

Abstract
We report the mutational analysis of an artificial oxygen transport protein, HP-7, which operates via a mechanism akin to human neuroglobin and cytoglobin. This protein destabilizes one of two heme-ligating histidine residues by coupling histidine side chain ligation with the burial of three charged glutamate residues on the same helix. Replacement of these glutamate residues with alanine, which is uncharged, increases the affinity of the distal histidine ligand by a factor of thirteen. Paradoxically, it also decreases heme binding affinity by a factor of five in the reduced state and sixty in the oxidized state. Application of a three-state binding model, in which an initial pentacoordinate binding event is followed by a protein conformational change to hexacoordinate, provides insight into the mechanism of this seemingly counterintuitive result: the initial pentacoordinate encounter complex is significantly destabilized by the loss of the glutamate side chains, and the increased affinity for the distal histidine only partially compensates. These results point to the importance of considering each oxidation and conformational state in the design of functional artificial proteins.
Introduction
Designed proteins containing bound cofactors, both natural and synthetic, hold great promise as inexpensive biocompatible catalysts useful in medicine, energy production and green industrial catalysis (1-3). Heme proteins were the first bioinorganic-cofactor containing proteins created over fifteen years ago (4, 5), and since then they have proven instructive as to the underlying engineering requirements which drive natural heme protein evolution (6-8).
The family of hexacoordinate hemoglobins are characterized by the property that they are bis-histidine-ligated in the oxidized state and exist in a mixed bis- and mono-histidine ligation state when reduced (9). The transient pentacoordination of the heme cofactor allows for the ligation of molecular oxygen. We have recently reported the design, bacterial expression, and biochemical analysis of the completely artificial hexacoordinate oxygen transport protein HP7 (10). This was the culmination of a series of design projects involving helical bundles which bind heme (11-16). The homodimeric protein HP7 is composed of two helix-loop-helix peptides in which the loops connect via a disulfide in a topology we have termed the ‘candelabra’ motif (12) (see Figure 1A, sequences in 1E).
Figure 1.
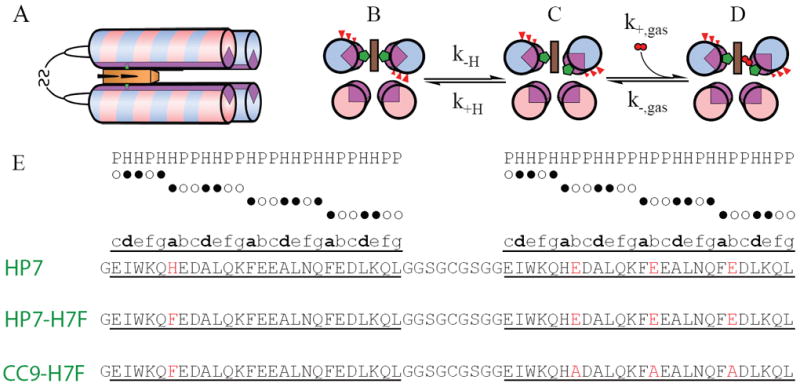
Structure, sequence and mechanism of HP7 and its mutants. (A) All three proteins are homodimers in a candelabra configuration in which the helix-loop-helix monomers are connected by a disulfide bond in the loop region. (B-D) Mechanism of gaseous ligand biding. State (B) is the entatic state. (E) Sequences of HP7, the distal heme binding site knockout mutant HP7-H7F, and the entatic state stabilizing mutant CC9-H7F. Helices are underlined and the b-position residues mutated in the transition from HP7 to CC9 are in red. The positions in the heptad repeat sequence of each helix is annotated above the sequence comparison. H = hydrophobic amino acid postion (●), P = polar amino acid position (○).
HP7 and its progenitors were designed in part using the principles of binary patterning, a simple alternating pattern of hydrophilic and hydrophobic helix-forming residues with the heptamer repeat sequence ●○○●●○○, where the first, third and fourth amino acids are non-polar (●) and the rest polar (○) (see Figure 1E) which imposes dimerization via hydrophobic sequestration (17-21). Each residue in the heptad repeat is designated with a letter, thus the hydrophobic residues are at the a, d and e positions of the heptad. There is a histidine residue at the sixth position of each helix, positioned to lie at the a-position of the heptad repeat (13). Two heme cofactors bind, one between each matching pair of histidines in the homodimer, and each binding event induces a large-scale helical rotation which brings three b position glutamate residues into the hydrophobic core of the protein. The burial of these residues creates an ‘entatic’ state (22): this stored energy is utilized to drive the detachment of the ligand histidine residue on that helix enabling the subsequent rotation of the helix to a relaxed state in which these b-position side chains are exposed to solvent, opening a coordination site for the ligation of the gaseous ligands oxygen and carbon monoxide (Figure 1B-D) (10). This design exercise turned out to be instructive as to the minimal engineering requirements for oxygen transport – a porphyrin cofactor with at least a transiently pentacoordinate ferrous iron buried within a hydrophobic cavity sufficiently large and stable as to restrict water penetration (11).
Thus in HP-7 as well as the natural hexacoordinate hemoglobins gaseous ligand affinity is gated by the presence of the pentacoordinate state. Many enzymes, especially those which utilize molecular oxygen as a substrate, contain a number of redox active cofactors in addition to the active site heme. These other cofactors, for example the heme cofactors in the cytochrome c oxidase, typically serve to ferry electrons into the O2-utilizing active site (23). These enzymes restrict oxygen binding at the other heme cofactors by maintaining them in the hexacoordinate state. Therefore it will prove important, in the future design of more complex artificial catalysts, to be able to effectively create high affinity cofactor binding sites with controllable distal ligand affinity. It is therefore important to understand the factors which determine distal side chain affinity and its relationship to overall cofactor binding affinity.
