

Abstract
Drug-induced lupus erythematosus (DILE) is a lupus-like syndrome temporally related to continuous drug exposure which resolves upon drug discontinuation. There are currently no standard diagnostic criteria for DILE. Findings include skin manifestations, arthritis, serositis, anti-nuclear and anti-histone antibodies positivity. Similarly to idiopathic lupus erythematosus, DILE can be divided into systemic (SLE), subacute cutaneous (SCLE) and chronic cutaneous lupus (CCLE). Systemic DILE presents as a milder version of idiopathic SLE, and the drugs most frequently implicated are hydralazine, procainamide and quinidine. Anti-TNFα therapies are the latest class of medications found to be associated, although rarely, with a “lupus-like” syndrome, which is however clinically distinct from classical DILE. Drug-induced SCLE is the most common form of DILE. It is very similar to idiopathic SCLE in terms of clinical and serologic characteristics. The most commonly implicated drugs are antihypertensive drugs and terbinafine, but in recent years also proton pump inhibitors and chemotherapeutic agents have been associated. Drug-induced CCLE is very rare and usually caused by fluorouracil agents and NSAIDS, but some cases have induced by pantoprazole and anti-TNFα agents.
Keywords: drug reactions, lupus erythematosus, drug-induced lupus erythematosus
Introduction
Systemic lupus erythematosus (SLE) is a common autoimmune disease, with an incidence in Europe and North America varying between 1 and 10 cases per 100 000 per year [1, 2]. It has been estimated that up to 10% of SLE cases are drug-induced. Drug-induced autoimmunity is idiosyncratic belonging to the category of “type B” drug reactions, which are unpredictable and may depend on many factors, such as genetic susceptibility, co-morbidities, interaction with other drugs and environmental factors [3]. Drug-induced lupus erythematosus (DILE) is a lupus-like syndrome temporally related to continuous drug exposure (from one month to as long as over a decade) which resolves after discontinuation of the drug [4]. DILE shows less predilections for women and Africans, and generally affects older patients than idiopathic SLE.
There are currently no standard diagnostic criteria for DILE, and in many cases patients with DILE do not fulfill the American College of Rheumatology (ACR) criteria for SLE. The four most common features (arthritis, serositis, antinuclear antibodies [ANA] and anti-histone antibodies) could be employed as diagnostic criteria; in addition the symptoms must have begun after initiation of the treatment with a drug and must resolve after discontinuation [5].
The pathogenesis of DILE remains unclear, and available data strongly suggest that there is no single mechanism responsible for the induction of autoimmunity by all lupus-inducing drugs. DILE does not present with the features of a typical drug hypersensitivity reaction. In particular, there is no evidence of drug-specific T cells or antibodies; the reaction occurs frequently months or years after exposure; development of DILE depends on the cumulative dose, and the recurrence of symptoms after rechallenge generally takes 1–2 days, indicating the absence of immune sensitization to the culprit drugs. Lupus-inducing drugs are commonly metabolized (oxidized) to reactive species by activated leucocytes, thus acquiring the capacity to bind to carrier proteins and become immunogenic. Alternatively, reactive drug metabolites could directly cause cell death via a non-immune mediated process or could alter degradation and clearance of apoptotic cells which leads to the loss of tolerance to self antigens. Disruption of central immune tolerance has also been hypothesized [6]. Finally, altered T-cell function due to hypomethylation has been suggested. Hypomethylation of DNA may alter T-cell gene expression profiles and T-cell function, making the T-cells autoreactive and promoting their activation [7].
Similarly to idiopathic lupus, DILE can be divided into systemic (SLE), subacute cutaneous (SCLE) and chronic cutaneous lupus (CCLE), both in the form of discoid and tumidus (LET).
Systemic DILE
Systemic DILE usually resembles a milder version of idiopathic SLE (Table 1). It is rare and it is characterized by typical general lupus-like symptoms with arthralgia, myalgia, fever, pleurisy and pericarditis. Central nervous system and renal involvement are usually absent. Skin involvement is generally less frequent and severe in DILE compared to SLE, and characterized by photosensitivity, purpura and erythema nodosum.
Table 1.
Characteristics of idiopathic, classical DILE, drug-induced SCLE and anti-TNFα DILE.
