. Molecular cytogenetic characterization of the 11q13 amplicon in head and neck squamous cell carcinoma. Cytogenet Genome Res. 2006;115(2):99–106. doi: 10.1159/000095228. [DOI] [PubMed] [Google Scholar]
- Gibcus JH, Kok K, Menkema L, Hermsen MA, Mastik M, Kluin PM, van der Wal JE, Schuuring E. High-resolution mapping identifies a commonly amplified 11q13.3 region containing multiple genes flanked by segmental duplications. Hum Genet. 2007;121(2):187–201. doi: 10.1007/s00439-006-0299-6. [DOI] [PubMed] [Google Scholar]
- Luo ML, Shen XM, Zhang Y, Wei F, Xu X, Cai Y, Zhang X, Sun YT, Zhan QM, Wu M. et al. Amplification and overexpression of CTTN (EMS1) contribute to the metastasis of esophageal squamous cell carcinoma by promoting cell migration and anoikis resistance. Cancer Res. 2006;66(24):11690–11699. doi: 10.1158/0008-5472.CAN-06-1484. [DOI] [PubMed] [Google Scholar]
- Jin Y, Jin C, Law S, Chu KM, Zhang H, Strombeck B, Yuen AP, Kwong YL. Cytogenetic and fluorescence in situ hybridization characterization of clonal chromosomal aberrations and CCND1 amplification in esophageal carcinomas. Cancer Genet Cytogenet. 2004;148(1):21–28. doi: 10.1016/S0165-4608(03)00213-9. [DOI] [PubMed] [Google Scholar]
- Ishizuka T, Tanabe C, Sakamoto H, Aoyagi K, Maekawa M, Matsukura N, Tokunaga A, Tajiri T, Yoshida T, Terada M. et al. Gene amplification profiling of esophageal squamous cell carcinomas by DNA array CGH. Biochem Biophys Res Commun. 2002;296(1):152–155. doi: 10.1016/S0006-291X(02)00836-7. [DOI] [PubMed] [Google Scholar]
- Komatsu Y, Hibi K, Kodera Y, Akiyama S, Ito K, Nakao A. TAOS1, a novel marker for advanced esophageal squamous cell carcinoma. Anticancer Res. 2006;26(3A):2029–2032. [PubMed] [Google Scholar]
- Kohlhammer H, Schwaenen C, Wessendorf S, Holzmann K, Kestler HA, Kienle D, Barth TF, Moller P, Ott G, Kalla J. et al. Genomic DNA-chip hybridization in t(11;14)-positive mantle cell lymphomas shows a high frequency of aberrations and allows a refined characterization of consensus regions. Blood. 2004;104(3):795–801. doi: 10.1182/blood-2003-12-4175. [DOI] [PubMed] [Google Scholar]
- Ronchetti D, Finelli P, Richelda R, Baldini L, Rocchi M, Viggiano L, Cuneo A, Bogni S, Fabris S, Lombardi L. et al. Molecular analysis of 11q13 breakpoints in multiple myeloma. Blood. 1999;93(4):1330–1337. [PubMed] [Google Scholar]
- Fenton JA, Pratt G, Rothwell DG, Rawstron AC, Morgan GJ. Translocation t(11;14) in multiple myeloma: Analysis of translocation breakpoints on der(11) and der(14) chromosomes suggests complex molecular mechanisms of recombination. Genes Chromosomes Cancer. 2004;39(2):151–155. doi: 10.1002/gcc.10304. [DOI] [PubMed] [Google Scholar]
- Tarsitano M, Palmieri S, Ferrara F, Riccardi C, Cavaliere ML, Vicari L. Detection of the t(11;14)(q13;q32) without CCND1/IGH fusion in a case of acute myeloid leukemia. Cancer Genet Cytogenet. 2009;195(2):164–167. doi: 10.1016/j.cancergencyto.2009.08.013. [DOI] [PubMed] [Google Scholar]
- Medeiros LJ, Carr J. Overview of the role of molecular methods in the diagnosis of malignant lymphomas. Arch Pathol Lab Med. 1999;123(12):1189–1207. doi: 10.5858/1999-123-1189-OOTROM. [DOI] [PubMed] [Google Scholar]
