

Abstract
The development of robust genetic tools to manipulate Borrelia burgdorferi, the etiologic agent of Lyme disease, now allows investigators to assess the role(s) of individual genes in the context of experimental Lyme borreliosis. This unit is devoted to the description of experimental approaches that are available for the molecular genetic analysis of B. burgdorferi with an emphasis placed on the cultivation, electrotransformation, the selection of desired mutants, and the genetic complementation of the mutants acquired. The intent here is to provide a consensus protocol that encapsulates the methodologies currently employed by the B. burgdorferi research community and describe pertinent issues that must be accounted for when working with these pathogenic spirochetal bacteria.
Introduction
Borrelia burgdorferi, the spirochetal bacterium that causes Lyme disease, is the most common tick-borne infection in the United States with over 35,198 cases reported in 2008. This value represented a 28% and 77% increase in incidence in the previous two years of reporting, respectively, indicating that Lyme borreliosis is a re-emerging disease. Early on, the infection is characterized by a skin lesion known as erythema migrans and a non-descript flu-like illness (Nadelman and Wormser, 1998; Steere et al., 2004). Patients with untreated Lyme disease experience multisystemic symptoms with the arthritis being the defining indicator of persistent infection. B. burgdorferi is a highly motile organism that is organized as a Gram-negative bacterium but lacks lipopolysaccharide (LPS) in its outer membrane. Interestingly, it has the ability to scavenge complex molecules for cellular envelope biosynthesis and essential nutrients for metabolism from its environment (Gherardini et al., 2010; Boylan et al., 2008). In the laboratory, in vitro cultivation of B. burgdorferi requires highly nutrient rich and undefined media. Furthermore, it is widely established that B. burgdorferi responds to changing environmental conditions in the arthropod and mammalian host environment.
B. burgdorferi genome was one of the first living systems whose genome was sequenced and annotated (Casjens et al., 2000; Fraser et al., 1997); however, one of the most striking features noted upon perusal of the genetic information was the lack of similarity of borrelial gene products with any other known virulence determinant or toxin within the existing database. This, coupled with the difficulties in the genetic manipulation of this pathogen, made it impossible to test whether putative virulence determinants contributed to borrelial pathogenesis. Subsequently, several independent groups developed genetic tools and methodologies to generate and characterize mutations in targeted genes, as well as suicide and shuttle vectors for cis and trans complementation, respectively (Li et al., 2007; Rosa et al., 2005, 2010; Weening et al., 2008). In the interim, the B. burgdorferi mutants characterized have provided important insight into the function of numerous borrelial gene products that affect the spirochetes pathogenic potential, physiologic capabilities, and gene regulatory systems (Skare et al., 2010; Barthold, et al., 2010; Rosa et al., 2010; Gherardini et al., 2010; Norris et al., 2010; Pal and Fikrig, 2010). Despite these important advances and continued refinement of the genetic modalities available, the ability to transform and complement infectious B. burgdorferi remains challenging.
This unit describes the cultivation (Basic Protocol 1), electrotransformation (Basic Protocol 1), and isolation of transformed clones (Basic Protocol 3) of B. burgdorferi. Subsequent complementation of confirmed mutants is outlined in Basic Protocol 3. We recognize that there are multiple methods to cultivate and genetically modify B. burgdorferi and have attempted to faithfully represent the “consensus” within the borrelial research community. Nevertheless, there are likely some approaches that have been unintentionally omitted within these protocols and, if so, this accidental oversight is not meant to imply that these methodologies are in any way insufficient in either their scope or utility.
CAUTION: Borrelia burgdorferi is a Biosafety Level 2 (BSL-2) infectious agent and should be handled in a BSL-2 approved biosafety cabinet (BSC). This includes in vitro grown cultures as well as infected animal tissues. Animal necropsies should also be conducted within BSC containment.
IMPORTANT NOTE: In addition to biosafety issues, the complex and nutrient rich media should be used under containment to prevent comtamination. It is important to note that the usage of antibiotic resistance in B. burgdorferi requires institutional approval as mandated by NIH.
Basic Protocol 1: in vitro cultivation of Borrelia burgdorferi
Borrelia burgdorferi requires a complex, undefined, nutrient rich media for in vitro cultivation. BSK-H media is available from Sigma but does not support growth of all infectious strains. As such, many researchers make their own BSK-II media. Because B. burgdorferi is such a fastidious organism, it is sensitive to subtle changes in the abundance of the unknown components, presumably provided by the abundant bovine serum albumin (BSA) or added rabbit serum, which may contribute to variations seen between purchased BSK-H and individually produced batches of BSK-II media. To circumvent this issue, lots of normal rabbit serum (NRS) and BSA must be screened for optimal motility and growth rate of B. burgdorferi. (Barbour, 1984). Because this media is rich, BSK-II is easily contaminated and should be manipulated following filtration media in a biological safety cabinet. Further safeguards for contamination include the addition of the antifungal amphotericin B, and the antimicrobials phosphomycin and rifampin, which do not adversely affect borrelial growth (Zuckert, 2007).