| Characteristics | Idiopathic SLE | Classic DILE | Drug-induced SCLE | Anti-TNFα DILE |
|---|---|---|---|---|
| Age of onset | Child-bearing years | Older | Older | Older |
| Female : male | 9 : 1 | 1 : 1 | 3 : 1 | 5 : 1 |
| Clinical course | Chronic, relapsing | Remits with drug discontinuation | Remits with drug discontinuation | Remits with drug discontinuation |
| Symptom severity | Mild to severe | Generally mild | Generally mild | Generally mild |
| Fever | 80% | 40% | Rare | 50% |
| Myalgia | 80% | 44–57% | Rare | 29% |
| Arthalgia/arthritis | 80% | 18–63% | Rare | 31–51% |
| Serositis | 20–40% | 5–50% | Rare | 3–24% |
| Mayor organ involvement (renal and neurologic) | Common | Rare | Rare | Rare (nephropathy 7%) |
| Cutaneous manifestations | 54–70% (malar rash, oral ulcers, photosensitivity) | <5–25% (photosensitivity, purpura) | > 99% (similar to idiopathic SCLE, bullous and EM-like lesions more frequent than in the idiopathic form) | 67% (photosensitivity) |
| ANA | >99% | >99% | >80% | >99% |
| ENA | up to 10% | |||
| Anti-Ro/SSA | up to 30% | <5% | >80% | |
| Anti-La/SSB | >45% | |||
| Anti-histone Ab | up to 50% | up to 95% | up to 33% | up to 57% |
| Anti-dsDNA Ab | 50–70% | <5% | <1% | 70–90% |
| Hypocomplementemia | 51% | <1% | 9% | 59% |
Other nonspecific skin features, including urticarial vasculitis, livedo reticularis and skin ulcers, may be part of the clinical presentation of systemic DILE [8]. Typical laboratory findings consist of mild cytopenia, an elevated erythrocyte sedimentation rate and the presence of ANA with a homogenous pattern. Anti-histone antibodies are classically associated with DILE; however multiple studies have revealed that they are present with significant frequency in several other autoimmune diseases, including idiopathic SLE. Zirwas et al. have demonstrated that the sensitivity of anti-histone antibodies for DILE is 67% and the specificity is 95%[9]. Their titer, together with ANA, gradually declines with the resolution of DILE. Anti-double stranded (ds) DNA and anti-extractable nuclear antigens (ENA) antibodies are rare [10, 11]. Usually months or years of exposure to the responsible drug are required for the development of DILE, which resolves within weeks of drug discontinuation. In contrast, exposure to low levels of certain drugs (antibiotics, NSAID, anti-convulsants and estrogens) for relatively short periods may exacerbate underlying SLE, which remains or recurs after withdrawal of the implicated drug. Over 80 drugs have been implicated in DILE, and the number is increasing constantly [11]. The most frequently drugs are hydralazine, procainamide, isoniazid, methyldopa, quinidine, minocycline, and chlorpromazine (Table 2). Minocycline, a tetracycline antibiotic, deserves special consideration because minocycline-induced DILE is characterized by typical DILE features but also by unusual cutaneous features (Raynaud phenomenon, polyarteritis nodosa-like lesions, erythema nodosum), hepatic manifestations and is rarely associated with positive anti-histone antibodies, while p-ANCA are present in 80% of cases [12]. The incidence of minocycline-induced lupus is approximately 15 cases/100 000 prescriptions and is more common in women [13]. Margolis et al. have shown a strong relationship between duration of exposure to minocycline (>300 days), total dose (>50 g) and occurrence of DILE, with an estimated threefold increased risk of developing lupus erythematosus [14]. Systemic DILE associated with interferon-α therapy has also been reported. It is characterized by a high frequency of mucocutaneous and renal involvement, with anti-dsDNA antibodies developing in 50% of cases [15]. Systemic DILE associated with interferon-β1 also has been described [16]. Recently Yokoyama et al. have reported two cases of systemic DILE induced by ticlopidine, a widely used drug-in people with ischemic vascular disease, characterized by the late-onset of symptoms [17].
Table 2.
Drugs implicated in drug-induced SLE.
| High risk | Moderate risk | Low risk | Very low risk | |
|---|---|---|---|---|
| Antiarrhythmics | • Procainamide (15–20%) | • Quinidine (<1%) | • Disopyramide | |
| • Propafenone | ||||
| Antihypertensives | • Hydralazine (5–8%) | • Methyldopa | • Clonidine | |
| • Captopril | • Enalapril | |||
| • Acebutol | • Labetalol | |||
| • Minoxidil | ||||
| • Pindolol | ||||
| • Prazosin | ||||
| Antipsychotics | • Chlorpromazine | • Chlorprothixene | ||
| • Lithium carbonate | ||||
| • Phenelzine | ||||
| Antibiotics | • Isoniazid | • Nitrofurantoin | ||
| • Minocycline | • Cefepime | |||
| Anticonvulsants | • Carbamazepine | • Ethosuximide | ||
| • Phenytoin | ||||
| • Primidone | ||||