- Janssen JW, Vaandrager JW, Heuser T, Jauch A, Kluin PM, Geelen E, Bergsagel PL, Kuehl WM, Drexler HG, Otsuki T. et al. Concurrent activation of a novel putative transforming gene, myeov, and cyclin D1 in a subset of multiple myeloma cell lines with t(11;14)(q13;q32) Blood. 2000;95(8):2691–2698. [PubMed] [Google Scholar]
- Wlodarska I, Meeus P, Stul M, Thienpont L, Wouters E, Marcelis L, Demuynck H, Rummens JL, Madoe V, Hagemeijer A. Variant t(2;11)(p11;q13) associated with the IgK-CCND1 rearrangement is a recurrent translocation in leukemic small-cell B-non-Hodgkin lymphoma. Leukemia. 2004;18(10):1705–1710. doi: 10.1038/sj.leu.2403459. [DOI] [PubMed] [Google Scholar]
- de Oliveira FM, Tone LG, Simoes BP, Falcao RP, Brassesco MS, Sakamoto-Hojo ET, dos Santos GA, Marinato AF, Jacomo RH, Rego EM. Acute myeloid leukemia (AML-M2) with t(5;11)(q35;q13) and normal expression of cyclin D1. Cancer Genet Cytogenet. 2007;172(2):154–157. doi: 10.1016/j.cancergencyto.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Jhang JS, Narayan G, Murty VV, Mansukhani MM. Renal oncocytomas with 11q13 rearrangements: cytogenetic, molecular, and immunohistochemical analysis of cyclin D1. Cancer Genet Cytogenet. 2004;149(2):114–119. doi: 10.1016/j.cancergencyto.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Wells RA, Hummel JL, De Koven A, Zipursky A, Kirby M, Dube I, Kamel-Reid S. A new variant translocation in acute promyelocytic leukaemia: molecular characterization and clinical correlation. Leukemia. 1996;10(4):735–740. [PubMed] [Google Scholar]
- Wells RA, Catzavelos C, Kamel-Reid S. Fusion of retinoic acid receptor alpha to NuMA, the nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat Genet. 1997;17(1):109–113. doi: 10.1038/ng0997-109. [DOI] [PubMed] [Google Scholar]
- Sukhai MA, Wu X, Xuan Y, Zhang T, Reis PP, Dube K, Rego EM, Bhaumik M, Bailey DJ, Wells RA. et al. Myeloid leukemia with promyelocytic features in transgenic mice expressing hCG-NuMA-RARalpha. Oncogene. 2004;23(3):665–678. doi: 10.1038/sj.onc.1207073. [DOI] [PubMed] [Google Scholar]
- Imagama S, Abe A, Suzuki M, Hayakawa F, Katsumi A, Emi N, Kiyoi H, Naoe T. LRP16 is fused to RUNX1 in monocytic leukemia cell line with t(11;21)(q13;q22) Eur J Haematol. 2007;79(1):25–31. doi: 10.1111/j.1600-0609.2007.00858.x. [DOI] [PubMed] [Google Scholar]
- Sarova I, Brezinova J, Zemanova Z, Gancarcikova M, Vydra J, Cermak J, Michalova K. A novel gene LRP5 on 11q13.2 is rearranged in two patients with acute myeloid leukemia. Leuk Res. 2011;35(11):200–20. doi: 10.1016/j.leukres.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Jesudasan RA, Rahman RA, Chandrashekharappa S, Evans GA, Srivatsan ES. Deletion and translocation of chromosome 11q13 sequences in cervical carcinoma cell lines. Am J Hum Genet. 1995;56(3):705–715. [PMC free article] [PubMed] [Google Scholar]
- Rao PH, Harris CP, Yan Lu X, Li XN, Mok SC, Lau CC. Multicolor spectral karyotyping of serous ovarian adenocarcinoma. Genes Chromosomes Cancer. 2002;33(2):123–132. doi: 10.1002/gcc.1221. [DOI] [PubMed] [Google Scholar]
