

Abstract
Naturally-occurring chemopreventive agent phenethyl isothiocyanate (PEITC), derived primarily from watercress, has been shown to inhibit cell growth and induce apoptosis in cancer cells. In this study, we examined the potential of PEITC in enhancing cisplatin-induced apoptosis in cervical cancer cells. HeLa cells were exposed to PEITC, cisplatin or both. Pretreatment of cells with PEITC strongly enhanced cisplatin-induced cytotoxicity. PEITC activated the mitogen-activated protein kinases, including JNK, ERK, and p38. The synergistic induction of apoptosis was significantly attenuated by MEK1/2 inhibitor U0126, but not by JNK or p38 inhibitor, suggesting that ERK activation is responsible for the synergistic effect. We found that NF-κB signaling pathway is not involved in the synergistic effect. Sulforaphane and benzyl isothiocyanate, two other members of the isothiocyanate family, also sensitize HeLa cells to apoptosis induced by cisplatin. Furthermore, we found that the synergistic effect was not seen in normal cells. Finally, we demonstrated that Noxa induction was associated with apoptosis induced by PEITC plus cisplatin. Taken together, this study shows that PEITC can sensitize cancer cells to apoptosis induced by cisplatin and this effect is mediated through ERK activation, suggesting the potential of PEITC to be used as an adjuvant with cisplatin in combination therapeutic treatments.
Keywords: Phenethyl isothiocyanate, cisplatin, apoptosis, ERK activation, sensitization
1 Introduction
Cisplatin (CDDP) is one of the most important anticancer drugs used in the treatment of human tumors, including head and neck, colorectal, ovarian, cervical, testicular, and small cell lung cancers [1]. The acquisition of resistance by cancer cells to cisplatin is one of the major hurdles in cisplatin-based chemotherapy. Mechanisms of resistance identified to date include reduced drug uptake, inactivation by glutathione and other antioxidants, and increased repair of cisplatin-induced DNA damage or enhanced DNA damage tolerance [2]. Various cisplatin-based combination therapies have been intensely evaluated for cancer treatment in recent years. A major remaining question is what type of drug would be the best candidate for the combination therapy with cisplatin. Many new chemotherapeutic strategies combine multiple agents, which result in improved tumor response and enhanced efficacy. However, the employment of multiple agents often leads to increased toxicity, causing a poor treatment outcome. Thus, combination chemotherapy must be optimized to increase the response of tumors to chemotherapy and, at the same time, diminish its toxicity. One strategy to overcome drug resistance and enhance chemotherapy efficacy is to use a combination of standard anticancer drugs with chemopreventive agents that are by themselves nontoxic at lower doses [3].
The chemopreventive agent Phenethyl isothiocyanate (PEITC) is present in high concentrations as its precursor gluconasturtiin in cruciferous vegetables, such as watercress. Upon chewing or chopping, PEITC is released as a product of hydrolysis mediated by myrosinase [4]. It has demonstrated strong chemopreventive activities in various carcinogen-induced cancer animal models [5-6]. Furthermore, accumulating evidence indicates that PEITC can inhibit cell growth and induce apoptosis in a variety of cultured cancer cells, suggesting its potential therapeutic value as an anticancer agent or an adjunct to current cancer therapies [7-9]. Despite the recent data showing that sulforaphane (SFN), another member of ITC family, and PEITC can sensitize various tumor cells to Fas, TRAIL, adriamycin, etoposide or docetaxel-induced apoptosis, not enough information is available on the mechanisms of the potential synergy between PEITC and standard chemotherapy agents [10-14]. A Phase I trial with PEITC showed that at the lower doses tested (40 and 80 mg daily for 30 days) it was well tolerated, and only minor toxicity with low-grade diarrhea was observed predominantly at the high doses (120 and 160 mg daily for 30 days) (unpublished results1). The dietary role of PEITC plus its relatively low toxicity in humans and its ability to induce apoptosis in cancer cells lend a strong support for a critical investigation of its combination treatments with other anticancer drugs to enhance the efficacy of cancer prevention and therapy.
In the present study, we investigated the mechanisms of apoptosis induction by PEITC and its synergism in combination with cisplatin in human cervical cancer HeLa cells. The data show that PEITC exhibits a synergistic effect with cisplatin on the induction of apoptosis. This synergistic effect was specifically blocked by MEK1/2 inhibitor U0126 treatment, suggesting that the effect involves ERK activation.
2 Materials and Methods
2.1 Materials
PEITC, benzyl isothiocyanate (BITC), and cisplatin were purchased from Sigma-Aldrich (St. Louis, MO). SFN was from Alexis Biochemicals (San Diego, CA). The p38 polyclonal and β-Actin monoclonal antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA); the anti-PARP monoclonal antibody from BD Biosciences (San Jose, CA); the phospho-specific ERK rabbit polyclonal antibody from Promega (Madison, WI); and the phospho-specific p38 and phosphor-specific JNK1/2 polyclonal antibodies from Cell Signaling Technology, Inc. (Beverly, MA). The antibodies to total forms of ERK1/2, p38, and JNK1/2 were obtained from Cell Signaling Technology, Inc. (Beverly, MA); and p38 inhibitor SB203580, MEK inhibitor U0126, and JNK inhibitor SP600125 were from Calbiochem (La Jolla, CA).
2.2 Cell culture
HeLa, C33A, and MCF-7 cells were obtained from American Type Culture Collection. NF-κB reporter stable cells (HeLa/NFkB-luc) were from Panomics, Inc. (Redwood City, CA). All cells were maintained in Dulbecco’s modified Eagle’s medium (Mediatech, Hernton, VA) containing 10% fetal bovine serum (Quality Biological, Inc., Gaithersburg, MD), 100 units/ml penicillin and 100 μg/ml streptomycin, and were maintained at 37°C in 5% CO2. Working solutions of ITCs were prepared in DMSO.
2.3 DAPI staining
DAPI (4’,6-diamidino-2-phenylindole) staining was performed as described previously [15]. In brief, prior to staining, the cells were fixed with 4% paraformaldehyde for 30 min at room temperature and then washed with PBS. DAPI was added to the fixed cells for 1 h, after which they were examined by fluorescence microscopy. Apoptotic cells were identified by condensation and fragmentation of nuclei.
2.4 Flow cytometry analysis of DNA content
HeLa cells were incubated with PEITC or vehicle control for 24 h. Cells were harvested and fixed in 70% chilled ethanol. Fixed cells were stained with propidium iodide (50 μg/ml). The stained cells were analyzed for DNA content on a Becton Dickinson Flow Cytometer (BD Biosciences, Franklin Lakes, NJ). The presence of a sub-G1 compartment of cells was indicative of apoptosis.