| • Trimethadione | ||||
| Antithyroidals | • Propylthiouracil | |||
| Anti-inflammatories | • D-penicillamine | • Phenylbutazone | ||
| • Sulfasalazine | • NSAIDs | |||
| Diuretics | • Chlorthalidone | |||
| • Hydrochlorothiazide | ||||
| Anticholesterolemics | • Atorvastatin Fluvastatin | |||
| • Lovastatin Pravastatin Simvastatin | ||||
| Proton pump inhibitors | • Lansoprazole | |||
| • Omeprazole | ||||
| • Pantoprazole | ||||
| Chemotherapeutic agents | • Taxanes | |||
| • Cyclofosfamide | ||||
| • Doxorubicin | ||||
| • Fluorouracil | ||||
| • Anastrozole | ||||
| • Bortezomib | ||||
| Antiaggregants | • Ticlopidine | |||
| Biologicals | • Etanercept | |||
| • Infliximab | ||||
| • Adalimumab | ||||
| • IL-2 | ||||
| • IFN-α | ||||
| • IFN-1b |
Drug-induced SCLE
Drug-induced SCLE is the most common form of DILE, with at least 128 cases reported in the English language literature [18, 19]. It presents clinically, histopathologically and immunologically in a manner similar to idiopathic SCLE, with the typical photosensitive symmetric, nonscarring annular polycyclic, or papulosquamous lesions, usually on sun-exposed areas (Figure 1). In general drug-induced SCLE has more limited skin lesions than idiopathic SCLE (Figure 2). The legs are more likely to be affected, usually with vasculitic skin lesions; in addition malar rash, bullous lesions and erythema multiforme-like changes are more common than in idiopathic SCLE [4]. In contrast with the idiopathic form, systemic involvement in drug-induced SCLE is very rare [19]. Most patients affected by drug-induced SCLE are female (72%) with a mean age of 58.0 years. The immunological profile includes the frequent presence of anti-Ro/SSA and/or anti-La/SSB, together with ANA and anti-histone antibodies. Anti-Ro/SS-A is just as prevalent in drug-induced and idiopathic SCLE; the majority of patients who are Ro/SS-A or La/SS-B positive do not become negative after disease resolution. Anti-histone antibodies are positive in one-third of the cases [18]. Clinical and serological findings of drug-induced SCLE are likely to differ from classical DILE [18, 19] (Table 1). The histopathologic findings of drug-induced SCLE do not differ from idiopathic SCLE, and tissue eosinophilia is not an indicator of drug-induced SCLE [20]. The most commonly implicated drugs in subacute cutaneous DILE are antihypertensive agents, like thiazide diuretics and calcium channel blockers, and terbinafine [21]. Thiazides diuretics tend to have the longest incubation period ranging from six months to five years; for channel blockers the mean incubation period is 3.2 years, while for terbinafine it is just five weeks [18, 22]. Other drugs implicated are beta blockers, angiotensin-converting enzyme inhibitors, antihistamines (ranitidine), immunomodulators (leflunomide and interferons), antiepileptics, statins, biologics (anti-TNFα), proton pump inhibitors such as lansoprazole, omeprazole and pantoprazole [23] (Figure 2). Chemotherapeutic agents such as taxanes (paclitaxel and docetaxel) have also been implicated in subacute cutaneous DILE and showed a rapid disease onset. Taxane may favor apoptosis leading to a release of nucleosomes which in turn can trigger a local autoimmune response [24]. Recently Guhl et al. reported a case of early-onset chemotherapy-induced SCLE in a patient treated with cyclophosphamide and doxorubicin for a relapse of breast carcinoma [25]. Anastrozole, a selective nonsteroidal aromatase inhibitor widely used as an adjuvant therapy for postmenopausal women with early hormone-sensitive breast cancer, has been also associated with onset of SCLE [26].
Figure 1.
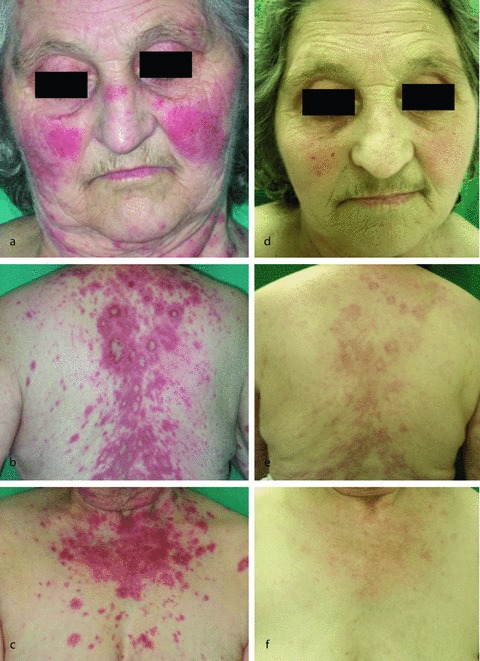
(a–c) A 70-year-old women presented with a photosensitive malar erythema since one month, and with macular erythematous lesions on the upper arms and trunk. She also complained of arthralgia, myalgia and low grade fever. Laboratory findings included a moderately elevated erythrocyte sedimentation rate and the presence of ANA with a homogenous pattern. Anti-dsDNA antibodies were absent; anti-histone antibodies were positive. The patients had been taking hydrochlorothiazide for hypertension for two years. (d–f) After drug discontinuation and one month of very low dose systemic steroids (prednisone 0.2 mg/kg), her skin lesions, and systemic symptoms progressively disappeared.
Figure 2.
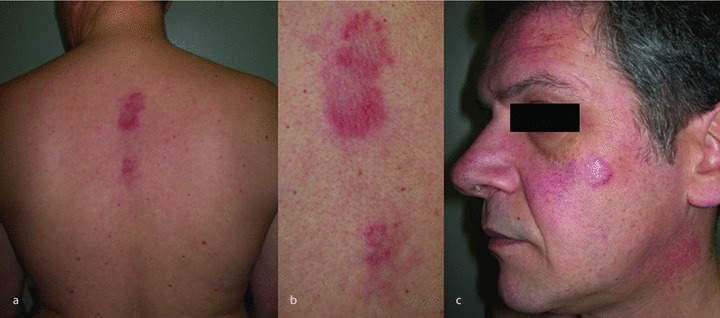
A 56-year-old man with chronic hepatitis C virus had annular-polycyclic lesions on his trunk, face and right knee with a chronic relapsing course for four years. The findings had appeared a few months after starting IFN-α therapy. He had no systemic symptoms.