Materials
Complete 1X Barbour-Stoenner-Kelly II (BSK-II) medium or BSK-H medium (Sigma) (see recipes)
Filter units (0.22 µm)
CO2 incubator
Type II A Biosafety Cabinet
Inoculation and In vitro Growth of B. burgdorferi
- Pipet 5–12 ml fresh complete BSK into a 15 ml conical tube while working in a biological safety cabinet to avoid exposure to B. burgdorferi and contamination of complete BSK media.Complete BSK refers to the supplementation of BSK-II or BSK-H supplemented with 6% normal rabbit serum (NRS).
- Cultures can be inoculated from frozen stock by two methods:
- New borrelial cultures can be started from a 1 ml frozen stock (10% DMSO final concentration) by scraping a portion of the frozen cells with a sterile spatula and transferring to complete BSK-II media.
- Thawing a 1 ml aliquot of late exponential cultures and transferring the entire volume to complete BSK media can start new borrelial cultures. Thawing and transfer should be done as quickly as possible in a 37°C water bath and the tube sterilized by immersion in 70% ethanol in a biosafety cabinet prior to inoculation. The aliquot should be diluted >15-fold in complete BSK media since high concentrations of DMSO do not support growth of B. burgdorferi.
-
B. burgdorferi is grown microaerobically under static conditions in with 1–5% CO2 at 32–37°C to the appropriate cell density. On average the doubling of B. burgdorferi is approximately 8–10 hours for wild type strains (range is between 5–16 hours depending on strain) (Table 1). Mutants may grow at different rates and different growth conditions can adversely affect doubling times.
In addition to their growth microaerobically, B. burgdorferi also grow anaerobically. Cultures grown under anaerobic conditions must be inoculated from a frozen stock under the same condition with media that has been equilibrated under anaerobic conditions.
For expansion of a cultivated sample, cultures can be passed into fresh media of the necessary volume for the planned experiments. Cultures intended for RNA or protein samples should be inoculated no higher than 1× 105 B. burgdorferi/ml to allow the cells to double numerous time prior to sampling in exponential growth phase or stationary growth phase in order to accurately reflect the experimental conditions imposed.
CO2 levels alters gene expression and protein synthesis in B. burgdorferi and loose caps are necessary for gas exchange in the complete BSK media. When this type of cultivation is utilized, it is important to employ secondary containment to prevent spills.
-
To examine the viability of a given culture, B. burgdorferi are examined using a dark field microscope to enumerate the number of live cells per field. A wet mount is made with 7–10 µl of culture or of an appropriate dilution for more dense cultures.
As with any sampling method, the calculation of cell density will be more accurate by counting more fields than less. On average at least 25 fields should be counted for an accurate density determination. If this method is used, the dark field microscope will need to be calibrated, based on limiting dilutions, in order to convert the values observed to a cellular density. Overall, >90% of the cell should be motile. As the cultures increase in density the media color will change in response to the acidification of the complete BSK media.
Cell density can also be determined using a Petroff Hausser Counting Chamber (Samuels, 1995).
-
The plasmid content of B. burgdorferi must be rigorously monitored since this bacterium contains a segmented genome and loses plasmids with passaging the cells into fresh media or other experimental manipulation, including transformation.
It is essential that the plasmid content of strains is assessed prior to in vivo analyses since some plasmids are required for optimal infectivity. A plasmid profile can be performed using PCR from boiled cells or purified genomic DNA using oligonucleotide primers specific for each strain B31 plasmid (Labandeira-Rey and Skare, 2001; Purser and Norris, 2000).
Table 1.