2.5 NF-κB luciferase assay
Stable HeLa/NF-κB-luc reporter cells were seeded into a 24-well plate (50,000 cells/well) in DMEM media with 10% fetal bovine serum (FBS) for 24 h. The cells were treated in triplicate with DMSO (vehicle), CDDP (10 μM), PEITC (5 μM), CDDP (10 μM) plus PEITC (5 μM) and 10 ng/ml TNF-α for 2, 4 and 6 h. The cells were then harvested, and the luciferase activity was measured using a Bright-Glo assay system from Promega (Madison, WI) according to manufacturer’s instructions; the data were collected using MicroLumat Plus LB96 V (Berthold technologies) and the attached Winglo version 1.25 software. The reproducibility of the results was verified with two independent experiments.
2.6 Caspase-3 activity assay
Caspase-3 activity was measured by detection of the cleavage of a colorimetric caspase-3 substrate, N-acetyl-Asp-Glu-Val-Asp (DEVD)-p-nitroaniline, using an assay kit (R&D Systems, Inc., Minneapolis, MN). In brief, cells were treated with drugs for different time. The attached and detached cells were collected, and lysed in ice-cold lysis buffer provided by the manufacturer. The same amount of protein extracts (100-200 μg) was incubated in a reaction buffer containing N-acetyl-DEVD-p-nitroaniline at 37°C for 2 to 4 h. The levels of the proteolytic fragment p-nitroanilide were measured as optical density at 405 nm with a plate reader. The data represent the mean ± SD of three independent experiments.
2.7 Western Blot analysis
For immunoblot analysis, cells were harvested in 300-500 μl of lysis buffer [(20 mM Hepes, pH 7.4, 2 mM EGTA, 50 mM b-glycerol phosphate, 1% Triton X-100, 10% glycerol, 1 mM dithiothreitol (DTT), 1 mM phenylsulfonyl fluoride (PMSF), 10 mg/ml leupeptin, 10 mg/ml aprotinin, 1 mM Na3VO4, and 5 mM NaF)]. The resulting protein samples were separated by 4-12% NuPAGE gels (Invitrogen, Carlsbad, CA) (30-40 mg/lane) and transferred onto PVDF membranes (Millipore, Bedford, MA). The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween 20 for 1 h at room temperature. After incubation of the membranes with appropriate antibodies, specific proteins were detected with enhanced chemiluminescence (ECL) reagents (Amersham Biosciences, Piscataway, NJ). β-Actin was used as a loading control for all the Western Blot analysis.
3 Results
3.1 PEITC induces apoptosis in HeLa cells
PEITC has been shown to induce apoptosis in various cancer cells [7-9]. We initially tested its effects on cell viability. HeLa cells were treated with different concentrations of PEITC. After 24 h of treatment, the number of live cells was determined by MTS assay, the IC50 for PEITC was around 13 μM (data not shown). We then investigated whether PEITC can cause apoptosis in HeLa cells. The apoptotic effects of PEITC were evaluated 24 h after treatment of HeLa cells with 15 μM PEITC. The PEITC-treated cells showed morphological alterations consistent with apoptosis, including shrinkage, membrane blebbing and detachment of cells, and DAPI stained cells showed evidence of nuclear condensation and fragmentation (Fig. 1A). The PEITC-induced cell death via apoptosis was further confirmed by FACS analysis. Fig. 1B shows a sub-G1 peak was evident in the treated cells but absent from the untreated control cells. We also determined caspase-3 activity in the PEITC-treated cells. As shown in Fig. 1C, a time-dependent increase of caspase-3 activity was seen in HeLa cells after treatment with 15 μM PEITC, with a 3.3-fold increase at 12 h. PARP cleavage was also evaluated in cells treated with PEITC. The results showed that PARP cleavage became evident at 8 h post-treatment and increased in a time-dependent manner (Fig. 1D).
Figure 1. PEITC treatment induces apoptosis in HeLa cells.
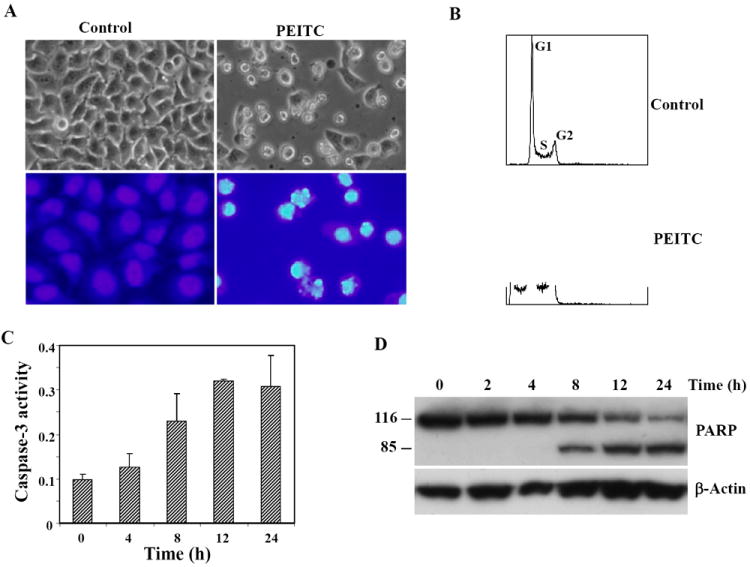
A, representative phase contrast microscopy (upper panels) and DAPI staining (lower panels) of untreated cells and cells treated with 15 μM PEITC for 24 h. Nuclei of apoptotic cells are fragmented and condensed. B, flow cytometry analysis of cells 24 h after treatment with 15 μM PEITC. AP denotes sub-G1 peak, indicative of apoptotic cells in PEITC-treated, but not in control, cells. C, time course of caspase-3 activation in PEITC-treated cells. Caspase-3 activity was analyzed by a caspase-3 colorimetric assay kit. D, cleavage of caspase substrate PARP in 15 μM PEITC-treated cells. Cells were harvested at the indicated times, and PARP cleavage was assessed by Western Blot analysis using an anti-PARP monoclonal antibody. β-actin was used as a loading control.
3.2 MAPKs activation is not involved in the PEITC-induced apoptosis in HeLa cells
The MAPKs signaling pathway has been shown to be activated by ITC treatment in different cell types [8, 9, 16-19]. To investigate whether PEITC treatment led to MAPKs activation in HeLa cells, lysates obtained at various times from PEITC-treated cells were subjected to Western Blot analysis using anti-phospho-ERK, phospho-JNK and phospho-p38 antibodies to detect phosphorylated (and therefore activated) MAPKs. As shown in Fig. 2A, PEITC treatment of HeLa cells resulted in a strong activation of ERK1/2, JNK1/2 and p38. Activation of these kinases was observed at as early as 2 h following treatment and was sustained over a period of 24 h. The same blots were subsequently stripped and reprobed with antibodies that recognize regular MAPKs; the results showed that PEITC treatment has no effect on the total MAPKs protein level. To examine whether MAPKs activation is required for PEITC-induced apoptosis, HeLa cells were pretreated with selective inhibitors of MAPKs, SP600125, U0126, or SB203580 for 30 min prior to addition of 15 μM PEITC. Apoptosis was determined by measuring caspase-3 activity and detecting PARP cleavage. As shown in Fig. 2B, none of the selective MAPKs inhibitors was able to suppress PEITC-induced caspase-3 activation or PARP cleavage. These results indicate that MAPKs activation is not involved in PEITC-induced apoptosis in HeLa cells.