Drug-induced CCLE and lupus erythematosus tumidus (LET)
Drug-induced CCLE is rare and usually refers to fluorouracil agents and NSAID (Figure 3). Moreover some cases have been triggered by pantoprazole and anti-TNFα agents [27, 28]. The patients usually show classic discoid skin lesions in photosensitive areas. LET is a rare form of CCLE presenting with single or multiple erythematous or violaceous indurated, urticarial plaques with smooth, non-scaling surface [29]. Rare cases of drug-induced LET have been attributed to anti-TNFα agents (infliximab and adalimumab), angiotensin-converting enzyme inhibitors and bortezomib, a proteasome inhibitor used for the treatment of multiple myeloma [30–33].
Figure 3.
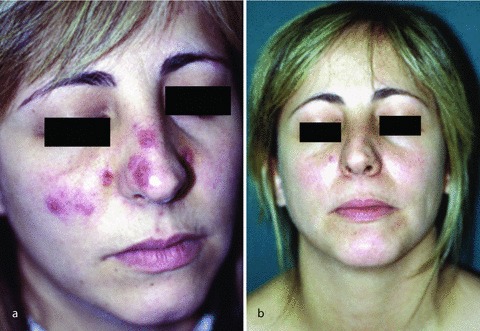
(a) A 28-year-old woman presented with pruritic, erythematous, scaling plaques on her nose and cheeks since 2 months. She had used ibuprofen daily for headache and dysmenorrhea for one year. She had no systemic symptoms. (b) Two months after drug discontinuation, her skin lesions disappeared without any treatment.
DILE due to anti-TNFα agents
Anti-TNFα therapies are the latest class of medications found to be associated with a “lupus-like” syndrome [34]. Most of the case reports of DILE secondary to anti-TNFα therapy occurred in patients receiving etanercept or infliximab, and only few cases with adalimumab, which may simply reflect fewer years of patient exposure to adalimumab than to infliximab or etanercept [28, 33, 35–38]. Two TNFα antagonists, golimumab and certolizumab pegol, have been more recently introduced, with the latest associated with one case of a lupus-like disorder [39].
In contrast to other forms of DILE, induction of ANA and/or anti-DNA antibodies in patients treated with anti-TNFα therapy is well established and quite common [40–50] (Table 3). ANA are more frequently observed in patients treated with infliximab in comparison with those receiving etanercept [51]. Bacquet-Deschryver et al. have shown similar findings in patients with rheumatoid arthritis or spondyloarthropathies, with appearance of anti-dsDNA antibodies in a very low proportion of patients, with no difference among the three biologic agents [50]. ANA titer cannot be used to predict recurrence of anti-TNFα DILE following rechallenge with another anti-TNFα agent [52]. Despite such high frequency of ANA antibodies due to anti-TNFα agents, relatively few cases of anti-TNFα DILE have been reported; DILE secondary to anti-TNFα has been documented in less than 0.5% of treated individuals in clinical trials [35]. DILE secondary to anti-TNFα agents is quite distinct from classical DILE (Table 1). Unlike classic DILE, anti-TNFα DILE more often affects women than men. The mean age of onset ranges from 46.2 to 50.9 years [52, 53]. Symptoms occur after prolonged anti-TNFα therapy (mean 40.6 weeks) and they are characterized, as in classic DILE, by generalized symptoms, musculoskeletal manifestations, lupus-like cutaneous features and the appearance of serum autoantibodies. Cutaneous involvement seems to be more common than in classic DILE and includes malar rash, photosensitivity, and subacute/chronic LE cutaneous features. Cutaneous lesions are more frequently observed in patients who received etanercept (44% vs. 12%), while serositis is more frequently observed in those treated with infliximab (24% vs. 3%) [54]. Visceral involvement is not rare, with evidence of renal disease in several cases [55, 56]. Anti-dsDNA antibodies positivity occurs more frequently in anti-TNFα DILE than in classic DILE while anti-histone antibodies are described in classic DILE more often than in anti-TNFα DILE. Hypocomplementemia and positive ENAs are also more common in anti-TNFα DILE. Cytopenias are the most common hematological disorders occurring in 2–61% of patients [28, 34]. As in classic DILE, ANA titers are frequently high. Pink et al. have proposed that the development of ANA in psoriatic patients treated with anti-TNFα may predict treatment failure, but Golberg et al. have proposed alternative explanations, as multiple therapies received by these patients could increase the chance of development of ANA [57, 58]. Finally, Iwata et al. have showed a correlation between elevated ANA titers and decreased therapeutic efficacy in the treatment of Behçet disease with infliximab [59].
Table 3.
ANA and anti-dsDNA antibodies in patients treated with anti-TNFα agents.