- Wong N, Lai P, Pang E, Leung TW, Lau JW, Johnson PJ. A comprehensive karyotypic study on human hepatocellular carcinoma by spectral karyotyping. Hepatology. 2000;32(5):1060–1068. doi: 10.1053/jhep.2000.19349. [DOI] [PubMed] [Google Scholar]
- Yamashita Y, Nishida K, Okuda T, Nomura K, Matsumoto Y, Mitsufuji S, Horiike S, Hata H, Sakakura C, Hagiwara A. et al. Recurrent chromosomal rearrangements at bands 8q24 and 11q13 in gastric cancer as detected by multicolor spectral karyotyping. World J Gastroenterol. 2005;11(33):5129–5135. doi: 10.3748/wjg.v11.i33.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuster MI, Han L, Le Beau MM, Davis E, Sawicki M, Lese CM, Park NH, Colicelli J, Gollin SM. A consistent pattern of RIN1 rearrangements in oral squamous cell carcinoma cell lines supports a breakage-fusion-bridge cycle model for 11q13 amplification. Genes Chromosomes Cancer. 2000;28(2):153–163. doi: 10.1002/(SICI)1098-2264(200006)28:2<153::AID-GCC4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Martin CL, Reshmi SC, Ried T, Gottberg W, Wilson JW, Reddy JK, Khanna P, Johnson JT, Myers EN, Gollin SM. Chromosomal imbalances in oral squamous cell carcinoma: examination of 31 cell lines and review of the literature. Oral Oncol. 2008;44(4):369–382. doi: 10.1016/j.oraloncology.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishmar D, Rahat A, Scherer SW, Nyakatura G, Hinzmann B, Kohwi Y, Mandel-Gutfroind Y, Lee JR, Drescher B, Sas DE. et al. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc Natl Acad Sci U S A. 1998;95(14):8141–8146. doi: 10.1073/pnas.95.14.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertson DG. Gene amplification in cancer. Trends Genet. 2006;22(8):447–455. doi: 10.1016/j.tig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Reshmi SC, Roychoudhury S, Yu Z, Feingold E, Potter D, Saunders WS, Gollin SM. Inverted duplication pattern in anaphase bridges confirms the breakage-fusion-bridge (BFB) cycle model for 11q13 amplification. Cytogenet Genome Res. 2007;116(1–2):46–52. doi: 10.1159/000097425. [DOI] [PubMed] [Google Scholar]
- Gajduskova P, Snijders AM, Kwek S, Roydasgupta R, Fridlyand J, Tokuyasu T, Pinkel D, Albertson DG. Genome position and gene amplification. Genome Biol. 2007;8(6):R120. doi: 10.1186/gb-2007-8-6-r120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquelle A, Pipiras E, Toledo F, Buttin G, Debatisse M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell. 1997;89(2):215–225. doi: 10.1016/S0092-8674(00)80201-9. [DOI] [PubMed] [Google Scholar]
- Huang X, Gollin SM, Raja S, Godfrey TE. High-resolution mapping of the 11q13 amplicon and identification of a gene, TAOS1, that is amplified and overexpressed in oral cancer cells. Proc Natl Acad Sci U S A. 2002;99(17):11369–11374. doi: 10.1073/pnas.172285799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada K, Oka M, Tangoku A, Hayashi H, Oga A, Sasaki K. Gains of 8q23-qter and 20q and loss of 11q22-qter in esophageal squamous cell carcinoma associated with lymph node metastasis. Cancer. 