Materials and Methods
Chemicals
Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA) and used without further purification. 15N-labelled ammonium chloride was purchased from Cambridge Isotope Labs (Cambridge, MA). Pyocyanine was purchased from Cayman Chemical (Ann Arbor, MI). Hemin was purchased from Fluka (Buchs, Switzerland). All other solvents and reagents were from either Fisher Scientific or Sigma.
Cloning, protein expression and purification
The CC9 gene was synthesized (Biomatik Corp., Cambridge, ON) with an N-terminal TEV cut site and inserted between the BamHI and XhoI restriction sites of pET32a (+) (Novagen) as previously described (12). Mutagenesis to produce CC9-H7F and HP7-H7F was accomplished using the Quikchange method (Stratagene Inc., La Jolla, CA). Mutagenized plasmid DNA was transformed into chemically competent DH5α E. coli, the resulting clones verified by sequencing, and a verified plasmid was transformed into BL21(DE3) cells for expression. Labelled and unlabeled proteins were expressed and purified as we have published previously (12). Apoprotein concentrations were determined optically using ε280 = 11.5 mM-1cm-1 on the basis of their tryptophan content (24).
Holoprotein complexes, containing 1.0 equivalent per homodimer, were prepared as before (12) by five consecutive additions of 0.2 equivalents of hemin in DMSO with at least ten minutes between additions. Proteins were then purified from any unbound heme by passage through a PD10 desalting columns pre-equilibrated with 250mM Boric Acid, 100mM KCl pH 9.0. Holoprotein solution concentrations were determined using experimentally derived Soret ε414 = 129 mM-1cm-1 for HP7-H7F and 118 mM-1cm-1 for CC9-H7F. If necessary, both holo- and apoproteins were concentrated using Centricon YM-10 spin concentrators (Millipore, Inc., Billerica, MA).
General biochemistry
Optical spectra were collected with a Hewlett-Packard (New York, NY) 8452A Diode array spectrophotometer equipped with a Quantum Northwest (Liberty Lake, WA) Peltier temperature controller. Experiments requiring anaerobic conditions were conducted in an anaerobic cuvette (25) equipped with a platinum working and Red Rod (Radiometer Analytical, Lyon, France) reference electrode. All experiments were performed at 20° C in 250mM Boric Acid, 100mM KCl pH 9.0 unless otherwise noted. Each binding, reduction potential and stopped flow experiment was performed at least three times and reported errors are standard deviations from the mean.
Circular dichroism spectropolarimetry
CD spectra were recorded on a JASCO J-810 sepectropolarimeter in a quartz cell of 0.1cm light-path, using a bandwidth of 1nm and a scan speed of 50nm/min. HP7-H7F and CC9-H7F were prepared at concentrations of 24.15μM and 14.75μM respectively in 50mM sodium phosphate, pH 7.5. Spectra were collected between 190nm and 280nm, with background correction. The spectra were observed as ellipticity(θobs, mdeg), and the mean residue ellipticity([θ],deg•cm2•dmol-1) were calculated as
| (1) |
Where MRW is the mean residue molecular weight, c is the sample concentration in mg/ml, and l is the light-path length of the cell in cm. Protein secondary structure percentages were calculated with the program K2d (26) using data points ranging from 200nm to 240nm.
Stopped-flow analysis distal histidine dissociation
Binding kinetics of CO with the ferrous protein-heme complexes were followed spectroscopically in rapid stopped-flow mixing experiments over gas concentrations from 2% to 50% saturation at 15° C using an Olis RSM 1000 spectrometer. Protein concentrations were 20-25 μM and ferrous complexes were prepared by carefully titrating anaerobic solutions of the holoproteins with a slight excess of dithionite as observed by visible spectroscopy and transferred to the stopped-flow loading syringe by canula. Binding kinetic data were fit with Eqn. 2, which assumes that CO binding rate, k+CO, is much greater than the sum of the distal histidine association and dissociation rates (9):
| (2) |
Where kobs is the fitted single exponential binding rate, k+H and k-H are the distal histidine-ferrous heme iron association and dissociation rates and k+CO is the CO association rate constant. This experiment determines the distal histidine ligand dissociation rate - at high [CO], kobs = k-H.
Flash photolysis analysis of distal histidine association
The rate constants for both CO and histidine binding to the pentacoordinate state were determined using laser flash photolysis: a 1-ns pulse-width frequency doubled YAG laser at 532 nm excites the preformed carbonmonoxyferrous complex, causing the detachment of the ligand CO. This transiently forms an unliganded pentacoordinate heme protein and the rates of these binding processes were determined by analyzing the multi-exponential rebinding traces taken as a function of CO concentration using the method of Hargrove (27):
| (3) |
| (4) |
Where γ1 and γ2 are the fitted first and second CO-dependent exponential rates and the kinetic constants are defined as in Equation 1. Protein concentrations were 20-25 μM and carbonmonoxyferrous complexes were prepared by titrating solutions of the holoproteins with an excess of dithionite as observed by visible spectroscopy under an atmosphere containing 10-100% CO mixed with argon.
Heme affinity measurements
Hemin stock solutions of approximately 0.5-1.0 mg/ml were prepared in DMSO and used within six hours. Stock solution concentrations were determined using the pyridine hemochrome assay (28). In oxidized binding experiments 0.5-2 μl aliquots of hemin solution, consisting of approximately 0.1 molar equivalents, were added via gastight syringe to a stirring 4 ml solution of 2-3 μM protein, with a ten minute equilibration delay between additions. Heme binding was monitored by loss of the absorption at 385 nm due to free hemin, and the concomitant appearance of a sharp Soret band at 412 nm, corresponding to heme bound to the protein via bis-histidine axial coordination. The number of heme binding sites was quantified for each protein from plots of the Soret maximum at 412 nm vs the number of equivalents added.