| Author | Disease | N | Drugs | ANA | dsDNA | ||
|---|---|---|---|---|---|---|---|
| Baseline (%) | End (%) | Baseline (%) | End (%) | ||||
| Hanauer et al. (2002) [40] | Crohn's | 188 | Infliximab (5mg/kg) | – | 56 | – | 34 |
| 385 | Infliximab (10 mg/kg) | – | 35 | – | 11 | ||
| Allanore et al. (2004) [41] | RA | 59 | Infliximab | 29 | 69 | 3 | 32 |
| Ferraro-Peyret et al. (2004) [42] | RA | 24 | Infliximab | 37.5 | 87.5 | 4.2 | 57 |
| AS | 15 | Infliximab | 13.3 | 66.7 | 13.3 | 31 | |
| Caramaschi et al. (2004) [43] | RA | 43 | Infliximab | 37 | 95 | 0 | 2.6 |
| 11 | Etanercept | 36 | 55 | 0 | 0 | ||
| Eriksson et al. (2005) [44] | RA | 53 | Infliximab | 24 | 69 | 2 | 45 |
| Sellam et al. (2005) [45] | SpA | 33 | Infliximab | 4 | 29 | 0 | 11 |
| Klareskog et al. (2005) [46] | RA | 549 | Etanercept | – | – | 0.4 | 2–4 |
| De Rycke et al. (2005) [47] | RA | 59 | Infliximab | 40 | 85 | 0 | 40 |
| SpA | 54 | Infliximab | 12 | 62 | 0 | 55 | |
| 20 | Etanercept | 15 | 30 | 0 | 15 | ||
| Atzeni et al. (2006) [48] | RA | 57 | Adalimumab | 7 | 28 | 0 | 7 |
| Poulalhon et al. (2007) [49] | Psoriasis | 28 | Infliximab | 12 | 72 | 0 | 68 |
| Bacquet-Deschryver et al. (2008) [50] | RA | 48 | Infliximab | 0 | 62.5 | 0 | 3 |
| 30 | Etanercept | 0 | 13.3 | 0 | 0 | ||
| 17 | Adalimumab | 0 | 29.4 | 0 | 0 | ||
| SpA | 44 | Infliximab | 0 | 47.7 | 0 | 0 | |
| 29 | Etanercept | 0 | 14.3 | 0 | 3.4 | ||
Abbr.: RA, rheumatoid arthritis; AS, ankylosing spondylitis; SpA, spondyloarthropathy; ANA, antinuclear antibodies; dsDNA, double-stranded deoxyribonucleic acid antibodies.
The mechanisms by which anti-TNFα therapy induces lupus remain unclear but are likely to differ from classic DILE. One hypothesis is that TNF inhibitors interfere with normal cell apoptosis, leading to a decreased clearance of autoreactive T and B cells and cellular debris, including nuclear material. Accumulation of nucleosomes and their breakdown products in a genetically susceptible host may result in the development of autoantibodies. Another hypothesis is that the suppression of the T-helper type 1 response by TNF blockers could enhance a T-helper type 2 response leading to SLE. Finally, bacterial infections which are increased with TNF blockers, may induce polyclonal B-lymphocyte activation and favor autoantibody production [3].
Conclusions
DILE is a reversible lupus-like condition due to exposure to an increasing number of drugs. Its symptoms are generally mild to moderate with resolution of both clinical and serological features over time following drug discontinuation. The possibility of drug-induction should always be considered in all patients with lupus erythematosus, because of the easy reversibility of the lesions. The management of DILE consists mainly of the discontinuation of the implicated drug. For severe or refractory cases the addition of systemic corticosteroids at the doses commonly used for the idiopathic form is the first-choice therapy. Some patients may need additional immunosuppressive therapy, including azathioprine, cyclophosphamide, methotrexate or mycophenolate. In the case of anti-TNFα DILE, if the clinical presentation of drug-induced lupus is mild and well tolerated, TNFα inhibitors do not need to be discontinued. The appearance of ANA is not a reason for stopping TNFα inhibitors in asymptomatic patients with psoriasis. There is limited evidence to support the switch to alternative TNFα antagonists in patients who develop anti-TNFα DILE [53].
Conflict of interest
None.
References
- 1.Pons-Estel GJ, Alarcón GS, Scofield L, Reinlib L, Cooper GS. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257–68. doi: 10.1016/j.semarthrit.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365:2110–21. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 3.Chang C, Gershwin ME. Drug-induced lupus erythematosus. Incidence, management and prevention. Drug Saf. 2011;34:357–74. doi: 10.2165/11588500-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Marzano AV, Vezzoli P, Crosti C. Drug-induced lupus: an update on its dermatological aspects. Lupus. 2009;18:935–40. doi: 10.1177/0961203309106176. [DOI] [PubMed] [Google Scholar]