Figure 2. MAPKs activation is not involved in the PEITC-induced apoptosis.
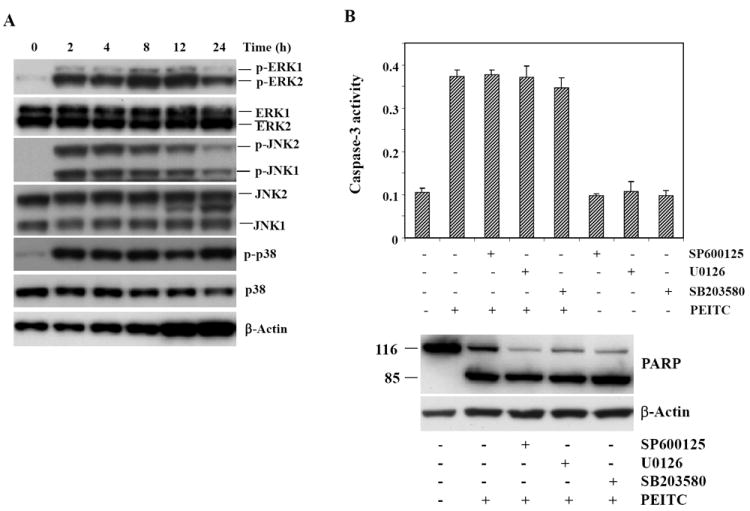
A, time course of MAPK activation by PEITC in HeLa cells. Cells were treated with 15 μM PEITC and harvested at the indicated times. Phosphorylation of JNK1/2, ERK1/2, and p38 was analyzed by Western Blotting with phospho-JNK, phospho-ERK, and phospho-p38 specific polyclonal antibodies. Total JNK, ERK, and p38 protein levels were also determined by Western Blot analysis using antibodies to the respective kinases. B, MAPKs activation is not involved in the PEITC-induced apoptosis in HeLa cells. Cells were pretreated with the MAPKs inhibitors (SP600125, U0126 and SB203580 for JNK, ERK and p38, respectively) for 1 h before the addition of 15 μM PEITC. After 24 h, the treated cells were harvested and assayed for caspase-3 activity with a colorimetric assay kit (top) and PARP cleavage assessed by Western Blot analysis using an anti-PARP monoclonal antibody (bottom).
3.3 The synergistic effect of PEITC and CDDP in apoptosis induction
Because both PEITC and cisplatin induce apoptosis in various cancer cells, we examined whether there is a synergistic effect on the induction of apoptosis by these agents. As shown in Fig. 3A, 24 h following treatment, the morphology of cells treated with 10 μM CDDP (the IC50 for CDDP in HeLa cells was around 12μM, data not shown) for or 5 μM PEITC alone showed no apoptotic changes; however, the cells treated with PEITC and CDDP in combination displayed typical features of apoptosis, such as cell shrinkage, membrane blebbing, and detachment of cells. After 24 h treatment, the combination of 5 μM PEITC and 10 μM CDDP caused a 4-fold increase in caspase-3 activity (Fig. 3A). In contrast, no substantial caspase-3 activity changes were observed in 10 μM cisplatin-treated cells, and only a 1.5-fold increase was observed in the 5 μM PEITC-treated cells. PARP cleavage was also examined in whole cell lysates collected 8 and 24 h after treatment and assessed by Western Blot analysis. At 24 h, the combination of 5 μM PEITC and 10 μM CDDP produced a large amount of the cleaved PARP, compared with that by either agent alone (Fig. 3A).
Figure 3. The synergistic effect of PEITC on CDDP-induced apoptosis in HeLa cells.
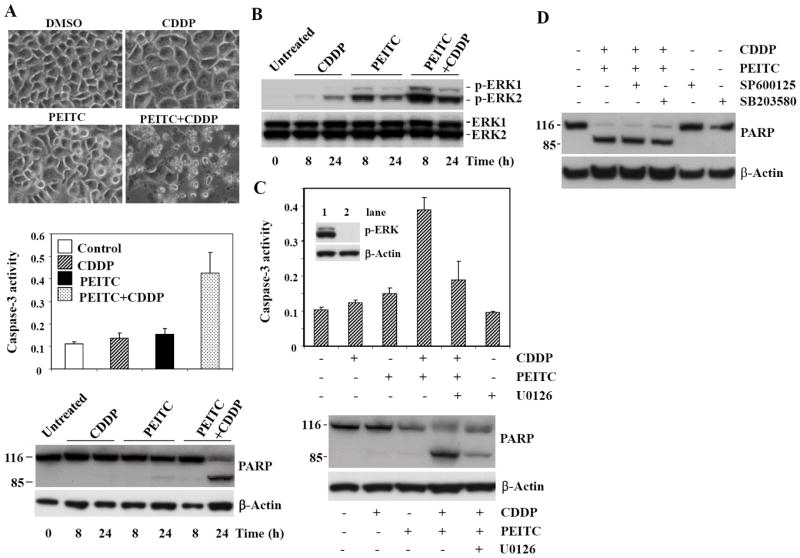
A, top: phase contrast photomicrographs of cells treated with DMSO (vehicle control), 10 μM CDDP, 5 μM PEITC, and 5 μM PEITC plus 10 μM CDDP for 24 h. Note the presence of massive cell death in cells treated with 5 μM PEITC plus 10 μM CDDP. Middle: caspase-3 activity analysis in cells treated with 5 μM PEITC, 10 μM CDDP, or the combination for 24 h. Bottom: Effect of PEITC plus CDDP on PARP cleavage. Subconfluent cells were incubated with 5 μM PEITC, 10 μM CDDP alone or the combination, PARP expression and cleavage was examined in whole cell lysates collected 8 and 24 h after treatment and assessed by Western Blot analysis using an anti-PARP monoclonal antibody. B, enhanced ERK activation after the combination treatment of PEITC plus CDDP in HeLa cells. Cells were treated with either 10 μM CDDP alone, 5 μM PEITC alone, or 5 μM PEITC plus 10 μM CDDP for 8 and 24 h. Western Blot analysis shows enhanced ERK activation in PEITC plus CDDP-treated cells. C, MEK1/2 inhibitor blocks apoptosis induced by the combination of PEITC and CDDP. Pretreatment with MEK1/2 inhibitor U0126 inhibits ERK (lane 2) and caspase-3 activation caused by the combination of PEITC and CDDP (top). Western Blot analysis shows reduced PARP cleavage in cells treated with U0126 prior to PEITC plus CDDP (bottom). D, PARP cleavage shows that JNK inhibitor SP600125 and p38 inhibitor SB203580 have no effect on apoptosis induced by the combination of PEITC and CDDP in HeLa cells.