Commonly used B. burgdorferi senus stricto strains.
| Strain | Description and Reference |
|---|---|
| B31 | Infectious low-passage wildtype strain isolated from Ixodes dammini and cloned by limiting dilution (Barbour, 1984; Burgdorfer et al., 1982). |
| 297 | Infectious low-passage wildtype strain isolated from human spinal fluid (Steere et al., 1983). |
| cN40 | Infectious clonal low-passage wildtype strain isolated from adult I. dammini (Barthold et al., 1990, 1988) |
| CA-2-87 | I. pacificus isolate from Northern California (Schwan et al., 1988) |
| JD-1 | Isolated and maintained in Ixodes ticks (Piesman et al., 1987) |
| B31 derivatives | |
| B31 MI | Derived from tick isolated B31 and passed through mouse (Casjens et al., 1997). |
| B31-A3 | Clonal B31 infectious and low-passage; plasmids and infectivity maintained with transformation of shuttle or suicide vectors (Elias et al., 2002) |
| MSK5 | Clonal infectious low-passage strain isolated from C3H/HeN mouse skin (Labandeira-Rey and Skare, 2001). |
| MSK7 | Clonal infectious low-passage strain isolated from C3H/HeN mouse skin, lacking lp28-4 (Labandeira-Rey and Skare, 2001). |
| MSK10 | Clonal low-passage strain unable to establish persistant infection; lacking lp28-1 isolated from C3H/HeN mouse skin (Labandeira-Rey and Skare, 2001). |
| ML23 | Clonal noninfectious low-passage strain lacking lp25 isolated from nonclonal P48 B31 culture (Labandeira-Rey and Skare, 2001). |
| 5A# | Clonal isolates with variable plasmid composition and infectivity (Purser and Norris, 2000). |
| 5A4 | Clonal infectious isolate containing all plasmids (Purser and Norris, 2000). |
| 5A10 & 5A14 | Clonal infectious isolate lacking lp25 and lp56 (Purser and Norris, 2000). |
| 5A18 | Clonal infectious isolate lacking lp28-4 and lp56 (Purser and Norris, 2000). |
| 5A4 NP1 | Clonal infectious isolate containing all plasmids(5A4) and bbe02 distrupted with PflgB-kanR (Kawabata et al., 2004) |
| 5A18 NP1 | 5A18 with bbe02 distrupted by a PflgB-kanR cassette (Kawabata et al., 2004) |
| 297 derivatives | |
| BbAH130 | Clonal infectious low-passage isolate from plating polyclonal 297 (Yang et al., 2004). |
| PL133 | Clonal infectious lowpassage strain isolated from ear punch biopsies of mice infected with polyclonal 297 (Revel et al., 2005). |
| BbDTR630 | Derived from PL133 with a PflgB-kanR cassette cloned into lp25 (Blevins et al., 2007). |
| 297-LK | Non-infectious clonal isolate derived from AH130 and is lacking lp28-1; lacI and PflgB-kan cloned into bbe02 on lp25 (Gilbert et al., 2007) |
| c155 | Infectious low-passage isolated from nonclonal 297 by two rounds of plating and colony extraction (Eggers et al., 2002). |
| CE162 | Clonal infectious low-passage derived from wildtype 297 (Caimano et al., 2004). |
| cN40 derivatives | |
| D10/E9 | Infectious low-passage clonal isolate (Parveen et al., 2006). |
Basic Protocol 2: Transformation of B. burgdorferi
Transformation of B. burgdorferi is highly inefficient and requires extremely large quantities of DNA relative to other bacteria, such as Escherichia coli and Salmonella. The borrelial cultures used for transformations should exhibit active motility and replicate at the expected rate for the given strain. Solutions used for transformation must be kept ice-cold and stored at 4°C. Pelleted B. burgdorferi must be handled carefully and quickly during these steps to ensure recovery of the bacteria and enhance the yield and viability of the samples prior to electroporation. It is important to note that this procedure is effectively identical to that published by Samuels et al. (1995) except that the one of the buffers used are different. The intention here is to provide an alternative protocol to an already effective procedure.
Materials
Dulbecco’s phosphate-buffered saline (DPBS)
8 mM HEPES, 272 mM Sucrose buffer, pH 7.4
Complete BSK
Sterile centrifuge bottles, conical tubes, pipets, and pipetman tips (aerosol-resistant)
0.2 cm cuvettes
Electroporator (Bio-rad Gene Pulser)
Prepare Electrocompetent B. burgdorferi Cells
Grow 500 ml of B. burgdorferi in complete 1X BSK media until it reaches a density of 5 × 107 spirochetes/ml. Viability (scored indirectly by the motility of the culture) should fall between 90–95%. Solutions used in step 3 and beyond must be ice-cold prior to use.
-
Split culture and centrifuge at 4000 × g for 20 minutes at 4°C in pre-chilled sterile bottles.
Make sure all subsequent centrifugations occur in pre-chilled sterile bottle or tubes.
Remove supernatants and keep pellets on ice.