- 5.Antonov D, Kazandjieva J, Etugov D, Gospodinov D, Tsankov N. Drug-induced lupus erythematosus. Clin Dermatol. 2004;22:157–66. doi: 10.1016/j.clindermatol.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 6.Rubin RL. Drug-induced lupus. Toxicology. 2005;209:135–47. doi: 10.1016/j.tox.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 7.Chang C, Gershwin ME. Drugs and autoimmunity – a contemporary review and mechanistic approach. J Autoimmun. 2010;34:J266–75. doi: 10.1016/j.jaut.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 8.Peroni A, Colato C, Zanoni G, Girolomoni G. Urticarial lesions: if not urticaria, what else? The differential diagnosis of urticaria: part II. Systemic diseases. J Am Acad Dermatol. 2010;62:557–70. doi: 10.1016/j.jaad.2009.11.687. [DOI] [PubMed] [Google Scholar]
- 9.Zirwas MJ, Kress DW, Deng JS. The utility of antihistone antibody screening in the diagnosis of drug-induced lupus erythematosus. Arch Dermatol. 2004;140:494–5. doi: 10.1001/archderm.140.4.494. [DOI] [PubMed] [Google Scholar]
- 10.Vasoo S. Drug-induced lupus: an update. Lupus. 2006;15:757–61. doi: 10.1177/0961203306070000. [DOI] [PubMed] [Google Scholar]
- 11.Dalle Vedove C, Del Giglio M, Schena D, Girolomoni G. Drug-induced lupus erythematosus. Arch Dermatol Res. 2009;301:99–105. doi: 10.1007/s00403-008-0895-5. [DOI] [PubMed] [Google Scholar]
- 12.Sturkenboom MC, Meier CR, Jick H, Stricker B. Minocycline and lupus-like syndrome in acne patients. Arch Intern Med. 1999;159:493–7. doi: 10.1001/archinte.159.5.493. [DOI] [PubMed] [Google Scholar]
- 13.Borchers AT, Keen CL, Gershwin ME. Drug-induced lupus. Ann N Y Acad Sci. 2007;1108:166–82. doi: 10.1196/annals.1422.019. [DOI] [PubMed] [Google Scholar]
- 14.Margolis DJ, Hoffstad O, Bilker W. Association or lack association between tetracycline class antibiotics used for acne vulgaris and lupus erythematosus. Br J Dermatol. 2007;157:540–6. doi: 10.1111/j.1365-2133.2007.08056.x. [DOI] [PubMed] [Google Scholar]
- 15.Borg FA, Isenberg DA. Syndromes and complications of interferon therapy. Curr Opin Rheumatol. 2007;19:61–6. doi: 10.1097/BOR.0b013e328010c547. [DOI] [PubMed] [Google Scholar]
- 16.Sladkova V, Mares J, Lubenova B, Hlustik P, Kanovsky P. Drug-induced systemic lupus erythematosus in interferon beta-1b therapy. Neuro Endocrinol Lett. 2011;32:4–6. [PubMed] [Google Scholar]
- 17.Yokoyama T, Usui T, Kiyama K, Nakashima R, Yukawa N, Kawabata D, Nojima T, Ohmura K, Fujii T, Mimori T. Two cases of late-onset drug-induced lupus erythematosus caused by ticlopidine in elderly men. Mod Rheumatol. 2010;20:405–9. doi: 10.1007/s10165-010-0289-3. [DOI] [PubMed] [Google Scholar]
- 18.Lowe G, Henderson CL, Grau RH, Hansen CB, Sontheimer RD. A systematic review of drug-induced subacute cutaneous lupus erythematosus. Br J Dermatol. 2011;164:465–72. doi: 10.1111/j.1365-2133.2010.10110.x. [DOI] [PubMed] [Google Scholar]
- 19.Marzano AV, Lazzari R, Polloni I, Crosti C, Fabbri P, Cugno M. Drug-induced subacute cutaneous lupus erythematosus: evidence for differences from its idiopathic counterpart. Br J Dermatol. 2011;165:335–41. doi: 10.1111/j.1365-2133.2011.10397.x. [DOI] [PubMed] [Google Scholar]
- 20.Hillesheim PB, Bahrami S, Brooke GJ, Jeffrey PC. Tissue eosinophilia. Not an indicator of drug-induced subacute cutaneous lupus erythematosus. Arch Dermatol. 2012;148:190–3. doi: 10.1001/archdermatol.2011.290. [DOI] [PubMed] [Google Scholar]
- 21.Gubinelli E, Cocuroccia B, Girolomoni G. Subacute cutaneous lupus erythematosus induced by nifedipine. J Cutan Med Surg. 2003;7:243–6. doi: 10.1007/s10227-002-0122-5. [DOI] [PubMed] [Google Scholar]
- 22.Lorentz K, Booken N, Goerdt S, Goebeler M. Subacute cutaneous lupus erythematosus induced by terbinafine: case report and review of literature. J Dtsch Dermatol Ges. 2008;6:823–7. doi: 10.1111/j.1610-0387.2008.06806.x. [DOI] [PubMed] [Google Scholar]
- 23.Dam C, Bygum A. Subacute cutaneous lupus erythematosus induced or exacerbated by proton pump inhibitors. Acta Derm Venereol. 2008;88:87–9. doi: 10.2340/00015555-0335. [DOI] [PubMed] [Google Scholar]
- 24.Chen M, Crowson AN, Woofter M, Luca MB, Magro CM. Docetaxel (taxotere) induced subacute cutaneous lupus erythematosus: report of 4 cases. J Rheumatol. 2004;31:818–20. [PubMed] [Google Scholar]
- 25.Guhl G, Diaz-ley B, Garcia-Garcia C, Fraga J, Garcia-Diez A. Chemotherapy-induced subacute lupus erythematosus. Lupus. 2009;18:859–60. doi: 10.1177/0961203309103050. [DOI] [PubMed] [Google Scholar]
- 26.Trancart M, Cavailhes A. Balme B, Skowron F. Anastrozole-induced subacute cutaneous lupus erythematosus. Br J Dermatol. 2008;158:628–9. doi: 10.1111/j.1365-2133.2007.08367.x. [DOI] [PubMed] [Google Scholar]