3.4 MEK1/2 inhibitor blocks PEITC plus CDDP-induced apoptosis
Previous findings have shown that ERK activation plays an active role in mediating cisplatin-induced apoptosis of HeLa cells [20]. Therefore we wanted to investigate ERK activation after the combination treatment of PEITC plus CDDP. As expected, higher ERK activation was evident in cells treated with PEITC plus CDDP, compared with those treated with PEITC or CDDP alone (Fig. 3B). To examine the role of the ERK signaling pathway in the synergistic interaction between PEITC and CDDP, we treated cells with the combination of agents in the absence or presence of specific MEK1/2 inhibitor U0126. Pretreating cells for 30 min with U0126 inhibited ERK activation (Fig. 3C, top, lane 2) and resulted in a significant attenuation of apoptosis caused by the combination of PEITC and CDDP, shown by both caspase-3 activity (Fig. 3C, top) and PARP cleavage (Fig. 3C, bottom). However, the synergistic effect could not be blocked by either the JNK inhibitor SP600125 or the p38 inhibitor SB203580 (Fig. 3D). These results suggest that the ERK signaling pathway contributes to the synergistic induction of apoptosis by PEITC. Interestingly, while PEITC alone activated the ERK pathway, ERK activation did not seem to play a role in the apoptosis induced by PEITC, because the inhibitor of MEK1/2 did not block apoptosis (Fig. 2B).
3.5 The role of NF-κB in the synergistic effect on apoptosis
NF-κB plays important roles in cell growth, differentiation, apoptosis, inflammation, and many other physiologic processes [21]. NF-κB mediates survival signals that inhibit apoptosis and promote cancer cell growth. It has been found that inhibition of NF-κB activation in tumor cells may increase the efficacy of chemotherapeutic agents [22]. Recent studies have indicated that both PEITC and SFN can inhibit NF-κB transcriptional activity [23-25]. We therefore investigated whether PEITC can inactivate NF-κB in HeLa cells and whether the inhibition of NF-κB contributes to its synergistic effect on cisplatin-induced apoptosis. Stably transfected HeLa/NF-κB-luc reporter cells were treated with DMSO (vehicle), CDDP (10 μM), PEITC (5 μM), CDDP (10 μM) plus PEITC (5 μM) and 10 ng/ml TNF-α (as a positive control) for 2, 4 and 6 h. The cells were then harvested, and the luciferase activity was measured using the Bright-Glo assay system (Promega). We found that 10 μM CDDP did not increase NF-κB activity at 2, 4 and 6 h after treatment, and there is no evidence for the difference in NF-κB activity between CDDP-treated and PEITC plus CDDP-treated groups, even though the inhibition of basal NF-κB activity was seen by PEITC (Fig. 4). Collectively, these results suggest that the NF-κB pathway do not contribute to the increased apoptosis induced by the combination of PEITC and cisplatin.
Figure 4. NF-κB Luciferase assay in treated-HeLa cells.
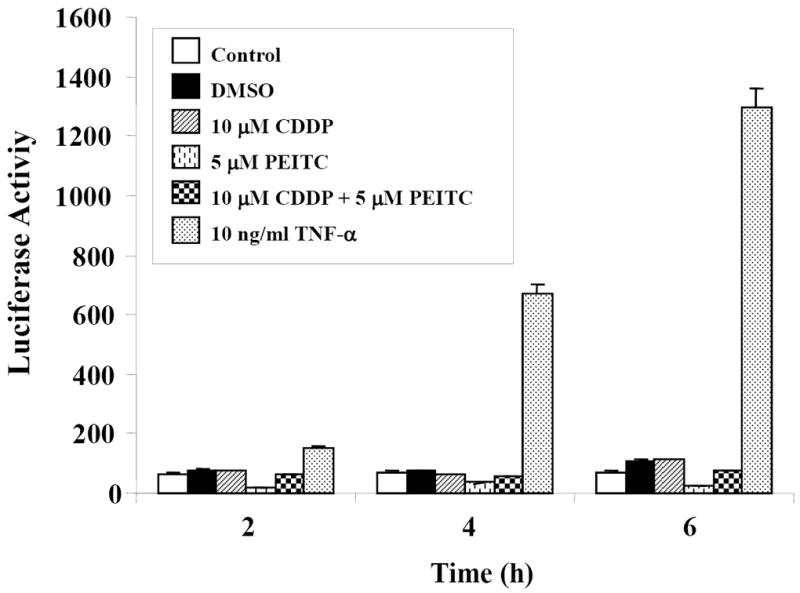
HeLa/NFκB-luc reporter cells were treated with DMSO (vehicle), CDDP (10 μM), PEITC (5 μM), CDDP (10 μM) plus PEITC (5 μM), and 10 ng/ml TNF-α for 2, 4 and 6 h. Cells were then harvested and the luciferase activity was measured using Bright-Glo assay system (Promega) according to manufacturer’s instructions. The reproducibility was verified with two independent experiments.
3.6 Both BITC and SFN synergize cisplatin-induced apoptosis in HeLa cells
Our results showed that SFN and BITC, two other members of the isothiocyanate family originated from broccoli and garden cress, also induce apoptosis in HeLa cells. As shown in Fig. 5A, both SFN and BITC caused a time-dependent cleavage of PARP protein. We observed that the combination of 5 μM BITC plus 10 μM CDDP or 20 μM SFN plus 10 μM CDDP produced a large amount of cleaved PARP, compared with that by either of the single agent (Fig. 5B, 5C), suggesting that, like PEITC, BITC and SFN can also synergize cisplatin-induced apoptosis in HeLa cells.
Figure 5. Pretreatment with BITC or SFN sensitizes HeLa cells to apoptosis induced by CDDP.