Resuspend the pellets in 40 ml of ice-cold dPBS and transfer to 50 ml pre-chilled conical tubes. It is important to quickly but gently resuspend the pellets. Spin at 4000 × g for 20 minutes at 4°C.
Remove supernatants and resuspend pellets in 40 ml of ice-cold dPBS. Spin at 4000 × g for 20 minutes at 4°C.
Remove supernatants and resuspend pellets in 10 ml of ice-cold HEPES/Sucrose buffer. Spin at 4000 × g for 20 minutes at 4°C.
Remove supernatants and pool pellets in 400 µl of ice-cold HEPES/Sucrose buffer.
-
Pipet cells into 100 µl aliquots containing approximately 1 × 109 B. burgdorferi cells and place on ice.
Cells need to be used the same day as prepared for transformation.The efficiency of snap frozen competent B. burgdorferi cells is greatly reduced; always use freshly made cells.
Transform cells
-
9
Use 0.2 cm pre-chilled cuvettes. Keep cuvettes on ice.
-
10Add between 4–10 µl of your DNA (5 µg/µl) to each cell aliquot and incubate on ice for 1 minute. An aliquot should be used as a negative control (i.e., no added DNA) to assess the quality of the competent cells and to determine background transformants as addressed in the plating and screening section.30 µg of precipitated DNA consistently results in the greatest number of positive transformants for shuttle vector and allelic exchange in wildtype B. burgdorferi. Multiple electroporations are performed with DNA concentrations ranging from 20 µg to 50 µg for infectious wildtype B. burgdorferi.
-
11
Transfer cells to a chilled cuvette. Make sure that there are no bubbles in the cuvette (shake or tap gently). Wipe off excess moisture from outside of cuvette.
-
12
Use the following settings: 2.5 kV, 25 µF, and 200 Ω. Time constants should fall between 4.0 and 5.0 milliseconds (ms).
-
13
Add 1 ml of complete BSK to the cuvette and re-suspend the cells. Transfer to 9 ml of complete BSK media to allow cells to recover for 18–20 hours at 1–5% CO2 and 32–34°C prior to antibiotic selection.
Basic Protocol 2: Antibiotic selection of B. burgdorferi transformants
Once B. burgdorferi has been electroporated with the desired construct and recovered overnight, it is necessary to distinguish an isolate that contains the desired genetic lesion or shuttle vector construct relative to non-transformed cells. This is facilitated by selection with an antibiotic resistance determinant that functions in B. burgdorferi. Currently there are 4 antibiotic resistant genes that are widely used for this spirochete; they confer resistance to kanamycin (kanR via aphI; (Bono et al., 2000)), streptomycin (strR via aadA [also confers resistance to spectinomycin in E. coli]; (Frank et al., 2003)), gentamicin (gentR via aacC1; ref’s), and erythromycin (ermR via ermC; (Sartakova et al., 2000)). Here we outline two different methods to obtain transformants with advantages and caveats indicated for each approach described.
A. Semisolid plating for B. burgdorferi transformed cells
Plating borrelial cells in a semisolid media is a traditional technique for isolating individual clones. Colonies appear, after approximately 10–17 days, as diffuse white spots in the red overlay. This approach is more problematic since it requires that the agarose overlay be within in a small temperature range in order to remain molten but not too hot to kill the borrelial cells and/or inactivate the antibiotics being used. It is also possible that the desired mutation may render the borrelial cell more sensitive to the osmotic tension of a semisolid matrix that would not be seen for cells grown in liquid broth.
Materials
1.7% molecular grade agarose
2X BSK-II (see solutions)
Normal Rabbit Serum (NRS)
Antibiotics (filter sterilized)
Autoclave
Water bath
Disposable Petri dish (sterile, 100 mm O.D. × 15 mm)
Disposable sterile pipets, pipet tips (aerosol resistant) and sterile 15 ml conical tubes
Parafilm
Autoclave 100 ml of 1.7% molecular grade agarose in a wide mouth bottle. Allow molten agarose to cool in a 48°C water bath.
Add 100 ml of prewarmed 2X BSK-II and 14 ml of prewarmed NRS to have enough mixture for 10 plates. Add the appropriate antibiotics and gently mix the solution; return to 48°C water bath. Once the antibiotics are added to media the temperature should not exceed 52°C.
To pour the underlay, pipet 10 ml of the complete BSK agarose mixture into a sterile petri dish promptly. Allow underlay to cool and polymerize in a sterile environment for approximately 15 minutes.
Spin 10 ml of the recovered transformed B. burgdorferi culture at 4000 × g for 10 minutes. Remove 9 ml of the resulting supernatant and re-suspend the cells in the remaining 1 ml of medium.