For reduced binding experiments, 9.0 ml of ~3 μM protein was made anaerobic by extended flushing with nitrogen, and the solution potential reduced to < -450 mV vs NHE by the addition of a small volume (< 20 μl) of sodium dithionite dissolved in degassed 100 mM KOH. The solution potential was monitored throughout the binding titration and kept below -450 mV by periodic additions of μl volumes of sodium dithionite. Hemin was added in approximately 0.1 molar equivalent aliquots with at least a ten minute equilibration time before spectra were collected. Kd values were obtained from plots of the Soret band absorbance measured at 436 nm vs. the concentration of hemin added and fit with the tight binding equation:
| (5) |
Where εunb is the molar absorption coefficient of unbound hemin at that wavelength, εbnd is the additional absorbance of bound hemin at that wavelength, [hem] is the hemin concentration, [prot] is the protein concentration and Kd is the dissociation constant for the reduced hemin.
Reduction potential determination
Redox titrations were performed in combination with optical analysis as described previously (13). Concentrated solutions of hemoprotein prepared in advance were diluted to 20-30 μM into a solution containing >100 μM of the corresponding apoprotein in order to eliminate the possibility of heme dissociation upon reduction. Reported reduction potentials are referenced to a standard hydrogen electrode. All redox titrations were performed anaerobically using μL additions of freshly prepared sodium dithionite to adjust the solution potential to more negative values and potassium ferricyanide to more positive values. Redox titrations were analyzed by monitoring the absorbance bands at 559 and 425 nm as the heme protein was reduced or oxidized. The data were analyzed with the Nernst equation using an n-value of 1.0:
| (6) |
where %R is the fraction of reduced heme, E is the solution potential, Em is the reduction midpoint potential and n is the number of electrons.
Nuclear magnetic resonance
All NMR experiments were performed at 20°C on Varian Inova spectrometer operating at a 600MHz and equipped with a triple resonance cryogenic probe capable of applying pulse field gradients in the z-direction. Data were processed using the program NMRPipe (29) and analyzed using Sparky (30). For structural specificity assays, sensitivity enhanced 1H-15N heteronuclear single quantum coherence (HSQC) spectra (31) were collected on 50-100 μM holo- and apoprotein samples with sweep widths of 10,000 Hz for 1H and 2000 Hz for 15N utilizing GARP decoupling of 15N during 1H acquisition. Chemical shifts are referenced to water at 4.77 ppm for 1H.
In order to determine the pKa of histidine side chains, imidazole 1H-15N multiple-bond correlation signals were collected using a non-sensitivity enhanced HSQC pulse sequence in which the INEPT (insensitive nuclei enhanced by polarization transfer) periods were set to 1/1JNH, where 1JNH is the one-bond 1H-15N coupling constant, in order to attenuate backbone amide signals (32, 33). Spectral widths were 8000 Hz for 1H and 12156 Hz for 15N. Lyophilized 15N-labelled apoprotein samples were dissolved in 25mM KaD2PO4 D2O buffer pD 5.0 (pH meter reading + 0.4 pH units), a spectrum taken, and the pD adjusted upwards by the addition of small amounts of KOD dissolved in D2O. The observed 15N chemical shifts were fit with the Henderson-Hasselbach equation:
| (7) |
where δ0 is the neutral chemical shift of 15N, δprot is the change in chemical shift due to protonation and pKa is the fitted acid dissociation constant.
Results and Discussion
Protein design
To simplify our analysis, we removed one binding site by mutating the histidine on the first helix to a phenylalanine, creating the protein HP7-H7F (see Table 1). This protein retains the heme binding site nearest the connecting loops (Figure 1A). This is the opposite binding site that is occupied when the HP7 homodimer is bound to a single heme. To examine the thermodynamic effects of the buried charges, all three b-position glutamates on the second helix were mutated to alanine, which has both neutral hydrophobicity and high helical propensity (34).
Table 1.
Characterization of the two HP7 variants
| λmax_Oxidized (mM-1cm-1) | λmax_Reduced (mM-1cm-1) | λ([θ]222 (deg cm2 dmol-1) | % α-helix | |
|---|---|---|---|---|
|
| ||||
| HP7H7F | 414 (129) | 428 (140) | -8307 | 42 |
| 530 (12.8) | 532 (12.1) | |||
| 558 (10.1) | 560 (20.9) | |||
|
| ||||
| CC9H7F | 414 (118) | 426 (139) | -15925 | 59 |
| 530 (11.1) | 530 (12.6) | |||
| 556 (9.1) | 560 (22.0) | |||
Both holoproteins display visible absorbance spectra indicative of bis-histidine coordination in both oxidation states (Figure 2A & C). It is important to confirm the helical bundle structure of each protein, as some designed helical bundle proteins have been found to form domain-swapped higher oligomeric states (35). Both apo- and holoprotein forms of both HP7-H7F and CC9-H7F were single homodimers as analyzed by native gel electrophoresis in both the holo- and apostates (data not shown). Circular dichroism spectra on the apo forms of both proteins demonstrate that both proteins are helical, with CC9-H7F having a larger helical character. This is unsurprising in that each homodimer contains six mutations from glutamic acid to alanine, which has a significantly higher helical propensity (36). Both had 15N-HSQC spectra indicative of molten globular structure (not shown). The addition of a single heme cofactor changes both spectra to a level of chemical shift dispersion indicative of a partial phase transition in which the two helices which ligate the heme become native-like and the two unliganded helices remain molten globular, consistent with what we have previously observed for HP7 (12).
Figure 2.