- 27.Correia O, Lomba Viana H, Azevedo R, Delgado L, Polonia J. Possible phototoxicity with subsequent progression to discoid lupus following pantoprazole administration. Clin Exp Dermatol. 2001;26:455–6. doi: 10.1046/j.1365-2230.2001.00857.x. [DOI] [PubMed] [Google Scholar]
- 28.Costa MF, Said NR, Zimmermann B. Drug-induced lupus due to anti-tumor necrosis factor α agents. Semin Arth Rheum. 2008;37:381–7. doi: 10.1016/j.semarthrit.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 29.Schmitt V, Meuth AM, Amler S, Kuehn E, Haust M, Messer G, Bekou V, Sauerland C, Metze D, Kopcke W, Bonsmann G, Kuhn A. Lupus erythematosus tumidus is a separate subtype of cutaneous lupus erythematosus. Br J Dermatol. 2009;162:64–73. doi: 10.1111/j.1365-2133.2009.09401.x. [DOI] [PubMed] [Google Scholar]
- 30.Bokle BC, Baltaci M, Weyrer W, Sepp NT. Bortezomib-induced lupus erythematosus tumidus. Oncologist. 2009;14:637–9. doi: 10.1634/theoncologist.2008-0197. [DOI] [PubMed] [Google Scholar]
- 31.Schneider SW, Staender S, Schluter B, Luger TA, Bonsmann G. Infliximab-induced lupus erythematosus tumidus in a patient with rheumatoid arthritis. Arch Dermatol. 2006;142:115–6. doi: 10.1001/archderm.142.1.115. [DOI] [PubMed] [Google Scholar]
- 32.Sohl S, Renner R, Winter U, Bodendorf M, Paasch U, Simon JC, Treudler R. Drug-induced lupus erythematosus tumidus during treatment with adalimumab. Hautarzt. 2009;60:826–9. doi: 10.1007/s00105-008-1699-4. [DOI] [PubMed] [Google Scholar]
- 33.Schepis C, Lentini M, Siragusa M, Batolo D. Ace-inhibitor-induced drug eruption resembling lymphocytic infiltration (of Jessner-Kanof) and lupus erythematosus tumidus. Dermatology. 2004;208:354–55. doi: 10.1159/000077849. [DOI] [PubMed] [Google Scholar]
- 34.Williams EL, Gadola S, Edwards CJ. Anti-TNF-induced lupus. Rheumatology (Oxford) 2009;48:716–20. doi: 10.1093/rheumatology/kep080. [DOI] [PubMed] [Google Scholar]
- 35.De Bant M, Sibilia J, Le Loet X, Prouzeau S, Fautrel B, Marcelli C, Boucquillard E, Siame JL, Mariette X. Club Rhumatismes et Inflammation. Systemic lupus erythematosus induced by anti-tumor necrosis factor alpha therapy: a French national survey. Arthritis Res Ther. 2005;7:R545–51. doi: 10.1186/ar1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soforo E, Baumgartner M, Francis L, Allam F, Phillips PE, Perl A. Induction of systemic lupus erythematosus with tumor necrosis factor blockers. J Rheumatol. 2010;37:1. doi: 10.3899/jrheum.081312. [DOI] [PubMed] [Google Scholar]
- 37.Ramos-Casals M, Brito-Zeron P, Soto MJ, Cuadrado MJ, Khamashta MA. Autoimmune diseases induced by TNF-targeted terapie. Best Pract Res Clin Rheumatol. 2008;22:847–61. doi: 10.1016/j.berh.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Al-Niaimi F. Adalimumab-induced lupus erythematosus. Eur J Dermatol. 2009;19:380. doi: 10.1684/ejd.2009.0668. [DOI] [PubMed] [Google Scholar]
- 39.Statkute L, Ruderman EM. Novel TNF antagonists for the treatment of rheumatoid arthritis. Expert Opin Investig Drugs. 2010;19:105–15. doi: 10.1517/13543780903438559. [DOI] [PubMed] [Google Scholar]
- 40.Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, Rachmilewitz D, Wolf DC, Olson A, Bao W, Rutgeerts P ACCENT I Study Group. Maintenance infliximab for Crohn's disease: the ACCENT I randomized trial. Lancet. 2002;359:1541–9. doi: 10.1016/S0140-6736(02)08512-4. [DOI] [PubMed] [Google Scholar]
- 41.Allanore Y, Sellam J, Batteux F, Job Deslandre C, Weill B, Kahan A. Induction of autoantibody in refractory rheumatoid arthritis patients treated by infliximab. Clin Exp Rheumatol. 2004;22:756–8. [PubMed] [Google Scholar]
- 42.Ferraro-Peyret C, Coury F, Tebib JG, Bienvenu J, Fabien N. Infliximab therapy in rheumatoid arthritis and ankylosing apondylitis-induced specific antinuclear and antiphospholipid autoantibodies without autoimmune clinical manifestations: a two-year prospective study. Arthritis Res Ther. 2004;6:R535–43. doi: 10.1186/ar1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caramaschi P, Biasi D, Colombatti M, Pieropan S, Martinelli N, Carletto A, Volpe A, Pacor LM, Mambara LM. Anti-TNFα therapy in rheumatoid arthritis and autoimmunity. Rheumatol Int. 2006;26:209–14. doi: 10.1007/s00296-004-0542-1. [DOI] [PubMed] [Google Scholar]