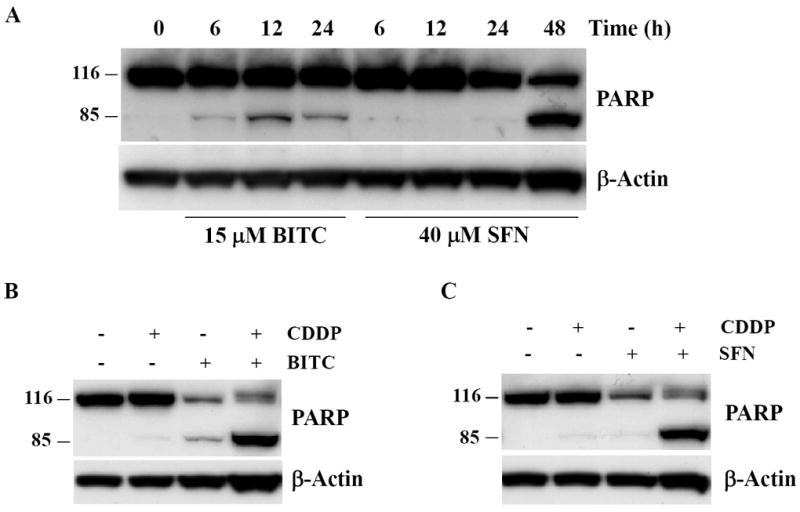
A, cells were treated with 15 μM BITC or 40 μM SFN for the indicated times. Cleavage of caspase substrate PARP was assessed by Western Blot analysis using an anti-PARP antibody. B, effect of the combined exposure to BITC (5 μM) and CDDP (10μM) on the cleavage of caspase substrate PARP. C, effect of the combined exposure to SFN (20 μM) and CDDP (10 μM) on the cleavage of caspase substrate PARP.
3.7 PEITC synergizes cisplatin-induced apoptosis in C33A cervical cancer and MCF-7 breast cancer cells but not normal human mammary epithelial MCF-10A cells
We examined whether the synergistic effect between PEITC and cisplatin is cell line-specific. First, we tested another human cervical cancer cell line, C33A cells were treated with 10 μM PEITC, 10 μM CDDP or the combination of PEITC and CDDP for 24 h, as shown in Fig. 6A, PEITC alone caused very little PARP cleavage, but the combination of PEITC and CDDP significantly increased the level of PARP cleavage. Next, we examined MCF-7 breast cancer cells, 5 μM PEITC caused an increase of PARP cleavage at 24 h after treatment, however, the combination of 5 μM PEITC plus 10 μM CDDP produced a highly significant increase of cleaved PARP (Fig. 6B). The synergistic effects were then evaluated in MCF-10A cells, a well-characterized immortalized normal (non-transformed) human mammary epithelial cell line. MCF-10A cells were treated with 5 μM PEITC, 10 μM CDDP or the combination of PEITC and CDDP. As shown in Fig. 6C, at the dose tested there was no cytotoxic effect by PEITC or synergistic effect between PEITC and CDDP in MCF-10A cells, suggesting that normal MCF-10A cells are more resistant to the cytotoxic effects of PEITC or PEITC plus CDDP than MCF-7 breast cancer cells.
Figure 6. Pretreatment with PEITC sensitizes C33A cervical cancer and MCF-7 breast cancer cells but not normal mammary epithelial MCF-10A cells to apoptosis induced by CDDP.
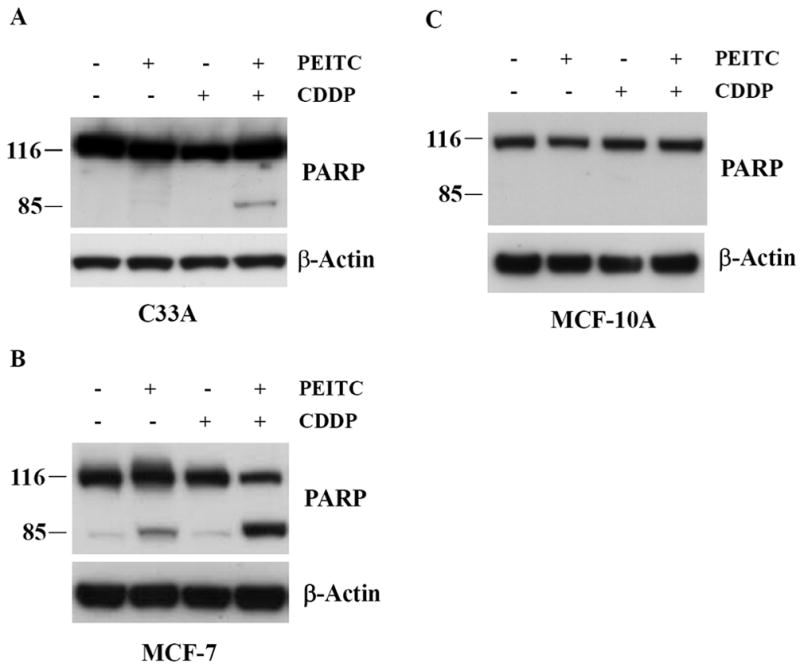
A, C33A cells were treated with 10 μM PEITC, 10 μM CDDP or combination of PEITC and CDDP for 24 h. Cleavage of caspase substrate PARP was assessed by Western Blot analysis. B, MCF-7 cells were treated with 5 μM PEITC, 10 μM CDDP or combination of PEITC and CDDP for 24 h. Cleavage of caspase substrate PARP was assessed by Western Blot analysis. C, effect of the combined exposure to PEITC (5 μM) and CDDP (10 μM) on caspase substrate PARP in MCF-10A cells.
3.8 Effects of PEITC and CDDP alone or in combination on Bcl-2 family members and other apoptosis-related proteins
Bcl-2 family proteins play central roles in the regulation of most, if not all, apoptotic pathways. The Bcl-2 homology 3 (BH3)-only members of this family, such as PUMA and Noxa, are pro-apoptotic; they are activated or induced in response to stress stimuli. These BH3-only proteins then interfere with the function of prosurvival Bcl-2 family members, thereby promoting the progression of apoptosis [26]. PEITC has been shown to regulate certain Bcl-2 family members in cancer cells [27, 28]. Therefore, we want to determine whether PEITC regulates the expression of several Bcl-2 family proteins in HeLa cells. Fig. 7A shows that the expression levels of Bcl-2, Bcl-xL and Bax were not altered in response to cisplatin, PEITC alone or the combination. Interestingly, we observed that cisplatin, PEITC alone, or the combined treatment for 24 h down-regulated the expression of the pro-apoptotic protein PUMA in HeLa cells. However, we found that 5 μM PEITC combined with 10 μM cisplatin induced a sustained expression of the pro-apoptotic protein Noxa (Fig. 7A), which correlated with the enhanced effect on apoptosis induction. In contrast, PEITC treatment alone increased Noxa protein expression only at 8 h, and cisplatin alone had no effect on Noxa. To further test the role of Noxa in apoptosis response to PEITC plus cisplatin, we examined if pretreating HeLa cells with MEK1/2 inhibitor U0126 would block Noxa induction, since U0126 can inhibit apoptosis induced by PEITC plus cisplatin (Fig. 3C). As shown in Fig. 7B, the presence of U0126 partially inhibited Noxa, and, as a control, we did not see Bax protein level change following U0126 pretreatment. These data suggest that Noxa induction is associated with apoptosis induced by PEITC or PEITC plus cisplatin. There are different apoptosis signaling pathways; one of the apoptosis signaling processes is initiated by ligation of cell surface death receptors, DR4 and DR5, by their cognate ligand TRAIL [29]. Others have reported that SFN enhances TRAIL-induced apoptosis through the induction of DR5 expression [10, 30]. As shown in Fig. 7A, we found that cisplatin, PEITC alone, or the combined treatment for 8 and 24 h failed to modify expression of the pro-apoptotic proteins DR4, DR5, or TRAIL.