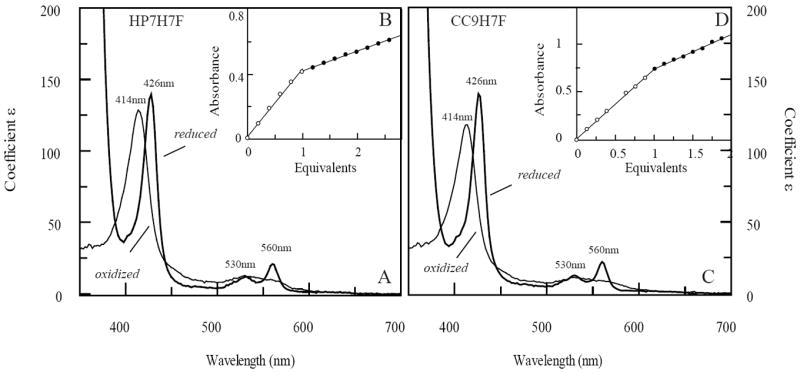
Absorption spectra and oxidized binding titrations of HP7-H7F and CC9-H7F with oxidized heme. (A) Oxidized and reduced spectra of heme-bound HP7-H7F. Extinction coefficients are calculated using the intercepts from the endpoint titrations in (B). (C) Oxidized and reduced spectra of heme-bound CC9-H7F, calculated using the data from (D).
Oxidized heme binding
In the oxidized state, both proteins bind heme too tightly to reliably extract a dissociation constant even at protein concentrations as low as 200 nM. This has enabled us to perform endpoint titrations confirming that both proteins bind a single heme cofactor (Figure 2) as well as calculate the extinction coefficient of the oxidized complex (Table 1).
Distal histidine affinity – histidine dissociation constant
In order to confirm that the triple mutation indeed stabilizes the affinity of the distal histidine, we must separately determine its association and dissociation rates. Figure 3A depicts the observed CO binding constants and their concentration dependence. For both proteins, CO binding to the ferrous state is monoexponential over more than five half-lives. Binding rates collected as a function of CO concentration demonstrate that CC9-H7F binds CO 22-fold more slowly than HP7-H7F (Table 2). As the protein is primarily in the hexacoordinate bis-histidine ligation state in solution (Figure 2), the rate limiting step for CO binding at high ligand concentrations is thus the detachment of the distal histidine from the heme iron (see Eqn. 2). Thus, histidine detachment is also 22-fold slower in CC9-H7F.
Figure 3.
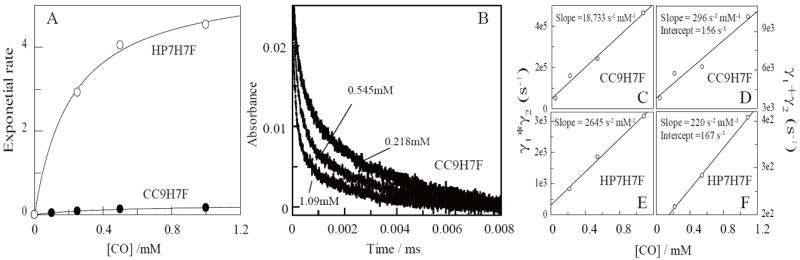
Kinetic analysis of CO and histidine binding to HP7-H7F and CC9-H7F. (A) Stopped-flow analysis of the rates of CO binding to reduced heme proteins as a function of ligand concentration. Lines are fits with eqn. 1. (B) Laser flash kinetic analysis of histidine and CO rebinding to CC9-H7F folfollowed at 419 nm. These data were fit with three exponentials, the first of which represents a [CO]-independent relaxation process. (C-F) Replots of the sums and products of the second two exponentials.
Table 2.
Thermodynamic parameters for two artificial heme proteins
| Heme Protein | Kd, red (μM) | Em (mV vs. NHE) | Kd, ox (nM) | pKa | k-his (s-1) | k+his (s-1) | KA,his | Kd, pent (μM) |
|---|---|---|---|---|---|---|---|---|
| HP7-H7F | 0.6±0.2 | -260±6 | 0.3±0.1 | 7.3±0.2 | 160±20 | 5.6±0.3 | 29±5 | 18±9 |
| CC9-H7F | 2.9±0.4 | -195±2 | 19±3 | 6.39±0.01 | 94±11 | 0.25±0.01 | 380±60 | 1100±300 |
Distal histidine affinity – CO-flash photolysis determination of k+His
Figure 3B depicts the rebinding kinetics of CO after laser induced dissociation in CC9-H7F. In CC9-H7F, HP7-H7F, and our original analysis of HP7 (10), a fast exponential process which is independent of the concentration of CO is observed. Similar behavior has been observed in mouse neuroglobin, and was ascribed to a relaxation process following a probable rearrangement caused by the change in heme iron planarity induced by detachment of the CO ligand (37, 38). In each case two slower exponential processes, each of which varies in rate and magnitude with CO concentration, were also observed.
Replots of the sums and products of these two exponential processes are depicted in Figure 3C-F. The distal histidine binding rates are similar in both proteins, with the triple mutation slowing this process by a factor of 1.7 (Table 2). Combining these rates with the histidine off-rates determined in the stopped flow experiments allows the calculation of the distal histidine affinity constants, KA, for both proteins – 30 for HP7-H7F increasing to 380 for CC9-H7F (as this is an intramolecular interaction, the association constant is dimensionless). Thus the removal of the residues which were implanted to create the entatic state in HP7 stabilizes the hexacoordinate state in the ferrous protein by 1.5 kcal/mol.
Reduced heme binding
Gibney and coworkers have determined the solution reduction potential of an isolated heme cofactor, -63 mV vs SHE, using two different designed heme proteins and a thermodynamic cycle (39). This value enables an alternate method for the determination of heme binding constants: experimentally determine the binding affinity in the reduced state, taking advantage of the weaker histidine-ferrous heme iron interaction (40), then the oxidized heme binding constant can be calculated in combination with the bound and unbound heme reduction potentials using a similar thermodynamic cycle (see Figure 4A). Binding titrations carried out under reducing conditions give reproducible binding curves (see Figure 4B) and the fitted ferrous heme dissociation constants were 0.6 ± 0.2 μM for HP7-H7F and 2.6 ± 0.6 μM for CC9-H7F.