- 44.Eriksson C, Engstrand S, Sundqvist KG, Rantapaa-Dahlqvist S. Autoantibody formation in patients with rheumatoid arthritis treated with anti-TNF alpha. Ann Rheum Dis. 2005;64:403–7. doi: 10.1136/ard.2004.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sellam J, Allanore Y, Batteux F, Deslandre CJ, Weill B, Kahan A. Autoantibody induction in patients with refractory spondyloarthropathy treated with infliximab and etanercept. Joint Bone Spine. 2005;72:48–52. doi: 10.1016/j.jbspin.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 46.Klareskog L, Wajdula J, Yeh P, Fatenejad S. Low autoantibody and anti-etanercept antibody formation and lack of impact on clinical outcomes after five years of treatment with etanercept in patients with rheumatoid arthritis. Arthritis Rheum. 2005;52:S348. [Google Scholar]
- 47.De Rycke L, Baeten D, Kruithof E, Van den Bosch F, Veys EM, De Keyser F. Infliximab, but not etanercept, induces IgM anti-dsDNA antibodies as main antinuclear reactivity: biological and clinical implications in autoimmune arthritis. Arthritis Rheum. 2005;52:2192–201. doi: 10.1002/art.21190. [DOI] [PubMed] [Google Scholar]
- 48.Atzeni F, Sarzi-Puttini P, Dell’Acqua D, De Portu S, Cecchini G, Cruini C, Carrabba M, Meroni PL. Adalimumab clinical efficacy is associated with rheumatoid factor and anti-cyclic citrullinated peptide antibody titer reduction: a one-year prospective study. Arthritis Res Ther. 2006;8:R3. doi: 10.1186/ar1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poulalhon N, Begon E, Lebbé C, Lioté F, Lahfa M, Bengoufa D, Morel P, Dubertret L, Bachelez H. A follow-up study in 28 patients treated with infliximab for severe recalcitrant psoriasis: evidence for efficacy and high incidence of biological autoimmunity. Br J Dermatol. 2007;156:329–36. doi: 10.1111/j.1365-2133.2006.07639.x. [DOI] [PubMed] [Google Scholar]
- 50.Bacquet-Deschryver H, Jouen F, Quillard M, Menard JF, Goeb V, Lequerre T, Mejjad O, Daragon A, Tron F, Le Loet X, Vittecoq O. Impact of three anti-TNFα biologics on existing and emergent autoimmunity in rheumatoid arthritis and spondylarthropathy patients. J Clin Immunol. 2008;28:445–55. doi: 10.1007/s10875-008-9214-3. [DOI] [PubMed] [Google Scholar]
- 51.Atzeni F, Turiel M, Capsoni F, Doria A, Meroni P, Sarzi-Puttini P. Autoimmunity and anti-TNF-a agents. Ann N Y Acad Sci. 2005;1051:559–69. doi: 10.1196/annals.1361.100. [DOI] [PubMed] [Google Scholar]
- 52.Subramanian S, Yajnik V, Sands BE, Cullen G, Korzenik JR. Characterization of patients with infliximab-induced lupus erythematosus and outcomes after retreatment with a second anti-TNF agent. Inflamm Bowel Dis. 2011;17:99–104. doi: 10.1002/ibd.21370. [DOI] [PubMed] [Google Scholar]
- 53.Williams VL, Cohen PR. TNF alpha antagonist-induced lupus-like syndrome: report and review of the literature with implications for treatment with alternative TNF alpha antagonists. Int J Dermatol. 2011;50:619–25. doi: 10.1111/j.1365-4632.2011.04871.x. [DOI] [PubMed] [Google Scholar]
- 54.Ramos-Casals M, Brito-Zeron P, Munoz S, Soria N, Galiana D, Bertolaccini L, Cuadrado MJ, Khamashta MA. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore) 2007;86:242–51. doi: 10.1097/MD.0b013e3181441a68. [DOI] [PubMed] [Google Scholar]
- 55.Stokes MB, Foster K, Markowitz GS, Ebrahimi F, Hines W, Kaufmen D. Development of glomerulonephritis during anti-TNF-alpha therapy for rheumatoid arthritis. Nephrol Dial Transplant. 2005;20:1400–6. doi: 10.1093/ndt/gfh832. [DOI] [PubMed] [Google Scholar]
- 56.Chadha T, Hernandez JE. Infliximab-related lupus and associated valvulitis: a case report and review of the literature. Arthritis Rheum. 2006;55:163–6. doi: 10.1002/art.21702. [DOI] [PubMed] [Google Scholar]
- 57.Pink AE, Fonia A, Allen MH, Smith CH, Barker JNWN. Antinuclear antibodies associate with loss of response to antitumour necrosis factor-a therapy in psoriasis: a retrospective, observational study. Br J Dermatol. 2010;162:780–5. doi: 10.1111/j.1365-2133.2009.09563.x. [DOI] [PubMed] [Google Scholar]
- 58.Golberg O, Osborne JE, Hutchinson PE. Antinuclear antibodies associate with loss of response to antitumour necrosis factor-a therapy in psoriasis but do not necessarily predict treatment failure. Br J Dermatol. 2011;164:459–60. doi: 10.1111/j.1365-2133.2010.10101.x. [DOI] [PubMed] [Google Scholar]
- 59.Iwata D, Namba K, Mizuuchi K, Kitaichi N, Kase S, Takemoto Y, Ohno S, Ishida S. Correlation between elevation of serum antinuclear antibody titer and decreased therapeutic efficacy in the treatment of Behçet's disease with infliximab. Graefes Arch Clin Exp Ophthalmol. 2012;250(7):1081–7. doi: 10.1007/s00417-011-1908-1. [DOI] [PubMed] [Google Scholar]