Figure 7. Noxa induction is associated with increased apoptosis caused by the combined treatment of PEITC and CDDP in HeLa cells.
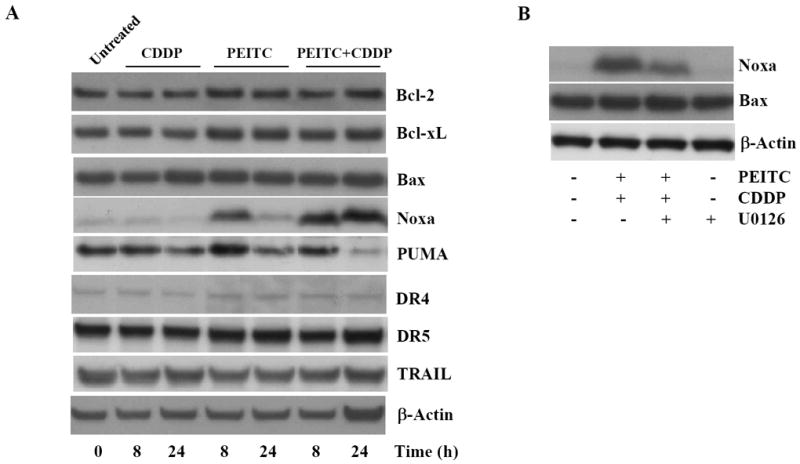
A, Immunoblot analysis of levels of Bcl-2, Bcl-xL, Bax, PUMA, Noxa, Bcl-xL, DR4, DR5, and TRAIL proteins in whole-cell lysates of cells after treatment with PEITC, CDDP or their combination for 8 to 24 h. B, MEK1 inhibitor U0126 partially inhibited Noxa protein level induced by the combined treatement of PEITC and CDDP.
4 Discussion
The major finding of the present study is that PEITC can synergize cisplatin-induced apoptosis in human cervical cancer cells. The significance of this study lies in the fact that cisplatin is routinely used in cancer treatment, but causes high toxicity, and the acquired resistance often diminishes its efficacy, resulting in treatment failure. One strategy to overcome resistance and reduce toxicity is the combination of standard anticancer drugs with naturally occurring compounds with known anticancer activity [3, 31, 32]. Recent studies have shown that SFN and PEITC can sensitize various tumor cells to Fas, TRAIL, adriamycin, etoposide or docetaxel-induced apoptosis [10-14], and that SFN can increases the efficacy of doxorubicin in mouse fibroblasts [33]. Supporting this notion, the present study shows that PEITC exhibits a synergistic effect on the induction of apoptosis in HeLa cells treated with cisplatin.
MAP kinase (MAPK) signaling pathways are important in regulating cell proliferation and cell survival in response to growth stimulation and stress. MAPKs consist of at least three signal transduction pathways (ERK, JNK and p38). Activation of the ERK pathway is involved in cell proliferation, while in contrast, JNK and p38 kinase pathways are primarily activated by stress signals, and activation of these pathways leads to inhibition of cellular proliferation and/or decreased cell survival [34]. Previous reports indicate a role of MAP kinases in the direct induction of apoptosis by PEITC [8, 9, 16]. Therefore, it is possible that induction of MAP kinase by PEITC may play a role in sensitizing cells to cisplatin-mediated apoptosis. While we observed activation of ERK, JNK and p38 upon treatment of HeLa cells with PEITC alone, inhibitors of these kinases did not block apoptosis. These results contradict the role of JNK activation in the induction of apoptosis by PEITC and do not support a recent study showing that PEITC-induced apoptosis in PC3 human prostate cancer cells is dependent on ERK1/2 activation [8, 9]. This discrepancy may be due to cell type differences. Previous studies have shown that ERK, JNK, and p38 are all activated in response to cisplatin treatment. However, inhibitor studies suggest that only the activation of ERK seems to be involved in regulating cell survival following exposure to cisplatin [20, 35-37]. In HeLa and A172 cells, ERK activation following cisplatin treatment correlates with increased sensitivity to cisplatin [20, 36], whereas in A2780 ovarian cancer cells, cisplatin-induced ERK activation is associated with increased resistance to its cytotoxicity [35, 37]. These studies imply that the effect of ERK activation following treatment of cells with cisplatin is cell type specific. Although MAPKs are not involved in the apoptosis induced by PEITC alone in HeLa cells, the decreased apoptosis by ERK inhibition with U0126 following PEITC plus cisplatin treatment, suggests that PEITC plus cisplatin-induced apoptosis was in part mediated through activation of the ERK pathway. This is in agreement with the previous observation that ERK activation plays an active role in mediating cisplatin-induced apoptosis in HeLa cells [20]. It is unclear how ERK activation is involved in the apoptosis induced by the combination of PEITC and CDDP and what are the downstream targets mediating its apoptotic effect. Recently we showed that PEITC, BITC, and SFN can bind selectively to tubulins, and their tubulin binding affinities correlate well with their potencies of inducing tubulin conformation changes, degradation and cell cycle arrest and apoptosis in human lung cancer A549 cells [38]. Because we observed that MEK1/2 inhibitor U0126 only partially inhibited apoptosis triggered by PEITC plus CDDP in HeLa cells, it is conceivable that other mechanisms, like tubulin degradation, may also contribute to chemosensitization induced by PEITC, and additional death signals triggered by CDDP treatment is necessary. Indeed, in a separate study we found that the sensitization of human non-small cell lung cancer A549 cells to cisplatin by PEITC, and BITC is correlated with their ability to deplete the tubulin [39].