Figure 4.
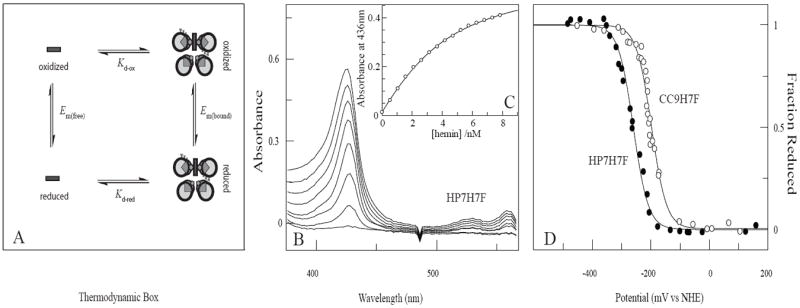
Thermodynamic cycle analysis of oxidized heme affinity. (A) The thermodynamic box used to determine Kd,ox. (B) Spectrometric titration of reduced heme binding to 3.0 μM HP7-H7F in 250mM Boric Acid, 100mM KCl pH 9.0. Some spectra have been omitted for clarity. (C) Equilibrium binding isotherm derived from the titration in (B). The line drawn is a fit with Eqn. 4 using a Kd,red of 600 nM. (D) Equilibrium potentiometric determination of the electron affinities HP7-H7F and CC9-H7F. Lines drawn are fits with Eqn. 1.
Reduction potentials
The potentiometric analysis of the two proteins is depicted in Figure 4C. HP7-H7F, at -260 ± 6 mV vs NHE, has a reduction potential 65 mV lower than CC9-H7F (see Table 2), and 40 mV higher than the progenitor protein HP7 complexed with two hemes (-300 mV vs NHE (10)). HP7 rotates two sets of three glutamates into the core upon binding these two hemes. HP7-H7F rotates only one set of three, and CC9-H7F does not bury any charged residues into the core. Thus, each set of three buried glutamates imparts a change in reduction potential of approximately -50 mV, or 1.2 kcal/mol.
The reduction potentials, coupled with the reduced state affinities, allows the calculation of oxidized affinities of 300 ± 100 pM and 19 ± 3 nM for HP7-H7F and CC9-H7F respectively. Given that the distal histidine binds more tightly to the heme iron in CC9-H7F than in HP7-H7F, the weaker energy of the former for heme binding is counterintuitive. This discrepancy might be explained by a large difference in bound cofactor reduction potentials, caused by some alteration in the local structure and/or electrostatics that moves the heme potential in the negative direction. The potentials, however, move in the direction predicted by simple electrostatics (positive), and thus the ferric heme binding energy difference between CC9-H7F and HP7-H7F is even larger.
NMR titration of the histidine pKa
Histidine residues can protonate, and the resultant histidine cation cannot bind heme. This proton competition effect is another possible explanation for the loss of binding affinity in the triple mutant protein (39). To determine whether this is the case, NMR was used to determine the pKa of the molten globular apoprotein histidine ligands. Both protein’s histidine pKa’s were well below the solution pH (9.0) used in the binding titrations (Figure 5), with that of CC9-H7F approaching the solution pKa for free histidine, therefore the observed differences in heme Kd are not a consequence of proton competition.
Figure 5.
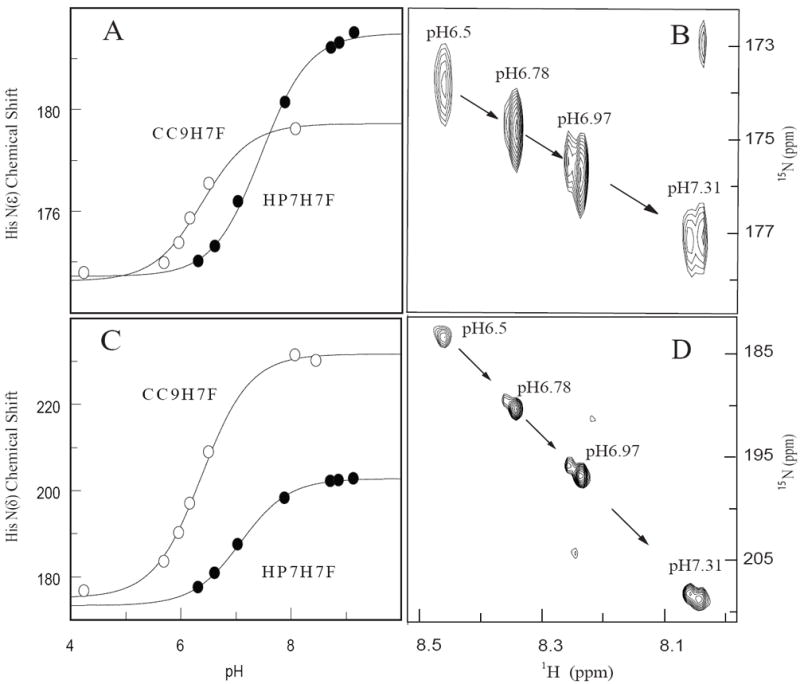
NMR determination of the pKas of the histidine ligands in apo-HP7-H7F and apo-CC9-H7F. (A) Overlay of multiple-bond correlated 1H-15N spectra as a function of pH, showing the titration of the imidazole Hε1-Nε2, signals of histidine side chains. (B) Fits of the 15Nε2 chemical shifts with eqn. 4. (C) Overlay of multiple-bond correlated Hε1-Nδ1 signals of histidine side chains. (D) Fits of the 15Nδ1 chemical shifts with eqn. 4.