2000;88(2):268–273. doi: 10.1002/(SICI)1097-0142(20000115)88:2<268::AID-CNCR4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Takeshita H, Ichikawa D, Komatsu S, Tsujiura M, Kosuga T, Deguchi K, Konishi H, Morimura R, Shiozaki A, Fujiwara H. et al. Prediction of CCND1 amplification using plasma DNA as a prognostic marker in oesophageal squamous cell carcinoma. Br J Cancer. 2010;102(9):1378–1383. doi: 10.1038/sj.bjc.6605657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D, Millon R, Velten M, Bronner G, Jung G, Engelmann A, Flesch H, Eber M, Methlin G, Abecassis J. Amplification of 11q13 DNA markers in head and neck squamous cell carcinomas: correlation with clinical outcome. Eur J Cancer. 1997;33(13):2203–2210. doi: 10.1016/S0959-8049(97)00198-6. [DOI] [PubMed] [Google Scholar]
- Miyamoto R, Uzawa N, Nagaoka S, Nakakuki K, Hirata Y, Amagasa T. Potential marker of oral squamous cell carcinoma aggressiveness detected by fluorescence in situ hybridization in fine-needle aspiration biopsies. Cancer. 2002;95(10):2152–2159. doi: 10.1002/cncr.10929. [DOI] [PubMed] [Google Scholar]
- Sugahara K, Michikawa Y, Ishikawa K, Shoji Y, Iwakawa M, Shibahara T, Imai T. Combination effects of distinct cores in 11q13 amplification region on cervical lymph node metastasis of oral squamous cell carcinoma. Int J Oncol. 2011;39(4):761–769. doi: 10.3892/ijo.2011.1094. [DOI] [PubMed] [Google Scholar]
- Huang X, Godfrey TE, Gooding WE, McCarty KS Jr, Gollin SM. Comprehensive genome and transcriptome analysis of the 11q13 amplicon in human oral cancer and synteny to the 7F5 amplicon in murine oral carcinoma. Genes Chromosomes Cancer. 2006;45(11):1058–1069. doi: 10.1002/gcc.20371. [DOI] [PubMed] [Google Scholar]
- Fortin A, Guerry M, Guerry R, Talbot M, Parise O, Schwaab G, Bosq J, Bourhis J, Salvatori P, Janot F. et al. Chromosome 11q13 gene amplifications in oral and oropharyngeal carcinomas: no correlation with subclinical lymph node invasion and disease recurrence. Clin Cancer Res. 1997;3(9):1609–1614. [PubMed] [Google Scholar]
- Xia J, Chen Q, Li B, Zeng X. Amplifications of TAOS1 and EMS1 genes in oral carcinogenesis: association with clinicopathological features. Oral Oncol. 2007;43(5):508–514. doi: 10.1016/j.oraloncology.2006.05.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genomic copy number alterations in ESCC cell lines detected by array-CGH.
Table S2. Regions of gain and the frequencies in the ESCC cell lines.
Table S3. Unbalanced breakpoints analyzed according to the copy number alterations.
Figure S1. High level amplifications and breakages of 11q13 in ten ESCC cell lines. (A) KYSE140, (B) KYSE410, (C) EC-GI-10, (D) HCE-4, (E) TE-6, (F) TE-8, (G) TE-9, (H) TE-10, (I) TE-11, (J) TE-15. Amplifications are mainly located within 67-72 Mb position. The smallest amplification region is similar to previously reported. More than two breakpoints are present in most of the cell lines, revealing these positions may be highly rearranged. (EPS 3433 kb)
Table S4. Splitting and amplification of 11q13.3-q13.4 in the primary ESCC tumors by FISH analyses.
Table S5. Clinico-pathological parameters of the examined ESCC patients.
Table S6. Relationship between region splittings and clinico-pathological features of ESCC.
Table S8. Relationship between region amplifications and clinico-pathological features of ESCC.