In this study, we examined the roles of several pro-apoptotic and anti-apoptotic proteins in the regulation of apoptosis induced by CDDP and PEITC alone, or the combined treatment. Among the proteins examined, Bcl-2, Bcl-xL, Bax, DR4, DR5, and TRAIL showed no protein level changes after treatments. A recent study by Xiao et al showed that the enhancement of docetaxel-induced apoptosis by PEITC in PC-3 and DU145 cells is associated with suppression of Bcl-2 and induction of Bax [13]. Noxa was up-regulated at early time points by PEITC and its expression was enhanced and sustained by cisplatin. We also showed that U0126, a MEK1/2 inhibitor, partially inhibited apoptosis and Noxa protein induced by PEITC plus cisplatin. These data suggest that Noxa induction is associated with apoptosis induced by PEITC or PEITC plus cisplatin. Noxa is mainly induced through p53 dependent pathways, at present, there are no reports of any direct link between ERK activation and Noxa induction. As one of the BH3-only proteins, Noxa is known to contribute directly to depolarization of the mitochondrial membrane, followed by cytochrome c release and apoptosis. Our previous study has shown that cytochrome c was released from mitochondrial in response to CDDP treatment in HeLa cells [20]. Other studies have reported that mitochondria are a target in PEITC-induced apoptosis in human prostate and bladder cancer cells [40, 41].
Our data suggests that the synergism shown in tumor cells is not observed in normal human mammary epithelial MCF-10A cells. The synergistic effects are also seen with SFN and BITC, suggesting that other ITCs derived from cruciferous vegetables may also potentially serve as adjuncts to chemotherapy in the treatment of human cancers. It should be noted that the plasma ITCs concentrations in the micromolar range are achievable following oral administration [42]. This study provides evidence supporting novel therapeutic strategies for human cancer by combining dietary PEITC with cisplatin. Studies using in vivo animal models and preclinical trials are needed to fully evaluate PEITC in combination with chemotherapeutic agents for human cancers.
Acknowledgments
We thank Michelle A. Lombard and Dr. Karen Creswell (Flow Cytometry and Cell Sorting Shared Resource at the Lombardi Comprehensive Cancer Center) for performing the FACS analysis. We also would like to thank Dr. Emily J. Greenspan for critical reading of the manuscript.
Abbreviations
- ITCs
isothiocyanates
- PEITC
Phenethyl isothiocyanate
- SFN
sulforaphane
- BITC
benzyl isothiocyanate
- CDDP
cisplatin
- PARP
poly(ADP-ribose) polymerase
- DMSO
dimethyl sulfoxide
- DAPI
4’,6-diamidino-2-phenylindole
- PBS
phosphate-buffered saline
- DMEM
Dulbecco’s modified Eagle’s medium
- MAPKs
mitogen-activated protein kinases
- ERK
extracellular signal-related kinase
- JNK
c-Jun N-terminal kinase
Footnotes
Leonard Liebes et al, unpublished results
References
- 1.Eastman A. Alkylating and platinum-based agents. Curr Opin Oncol. 1990;2:1109–1114. doi: 10.1097/00001622-199012000-00014. [DOI] [PubMed] [Google Scholar]
- 2.Siddik Z. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 3.Sarkar F, Li Y. Using chemopreventive agents to enhance the efficacy of cancer therapy. Cancer Res. 2006;66:3347–3350. doi: 10.1158/0008-5472.CAN-05-4526. [DOI] [PubMed] [Google Scholar]
- 4.Tookey H, VanEtten C, Daxenbichler M. Glucosinolates. In: Liener IE, editor. Toxic Constituents of Plan Foodstuffd. New York: Academic Press; 1980. pp. 103–142. [Google Scholar]
- 5.Conaway C, Yang Y, Chung F. Isothiocyanates as cancer chemopreventive agents: their biological activities and metabolism in rodents and humans. Curr Drug Metab. 2002;3:233–255. doi: 10.2174/1389200023337496. [DOI] [PubMed] [Google Scholar]
- 6.Conaway C, Wang C, Pittman B, Yang Y, et al. Phenethyl isothiocyanate and sulforaphane and their N-acetylcysteine conjugates inhibit malignant progression of lung adenomas induced by tobacco carcinogens in A/J mice. Cancer Res. 2005;65:8548–8557. doi: 10.1158/0008-5472.CAN-05-0237. [DOI] [PubMed] [Google Scholar]
- 7.Keum Y, Jeong W, Kong A. Chemoprevention by isothiocyanates and their underlying molecular signaling mechanisms. Mutat Res. 2004;555:191–202. doi: 10.1016/j.mrfmmm.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 8.Xiao D, Singh S. Phenethyl isothiocyanate-induced apoptosis in p53-deficient PC-3 human prostate cancer cell line is mediated by extracellular signal-regulated kinases. Cancer Res. 2002;62:3615–3619. [PubMed] [Google Scholar]
- 9.Hu R, Kim B, Chen C, Hebbar V, et al. The roles of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adenocarcinoma cells. Carcinogenesis. 2003;24:1361–1367. doi: 10.1093/carcin/bgg092. [DOI] [PubMed] [Google Scholar]
- 10.Kim H, Kim E, Eom Y, Kim W, et al. Sulforaphane sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-resistant hepatoma cells to TRAIL-induced apoptosis through reactive oxygen species-mediated up-regulation of DR5. Cancer Res. 2006;66:1740–1750. doi: 10.1158/0008-5472.CAN-05-1568. [DOI] [PubMed] [Google Scholar]
- 11.Jin C, Moon D, Lee J, Heo M, et al. Sulforaphane sensitizes tumor necrosis factor-related apoptosis inducing ligand (TRAIL)-mediated apoptosis through downregulation of ERK and Akt in lung adenocarcinoma A549 cells. Carcinogenesis. 2007;28:1058–1066. doi: 10.1093/carcin/bgl251. [DOI] [PubMed] [Google Scholar]
- 12.Pullar J, Thomson S, King M, Turnbull C, et al. The chemopreventive agent phenethyl isothiocyanate sensitizes cells to Fas-mediated apoptosis. Carcinogenesis. 2004;25:765–772. doi: 10.1093/carcin/bgh063. [DOI] [PubMed] [Google Scholar]