Split the 1 ml concentrate of cells between two 15 ml conical tubes.
Pipet 10 ml of complete BSK agarose mixture and quickly mix by pipeting with the 0.5 ml aliquot of transformed cells in the 15 ml conical tubes.
-
Transfer cells in the complete BSK agarose mixture to a sterile petri dish containing a polymerized underlay. Gently swirl the overlay to allow an even spread of cell on the underlay.
NOTE: the agarose overlay will solidify rapidly so it is important to evenly distribute it over the surface of the existing overlay as quickly as possible.
Allow the plates to completely solidify for 15 minutes in a sterile environment. Close plates and wipe the outer edge of plate with ethanol. Wrap the plates with parafilm.
Incubate inverted plates for borrelial growth. Depending on the strain and/or mutation introduced, colonies will appear as early as 7 days and out to 21 days or longer.
Individual colonies can be removed by using a sterile barrier tip to pull agarose plugs from the semisolid matrix. Transfer the plug to 5 ml of fresh complete media with the appropriate selection.
Following expansion in complete BSK, an aliquot of the putative mutant or shuttle vector transformant should be frozen in complete BSK with 10% DMSO at −80°C and subsequently verified by appropriate molecular approaches (i.e., PCR, Southern and/or Western blotting [if an antibody reagent is available]).
B. Liquid plating of transformed B. burgdorferi cells
Liquid plating of transformed B. burgdorferi cells is widely used because of the ease of the technique (i.e., no molten agarose and temperature issue to contend with). Effectively, the total volume of the culture is distributed in small volumes over 96-well plates (Yang et al., 2004). This, coupled with the inefficiency of the borrelial transformation generally assures that, if a transformant is obtained, it is likely to be clonal. Nevertheless, there is a real possibility that an individual liquid sample may contain multiple transformants.
Materials
Complete 1X BSK
0.5% Phenol red (filter sterilized)
Antibiotics (filter sterilized)
Multichannel pipet
Sterile 96-well plate, basin, pipet tips (aerosol resistant)
Plate tape (Petri Seal™)
Following transformation and 16–18 hours of recovery, 35 ml of complete BSK supplemented with Phenol red (0.002% final concentration) and the appropriate antibiotics is added.
Transfer 40 ml into a sterile basin and, using a multichannel pipet, transfer 180 µl aliquots per well over two 96-well plates.
Incubate the remaining 5 ml of the transformed culture, along with the 96 well plates, at 1–5% CO2 and 32–34°C.
Generally speaking, if the transformation is successful, live, motile cells should be visible in the 5 ml culture approximately 10 days after electroporation. A few days following their visualization in the 5 ml culture, individual wells should begin to change color, due to borrelial growth and concomitant acidification of the complete BSK media. This generally occurs 12–16 days after transformation, but may take up to (or longer than) 21 days. Positive wells can then be verified by dark field microscopy to confirm the presence of live motile cells.
Transfer 10 µl of culture from a positive well into 10 ml of complete BSK media and grow under appropriate selection. Add DMSO to a final concentration of 10% to the remaining volume of the well (i.e., 170 µl) and store at −80°C. Alternatively, a final concentration of 20–25% glycerol can used in place of DMSO.
Following expansion in complete BSK, the mutant or shuttle vector transformant should be verified by appropriate molecular approaches (i.e., PCR, Southern and/or Western blotting [if an antibody reagent is available]).
Basic Protocol 3: B. burgdorferi Complementation—recombinase based method
Once the mutants have been isolated and confirmed, it is important to genetically complement them to ensure that the mutation isolated is attributable to the mutation introduced and not the result of a second site mutation. There are several methods to carry out genetic complementation that involve conventional restriction digested and ligated constructs, either of which can be designed to complement in both a trans or cis acting sites or at a heterologous location within the genome (Li et al., 2007; Rosa et al., 2005; Weening et al., 2008). This approach is dependent on specific details that are key to any recombinant construct (i.e., presence or absence of restriction sites on the gene of interest relative to the cloning backbone) and, as such, will not be described here. Instead, the focus of this protocol will be to outline how existing recombinase-dependent constructs, which require no restriction digestion or ligation steps, have been used to complement B. burgdorferi mutants (Figure 1 and (Weening et al., 2008).
Figure 1.