Based on the crystal (14) and NMR (41) structures of the progenitor protein BB in its apo form, the apoprotein forms of HP7-H7F and CC9-H7F can be expected to take a conformation in which the histidine side chains are buried in the protein core and the b-position residues exposed on the surface. Given this solvent exposure, it is unsurprising that the removal of six negatively charged side chains has such a small effect on the histidine pKa. In the heme-bound state, however, b-position residues rotate into the low-dielectric hydrophobic core (Figure 1B) and electrostatic coupling has a larger effect, as demonstrated by the change in reduction potentials between the two proteins.
The origin of the difference in heme affinities in the two proteins
Heme binding in hexacoordinate hemoglobins is a three state process consisting of the apoprotein, a pentacoordinate bound state and a hexacoordinate bound state (Figure 6A-C), with the latter two states considered bound states. Binding affinity in such a case was first examined by Morrison and coworkers in their analysis of slow, tight binding enzyme inhibition (42). Extending this model to the hexacoordinate hemoglobins gives the relation:
| (8) |
where the ratio k1/k2 is Kd,pent, the dissociation constant which reflects the two-state energy difference between the apo state and the pentacoordinate state, and the term in brackets represents the additional binding energy conferred by distal histidine ligation. Combining the experimental ferrous binding constant with the distal histidine ligation rate constants allows the calculation of Kd,pent (Table 2).
Figure 6.
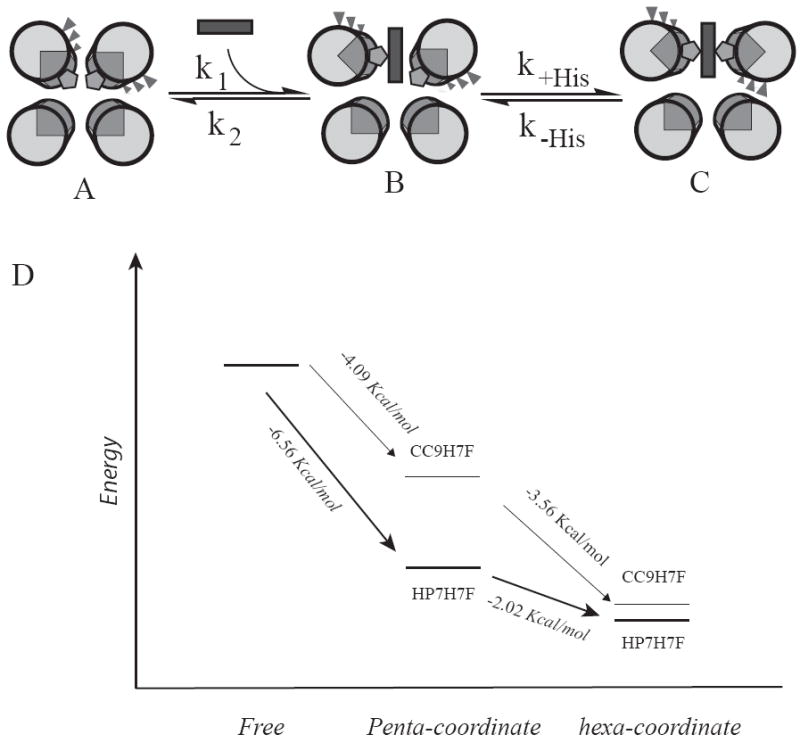
Comparative energy levels of the binding states of the two proteins. (A-C) Detail of the binding mechanism: heme binding initially occurs via a pentacoordinate encounter complex (B) which is followed by a protein conformational change driven by distal histidine attachment, resulting in the hexacoordinate, bis-histidine bound state (C). (D) Energy level diagram depicting the relative energies of each of these states in the two proteins. The absolute energies of the free states are unknown and are set equal to each other for comparative purposes.
These values explain the increase in dissociation constant in CC9-H7F – Kd,pent is increased by more than a factor of 60 by the triple mutation. An energy diagram which relates the relative energies of each of these states is depicted in Figure 5D. This graphically demonstrates that the increased affinity of the distal histidine incompletely compensates for the large loss in interaction energy of the pentacoordinate state. The structural origin of this loss in affinity is unclear. One possibility is that in the pentacoordinate state the b-position glutamate side chains on the helix with the distal (detached) histidine form Van der Waals interactions with the bound heme, and the removal of these side chains therefore weakens the shape complementarity of the binding site. Another possibility is similar behavior in helix containing the proximal (attached) histidine ligand. These differing possibilities point to the limitations inherent in the homodimeric candelabra scaffold. It may be that a single chain protein (43), which allows the independent optimization of each helix independent of the other will allow both heme binding and distal histidine coordination to be optimized independently.
Conclusion
The detailed analysis presented here, of the effects of the mutational manipulation of the entatic state energy in an artificial oxygen transport protein, point to the difficulties inherent in the design and optimization of a functional protein with several conformational and oxidation states. Recent progress in the protein design field has made the creation of a natively structured, high affinity ferric heme protein relatively straightforward, but functional proteins will necessarily require high cofactor affinity in, at the least, both the ferric and ferrous oxidation states of the heme cofactor. Early successes in the design of ferric heme binding proteins took advantage of the strong ferric iron-histidine interaction, estimated to be as high as six kcal/mol (44). These interactions are much weaker, however, in the ferrous state (40), and high affinity binding of reduced heme and porphyrin cofactors in designed proteins will require a much more sophisticated combination of stabilizing interactions such as binding site complementarity, electrostatic pairing, and heme ligand rotamer optimization (16, 45).
Acknowledgments
The authors thank Brian Gibney of the Department of Chemistry, Brooklyn College and Mark Hargrove, of the Department of Biophysics, Iowa State University for many helpful discussions. We thank Hsin Wang, of the Department of Chemistry, the City College of New York, for assistance with NMR measurements.