- 13.Xiao D, Singh S. Phenethyl Isothiocyanate Sensitizes Androgen-Independent Human Prostate Cancer Cells to Docetaxel-Induced Apoptosis In Vitro and In Vivo. Pharm Res. 2010;27:722–731. doi: 10.1007/s11095-010-0079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kallifatidis G, Labsch S, Rausch V, Mattern J, et al. Sulforaphane Increases Drug-mediated Cytotoxicity Toward Cancer Stem-like Cells of Pancreas and Prostate. Mol Ther. 2010 Oct 12; doi: 10.1038/mt.2010.216. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Gorospe M, Huang Y, Holbrook N. p27Kip1 overexpression causes apoptotic death of mammalian cells. Oncogene. 1997;15:2991–2997. doi: 10.1038/sj.onc.1201450. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Wang W, Kong A, Tan T. Molecular mechanisms of c-Jun N-terminal kinase-mediated apoptosis induced by anticarcinogenic isothiocyanates. J Biol Chem. 1998;273:1769–1775. doi: 10.1074/jbc.273.3.1769. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y, Conaway C, Chiao J, Wang C, et al. Inhibition of benzo(a)pyrene-induced lung tumorigenesis in A/J mice by dietary N-acetylcysteine conjugates of benzyl and phenethyl isothiocyanates during the postinitiation phase is associated with activation of mitogen-activated protein kinases and p53 activity and induction of apoptosis. Cancer Res. 2002;62:2–7. [PubMed] [Google Scholar]
- 18.Xiao D, Choi S, Lee Y, Singh S. Role of mitogen-activated protein kinases in phenethyl isothiocyanate-induced apoptosis in human prostate cancer cells. Mol Carcinog. 2005;43:130–140. doi: 10.1002/mc.20099. [DOI] [PubMed] [Google Scholar]
- 19.Xu C, Shen G, Yuan X, Kim J, et al. ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis. 2006;27:437–445. doi: 10.1093/carcin/bgi251. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Martindale J, Holbrook N. Requirement for ERK activation in cisplatin-induced apoptosis. J Bio Chem. 2000;275:39435–39443. doi: 10.1074/jbc.M004583200. [DOI] [PubMed] [Google Scholar]
- 21.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 22.Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- 23.Heiss E, Herhaus C, Klimo K, Hartsch H, et al. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J Biol Chem. 2001;276:32008–32015. doi: 10.1074/jbc.M104794200. [DOI] [PubMed] [Google Scholar]
- 24.Jeong W, Kim I, Hu R, Kong A. Modulatory properties of various natural chemopreventive agents on the activation of NF-kappaB signaling pathway. Pharm Res. 2004;21:661–670. doi: 10.1023/b:pham.0000022413.43212.cf. [DOI] [PubMed] [Google Scholar]
- 25.Xu C, Shen G, Chen C, Gelinas C, et al. Suppression of NF-kappaB and NF-kappaB-regulated gene expression by sulforaphane and PEITC through IkappaBalpha, IKK pathway in human prostate cancer PC-3 cells. Oncogene. 2005;24:4486–4495. doi: 10.1038/sj.onc.1208656. [DOI] [PubMed] [Google Scholar]
- 26.Willis S, Adams J. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17:617–625. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao D, Zeng Y, Choi S, Lew K, et al. Caspase-dependent apoptosis induction by phenethyl isothiocyanate, a cruciferous vegetable-derived cancer chemopreventive agent, is mediated by Bak and Bax. Clin Cancer Res. 2005;11:2670–2679. doi: 10.1158/1078-0432.CCR-04-1545. [DOI] [PubMed] [Google Scholar]
- 28.Satyan K, Swamy N, Dizon D, Singh R, et al. Phenethyl isothiocyanate (PEITC) inhibits growth of ovarian cancer cells by inducing apoptosis: role of caspase and MAPK activation. Gynecol Oncol. 2006;103:261–270. doi: 10.1016/j.ygyno.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 29.Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer. 2002;2:420–430. doi: 10.1038/nrc821. [DOI] [PubMed] [Google Scholar]
- 30.Matsui T, Sowa Y, Yoshida T, Murata H, et al. Sulforaphane enhances TRAIL-induced apoptosis through the induction of DR5 expression in human osteosarcoma cells. Carcinogenesis. 2006;27:1768–1777. doi: 10.1093/carcin/bgl015. [DOI] [PubMed] [Google Scholar]
- 31.Ermakova S, Kang B, Choi B, Choi H, et al. (-)- Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 2006;66:9260–9269. doi: 10.1158/0008-5472.CAN-06-1586. [DOI] [PubMed] [Google Scholar]
- 32.Tyagi A, Agarwal C, Chan D, Agarwal R. Synergistic anti-cancer effects of silibinin with conventional cytotoxic agents doxorubicin, cisplatin and carboplatin against human breast carcinoma MCF-7 and MDA-MB468 cells. Oncol Rep. 2004;11:493–499. [PubMed] [Google Scholar]
- 33.Fimognari C, Nusse M, Lenzi M, Sciuscio D, et al. Sulforaphane increases the efficacy of doxorubicin in mouse fibroblasts characterized by p53 mutations. Mutat Res. 2006;601:92–101. doi: 10.1016/j.mrfmmm.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 35.Persons D, Yazlovitskaya E, Cui W, Pelling J. Cisplatin-induced activation of mitogen-activated protein kinases in ovarian carcinoma cells: inhibition of extracellular signal-regulated kinase activity increases sensitivity to cisplatin. Clin Cancer Res. 1999;5:1007–1014. [PubMed] [Google Scholar]
- 36.Choi B, Choi C, Oh H, Kim Y, et al. Role of ERK activation in cisplatin-induced apoptosis in A172 human glioma cells. Neurotoxicology. 2004;25:915–924. doi: 10.1016/j.neuro.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Hayakawa J, Ohmichi M, Kurachi H, Ikegami H, et al. Inhibition of extracellular signal-regulated protein kinase or c-Jun N-terminal protein kinase cascade, differentially activated by cisplatin, sensitizes human ovarian cancer cell line. J Biol Chem. 1999;274:31648–31654. doi: 10.1074/jbc.274.44.31648. [DOI] [PubMed] [Google Scholar]
- 38.Mi L, Gan N, Cheema A, Dakshanamurthy S, et al. Cancer preventive isothiocyanates induce selective degradation of cellular alpha- and beta-tubulins by proteasomes. J Biol Chem. 2009;284:17039–17051. doi: 10.1074/jbc.M901789200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Pasqua A, Hong C, Wu M, McCracken E, et al. Sensitization of non-small cell lung cancer cells to cisplatin by naturally occurring isothiocyanates. Chem Res Toxicol. 2010;23:1307–1309. doi: 10.1021/tx100187f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang L, Zhang Y. Mitochondria are the primary target in isothiocyanate-induced apoptosis in human bladder cancer cells. Mol Cancer Ther. 2005;4:1250–1259. doi: 10.1158/1535-7163.MCT-05-0041. [DOI] [PubMed] [Google Scholar]
- 41.Xiao D, Lew K, Zeng Y, Xiao H, et al. Phenethyl isothiocyanate-induced apoptosis in PC-3 human prostate cancer cells is mediated by reactive oxygen species-dependent disruption of the mitochondrial membrane potential. Carcinogenesis. 2006;27:2223–2234. doi: 10.1093/carcin/bgl087. [DOI] [PubMed] [Google Scholar]
- 42.Liebes L, Conaway C, Hochster H, Mendoza S, et al. High-performance liquid chromatography-based determination of total isothiocyanate levels in human plasma: application to studies with 2-phenethyl isothiocyanate. Anal Biochem. 2001;291:279–289. doi: 10.1006/abio.2001.5030. [DOI] [PubMed] [Google Scholar]