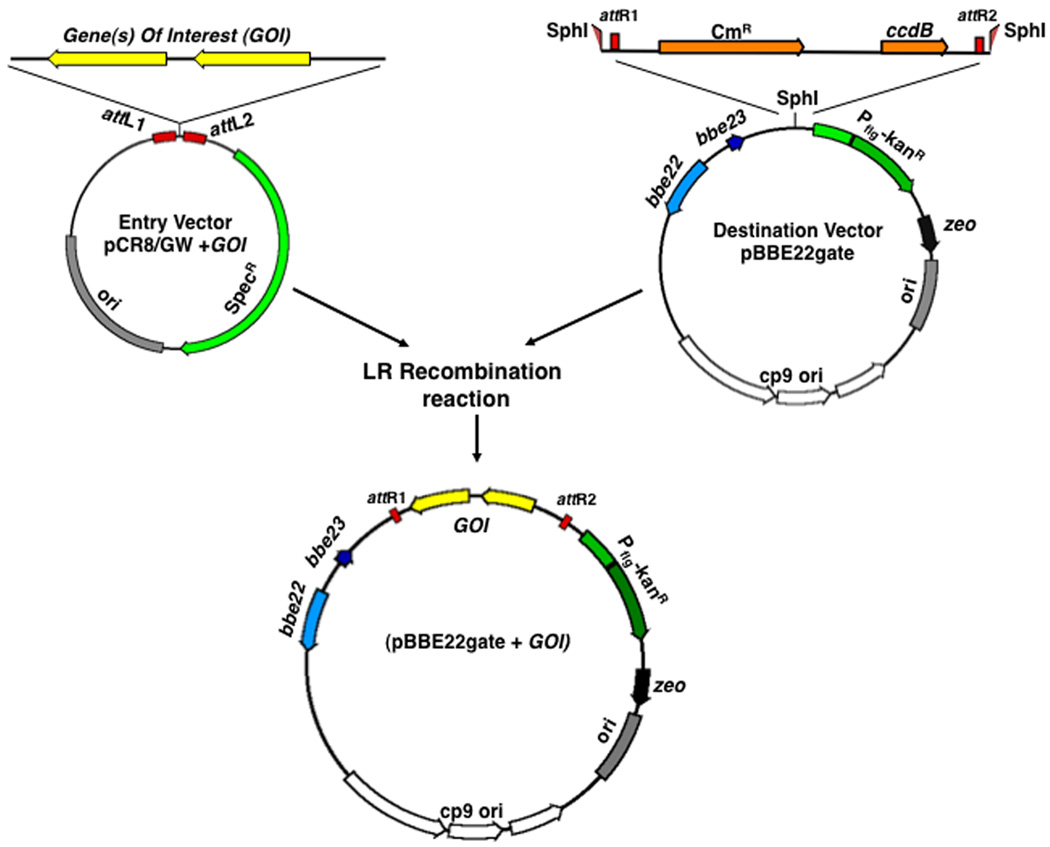
B. burgdorferi adapted Gateway recombinase cloning (Invitrogen). The gene(s) of interest (GOI) are cloned into an entry vector (kanR) and, via a ligation-independent, recombinase-specific reaction at the indicated att sites, recombine with a chloramphenicol resistant destination vector. The desired recombined construct carries the gene(s) of interest, retains kanR, and loses the chloramphenicol resistance (CmR) determinant.
Borrelial Gateway trans Complementation
Materials
PCR fragment of a gene of interest
Entry Vector pCR8 or one of many pENTR vectors (Invitrogen)
Borrelial destination shuttle vector (Table 2)
LR Clonase™ II (Invitrogen)
Proteinase K
ccdB survival T1R E. coli competent cells (Invitrogen)
DH5α competent cells
Table 2.