Funding: RLK gratefully acknowledges support by the following grants: MCB-0920448 from the National Science Foundation, infrastructure support from P41 GM-66354 to the New York Structural Biology Center and the NIH National Center for Research Resources to CCNY (NIH 5G12 RR03060). PLD gratefully acknowledges support from NIH grant GM-41048. IA and CN gratefully acknowledge support from the NIH’s Minority Access to Research Careers program (T34 GM007639). ACM gratefully acknowledges support from the Center for Exploitation of Nanostructures in Sensor and Energy Systems (CENSES) under NSF Cooperative Agreement Award Number 0833180. JLRA was supported by a Royal Society Fellowship.
Abbreviations used
- CO
carbon monoxide
- HSQC
heteronuclear single quantum coherence
- NHE
normal hydrogen electrode
Biliography
- 1.Sheng H, Yang W, Fukuda S, Tse HM, Paschen W, Johnson K, Batinic-Haberle I, Crapo JD, Pearlstein RD, Piganelli J, Warner DS. Long-term neuroprotection from a potent redox-modulating metalloporphyrin in the rat. Free Radic Biol Med. 2009;47:917–923. doi: 10.1016/j.freeradbiomed.2009.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koder RL, Dutton PL. Intelligent design: the de novo engineering of proteins with specified functions. Dalton Transactions. 2006;25:3045–3051. doi: 10.1039/b514972j. [DOI] [PubMed] [Google Scholar]
- 3.Lu Y. Design and engineering of metalloproteins containing unnatural amino acids or non-native metal-containing cofactors. Current Opinion in Chemical Biology. 2005;9:118–126. doi: 10.1016/j.cbpa.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 4.Robertson DE, Farid RS, Moser CC, Urbauer JL, Mulholland SE, Pidikiti R, Lear JD, Wand AJ, Degrado WF, Dutton PL. Design and Synthesis of Multi-Heme Proteins. Nature. 1994;368:425–431. doi: 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- 5.Choma CT, Lear JD, Nelson MJ, Dutton PL, Robertson DE, Degrado WF. Design of a Heme-Binding 4-Helix Bundle. Journal of the American Chemical Society. 1994;116:856–865. [Google Scholar]
- 6.Cowley AB, Kennedy ML, Silchenko S, Lukat-Rodgers GS, Rodgers KR, Benson DR. Insight into heme protein redox potential control and functional aspects of six-coordinate ligand-sensing heme proteins from studies of synthetic heme peptides. Inorganic Chemistry. 2006;45:9985–10001. doi: 10.1021/ic052205k. [DOI] [PubMed] [Google Scholar]
- 7.Reedy CJ, Gibney BR. Heme protein assemblies. Chemical Reviews. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 8.Cowley AB, Benson DR. Weak-field anions displace the histidine ligand in a synthetic heme peptide but not in N-acetylmicroperoxidase-8: Possible role of heme geometry differences. Inorganic Chemistry. 2007;46:48–59. doi: 10.1021/ic060682c. [DOI] [PubMed] [Google Scholar]
- 9.Trent JT, Hvitved AN, Hargrove MS. A model for ligand binding to hexacoordinate hemoglobins. Biochemistry. 2001;40:6155–6163. doi: 10.1021/bi0100790. [DOI] [PubMed] [Google Scholar]
- 10.Koder RL, Anderson JLR, Solomon LA, Reddy KS, Moser CC, Dutton PL. Design and engineering of an O2 transport protein. Nature. 2009;458:305–309. doi: 10.1038/nature07841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson JLR, Koder RL, Moser CC, Dutton PL. Controlling complexity and water penetration in functional de novo protein design. Biochemical Society Transactions. 2008;36:1106–1111. doi: 10.1042/BST0361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koder RL, Valentine KG, Cerda JF, Noy D, Smith KM, Wand AJ, Dutton PL. Native-like structure in designed four helix bundles driven by buried polar interactions. Journal of the American Chemical Society. 2006;128:14450–14451. doi: 10.1021/ja064883r. [DOI] [PubMed] [Google Scholar]
- 13.Huang SS, Koder RL, Lewis M, Wand AJ, Dutton PL. The HP-1 maquette: From an apoprotein structure to a structured hemoprotein designed to promote redox-coupled proton exchange. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:5536–5541. doi: 10.1073/pnas.0306676101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang SS, Gibney BR, Stayrook SE, Dutton PL, Lewis M. X-ray Structure of a Maquette Scaffold. J Mol Biol. 2003;326:1219–1225. doi: 10.1016/s0022-2836(02)01441-9. [DOI] [PubMed] [Google Scholar]
- 15.Discher BM, Koder RL, Moser CC, Dutton PL. Hydrophilic to Amphiphilic Design in Redox Protein Maquettes. Current Opinion in Chemical Biology. 2003;7:741–748. doi: 10.1016/j.cbpa.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 16.Negron C, Fufezan C, Koder RL. Helical Templates for Porphyrin Binding in Designed Proteins. Proteins-Structure Function And Bioinformatics. 2009;74:400–416. doi: 10.1002/prot.22143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hecht MH, Das A, Go A, Bradley LH, Wei YN. De novo proteins from designed combinatorial libraries. Protein Science. 2004;13:1711–1723. doi: 10.1110/ps.04690804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamtekar S, Schiffer JM, Xiong HY, Babik JM, Hecht MH. Protein Design by Binary Patterning of Polar and Nonpolar Amino-Acids. Science. 1993;262:1680–1685. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 19.Moffet DA, Hecht MH. De novo proteins from combinatorial libraries. Chemical Reviews. 2001;101:3191–3203. doi: 10.1021/cr000051e. [DOI] [PubMed] [Google Scholar]