Plasmids for genetic manipulation of B. burgdorferi
| Plasmid | Selectiona | Description and Source |
|---|---|---|
| Shuttle vectors | ||
| pBSV2 | kanR | Shuttle vector containing three ORF’s from cp9 required for plasmid replication and distribution (Stewart et al., 2001). |
| pBBE22 | kanR | pBSV2 containing bbe22/pncA fragment to restore infectivity to strains lacking lp25 (Purser et al., 2003). |
| pBBE22gate | kanR | Modified pBBE22 containing Gateway destination vector attR1 and attR2 sites from Invitrogen’s pDEST17 (Weening et al., 2008). |
| pKFSS1 | strepR | Streptomycin resistant borrelial shuttle vector derived from pBSV2 (Frank et al., 2003). |
| pKFSS1gate | strepR | Modified pKFSS1 containing Gateway destination vector attR sites coned into the SphI site within the MCS (unpublished) |
| pCADDY | strepR | Shuttle vector derived from pBBE22; replacing the kanamycin resistance with streptomycin resistant gene (Hyde et al., 2009). |
| pCADDYgate | strepR | Derived from pBBE22gate (Weening et al., 2008); replacing the kanamycin resistance with streptomycin resistant determinant (unpublished) |
| pJD1 | strepR | pKFSS1 derivative with reduced PflgB-aadA region (Blevins et al., 2007). |
| pJD7 | strepR | pJD1 derivative with terminator inserted and Plac removed (Blevins et al., 2007). |
| pJD44 | kanR | pJD7 derived plasmid replaced specR/strepR with kanR (Blevins et al., 2007) |
| pBSV2G | gentR | Gentamicin resistant shuttle vector derived from pBSV2 (Elias et al., 2003). |
| pBSV2Ggate | gentR | Modified pBSV2G containing Gateway destination vector attR sites at SphI site in MCS (unpublished) |
| pBSV2SynG | courR | Shuttle vector derived from pBSV2 encoding a synthetic gyrB (Elias et al., 2003) |
| pCE310 | kanR | Shuttle vector with cp32 loci required for replication and distribution (Eggers et al., 2002). |
| pCB52 | kanR | Contains replicon and telomere ends from lp17; replicates as circular plasmids in E. coli and as a linear plasmid in B. burgdorferi (Beaurepaire and Chaconas, 2005). |
| pBSV17 | kanR | Shuttle vector derived from pBSV2 with the gene required for replication from lp17 (Beaurepaire and Chaconas, 2005). |
| pGK12 | ermR | Broad-host plasmid able to propagate in Gram-positive and Gram-negative bacteria including B. burgdorferi (Sartakova et al., 2003). |
| Allelic exchange vectors | ||
| pJH333 | gentR | A multicloning site and PflgB-gentR cassette flanked by DNA regions for allelic exchange into the borrelial chromosome between bb0445 and bb0446; based on the construct reported by Li et al., 2007 (unpublished) |
| pMC1667 | noneb | Suicide vector with multicloning site flanked by cp26 regions forallelic exchange (Dunham-Ems et al., 2009). |
| pMC1916 | gentR | Suicide vector derived from pMC1667 with PflgB-gentR and PflaB-gfp in divergent orientation are flanked by cp26 regions for allelic exchange (Dunham-Ems et al., 2009). |
| pSPC-G | gentR | Suicide vector with PflaB-gentR and PflaB-gfp in divergent orientation are flanked by cp26 regions for allelic exchange (Kenedy et al., 2009). |
Selection is in reference to antibiotic resistance in Borrelia burgdorferi only; antibiotic resistance for usage in E. coli is not indicated
No selectable marker is incorporated for usage in B. burgdorferi; plasmid does confer resistance to kanamycin and ampicillin for selection in E. coli
Transformed destination vectors, like pBBE22gate, must be maintained in ccdB survival T1R E. coli cells (Invitrogen).
Generate an entry vector by TOPO cloning a PCR fragment of a gene of interest into pCR8 or one of many pENTR vectors available from Invitrogen.
-
Combine 150 ng of the entry vector and destination vector into an eppendorf tube and incubate at 55°C for 10 minutes. Quickly chill the sample on ice.
Entry and destination vectors should not have the same selectable marker to prevent recovering cells with the entry vector and the newly recombined plasmid.
Add 2 µl of LR Clonase™ II enzyme mix to the sample. Briefly vortex and spin. Incubate for 1 hour at room temperature followed by an overnight incubation at 15°C.
Add 1µl of proteinase K (2 µg/µl) to end the reaction. Incubate at 37°C for 10 minutes.
Transform into DH5α E. coli strain. Select for positive transformants with antibiotic resistance encoded on the destination vector. Positive transformants can be screened by PCR and for sensitivity to chloramphenicol.
Purify plasmid from E. coli.
Sequence insert to make sure correct.
Transform B burgdorferi.
Support Protocol: DNA methylation of transformed DNA
The borrelial genome contains two restriction modification systems, bbe02 and bbq67, that inhibit the recovery of positive transformants. Methylation of DNA prior to transformation improves transformation efficiency of shuttle vectors in strains encoding bbq67 (Chen et al., 2008). In theory the methylation of suicide vectors should also have a increased transformation efficiency relative to unmethylated DNA. For reasons that are not entirely clear, it appears that DNA methylation does not overcome barriers imposed by bbe02 (Chen et al., 2008).
Materials
CpG Methyltransferase (M.SssI; New England Biolabs)
NEBbuffer 2 (New England Biolabs)
S-adenosylmethionine (SAM)
Phenol-chloroform-isoamyl alcohol
Add 1unit/µg M.SssI, 1X NEBbuffer 2 and 160µM S-adenosylmethionine (SAM) to the DNA for electroporation.