- 20.Wei YN, Kim S, Fela D, Baum J, Hecht MH. Solution structure of a de novo protein from a designed combinatorial library. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13270–13273. doi: 10.1073/pnas.1835644100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei YN, Liu T, Sazinsky SL, Moffet DA, Pelczer I, Hecht MH. Stably folded de novo proteins from a designed combinatorial library. Protein Science. 2003;12:92–102. doi: 10.1110/ps.0228003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vallee BL, Williams RJP. Metalloenzymes - entatic nature of their active sites. Proceedings Of The National Academy Of Sciences Of The United States Of America. 1968;59:498. doi: 10.1073/pnas.59.2.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brunori M, Giuffre A, Sarti P. Cytochrome c oxidase, ligands and electrons. Journal Of Inorganic Biochemistry. 2005;99:324–336. doi: 10.1016/j.jinorgbio.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 24.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to Measure and Predict the Molar Absorption-Coefficient of a Protein. Protein Science. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dutton PL. Redox potentiometry: determination of midpoint potentials of oxidation-reduction components of biological electron-transfer systems. Methods in Enzymology. 1978;54:411–435. doi: 10.1016/s0076-6879(78)54026-3. [DOI] [PubMed] [Google Scholar]
- 26.Andrade MA, Chacon P, Merelo JJ, Moran F. Evaluation of secondary structure of proteins from UV circular-dichroism spectra using an unsupervised learning neural network. Protein Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- 27.Hargrove MS. A flash photolysis method to characterize hexacoordinate hemoglobin kinetics. Biophysical Journal. 2000;79:2733–2738. doi: 10.1016/S0006-3495(00)76512-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berry EA, Trumpower BL. Simultaneous determination of hemes-A, hemes-B, and hemes-C from pyridine hemochrome spectra. Analytical Biochemistry. 1987;161:1–15. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- 29.Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A. NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 30.Goddard TD, Kneller DG. Sparky. The University of California; San Francisco: 2007. [Google Scholar]
- 31.Kay LE, Keifer P, Saarinen T. Pure Absorption Gradient Enhanced Heteronuclear Single Quantum Correlation Spectroscopy with Improved Sensitivity. J Am Chem Soc. 1992;114:10663–10665. [Google Scholar]
- 32.Pelton JG, Torchia DA, Meadow ND, Roseman S. Tautomeric states of the active-site histidines of phosphorylated and unphosphorylated III(GLC), a signal-transducing protein from escherichia coli, using 2-dimensional heteronuclear NMR techniques. Protein Science. 1993;2:543–558. doi: 10.1002/pro.5560020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nanda V, Koder RL. Designing artificial enzymes by intuition and computation. Nature Chemistry. 2010;2:15–24. doi: 10.1038/nchem.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lyu PC, Liff MI, Marky LA, Kallenbach NR. Side-Chain Contributions To The Stability Of Alpha-Helical Structure In Peptides. Science. 1990;250:669–673. doi: 10.1126/science.2237416. [DOI] [PubMed] [Google Scholar]
- 35.Ogihara NL, Ghirlanda G, Bryson JW, Gingery M, DeGrado WF, Eisenberg D. Design of three-dimensional domain-swapped dimers and fibrous oligomers. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2001;98:1404–1409. doi: 10.1073/pnas.98.4.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dunbrack RL, Karplus M. Backbone-Dependent Rotamer Library for Proteins - Application to Side-Chain Prediction. Journal of Molecular Biology. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- 37.Ascenzi P, Bocedi A, de Sanctis D, Pesce A, Bolognesi M, Marden MC, Dewilde S, Moens L, Hankeln T, Burmester T. Neuroglobin and cytoglobin - Two new entries in the hemoglobin superfamily. Biochem Mol Biol Educ. 2004;32:305–313. doi: 10.1002/bmb.2004.494032050386. [DOI] [PubMed] [Google Scholar]
- 38.Du WH, Syvitski R, Dewilde S, Moens L, La Mar GN. Solution H-1 NMR characterization of equilibrium heme orientational disorder with functional consequences in mouse neuroglobin. Journal of the American Chemical Society. 2003;125:8080–8081. doi: 10.1021/ja034584r. [DOI] [PubMed] [Google Scholar]
- 39.Reddi AR, Reedy CJ, Mui S, Gibney BR. Thermodynamic investigation into the mechanisms of proton-coupled electron transfer events in heme protein maquettes. Biochemistry. 2007;46:291–305. doi: 10.1021/bi061607g. [DOI] [PubMed] [Google Scholar]
- 40.Reedy CJ, Kennedy ML, Gibney BR. Thermodynamic characterization of ferric and ferrous haem binding to a designed four-alpha-helix protein. Chemical Communications. 2003:570–571. doi: 10.1039/b211488g. [DOI] [PubMed] [Google Scholar]
- 41.Skalicky JJ, Gibney BR, Rabanal F, Urbauer RJB, Dutton PL, Wand AJ. Solution structure of a designed four-alpha-helix bundle maquette scaffold. Journal of the American Chemical Society. 1999;121:4941–4951. [Google Scholar]
- 42.Sculley MJ, Morrison JF. The determination of kinetic constants governing the slow,tight-binding inhibition of enzyme-catalyzed reactions. Biochimica et Biophysica Acta. 1986;874:44–53. [Google Scholar]
- 43.Bender GM, Lehmann A, Zou H, Cheng H, Fry HC, Engel D, Therien MJ, Blasie JK, Roder H, Saven JG, DeGrado WF. De novo design of a single-chain diphenylporphyrin metalloprotein. Journal Of The American Chemical Society. 2007;129:10732–10740. doi: 10.1021/ja071199j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shifman JM, Moser CC, Kalsbeck WA, Bocian DF, Dutton PL. Functionalized de novo designed proteins: Mechanism of proton coupling to oxidation/reduction in heme protein maquettes. Biochemistry. 1998;37:16815–16827. doi: 10.1021/bi9816857. [DOI] [PubMed] [Google Scholar]
- 45.Braun P, Goldberg E, Negron C, von Jan M, Xu F, Nanda V, Koder RL, Noy D. Design principles for chlorophyll-binding sites in helical proteins. Proteins-Structure Function And Bioinformatics. 2011;79:463–476. doi: 10.1002/prot.22895. [DOI] [PMC free article] [PubMed] [Google Scholar]