Incubate at 37°C for 4 hours.
Heat inactivate at 65°C for 20 minutes
Purify and isolate DNA by phenol-chloroform-isoamyl alcohol extraction
Resuspend precipicated at 5 µg/µl final concentration
Solutions
Barbour-Stoenner-Kelly (BSK) media
Materials
10X CMRL 1066 (without L-glutamine)
Neopeptone
Yeastolate
Glucose
HEPES
Citric Acid Trisodium Salt
Sodium Pyruvate
N-acetyl-D-glucosamine
Sodium bicarbonate
Bovine Serum Albumin (BSA) – Invitrogen
Normal Rabbit Serum (NRS) – Pel-Freez Biologicals, Inc.
5M NaOH
Rifampicin
Phosphomycin
Amphotericin B
Filter units (0.22 µm)
Recipe for 1 liter of BSK-II media:
| 9.82g | 10X CMRL 1066 (without L-glutamine) |
| 5.0g | Neopeptone |
| 2g | Yeastolate |
| 6g | HEPES |
| 5g | Glucose |
| 0.7g | Citric Acid Trisodium Salt |
| 0.8g | Sodium Pyruvate |
| 0.4g | N-acetyl-D-glucosamine |
| 2.2g | Sodium bicarbonate |
| 50g | BSA |
The recipe for BSK-H was developed by Pollack et al and is more cumbersome to produce in the laboratory relative to BSK-II media (Pollack et al., 1993). It can be purchased from Sigma.
- Mix components of BSK media into 500 ml of glass distilled H2O with the BSA added to the solution last. Allow solution to mix well until BSA has dissolved into solution.Several lots of BSA should be screened for optimal motility and growth rate. Dramatic differences can be observed by microscopic and growth curve analysis between different lots of BSA.
Adjust pH to 7.6 with 5M NaOH.
Add rifampicin (50 µg/ml final concentration), phosphomycin (100 µg/ml final concentration), and amphotericin B (5 µg/ml final concentration).
Incubate media in a 45°C water bath and allow media to equilibrate for approximately 1 hour to ensure the BSA is completely dissolved.
Filter media with a 0.22 µm filter unit.
Media can be stored at room temperature for 4–6 weeks and at 4°C for at least 1–2 years without diminished growth capacity (if frozen the media should be good indefinitely).
-
Prior to cultivation of B. burgdorferi add NRS to media in a 6% final concentration. This is referred to as complete BSK media (either BSK-II or BSK-H depending on the recipe used).
Note: 2X media can be made by reducing the total volume to 500 ml and used in semi-solid plating of B. burgdorferi (see Basic Protocol 3).
Dulbecco’s phosphate-buffered saline (dPBS
8 g NaCl
0.2 g KCl
1.15 g Na2HPO4
0.2 g KH2PO4
1L glass distilled water
sterilize by autoclaving under slow, liquid exhaust
Commentary
Background Information
The development of genetic tools and analyses in B. burgdorferi in recent years has provided the means to evaluate the roles of individual gene in borrelial physiology and pathogenesis. The construction and characterization of antibiotic resistance determinants that functioned in B. burgdorferi was key in the development of shuttle vectors and allelic exchange mutagenesis that revolutionized our ability to apply molecular Koch’s postulates to Lyme borreliosis (Rosa et al., 2005, 2010). This, coupled with the identification of restriction/modification systems, bbe02 and bbq67, respectively, that mapped to plasmids lp25 and lp56 in the B. burgdorferi strain B31 genome, resulted in the isolation of plasmid-less strains that lacked these enzymes and were more readily transformable (Lawrenz et al., 2002). Unfortunately, strains missing lp25 are non-infectious in mice unless the lp25-encoded nicotinamidase (bbe22 or pncA) is added back to these strains (Labandeira-Rey et al., 2003; Labandeira-Rey and Skare, 2001; Purser et al., 2003; Purser and Norris, 2000). In addition, a gene required for tick colonization, bbe16 or bptA, also maps to lp25 (Revel et al., 2005). Therefore, to complete the infectious lifecycle, both bbe22 (pncA) and bbe16 (bptA) are needed.
Based on these observations, one approach is to isolate a mutant in an lp25 deficient strain via allelic exchange and then complement back the bbe22 region to assess the role of the inactivated gene in Lyme borreliosis and, separately, together with intact version of the inactivated gene on a shuttle vector to genetically complement the mutant (Labandeira-Rey et al., 2003; Labandeira-Rey and Skare, 2001; Purser et al., 2003; Seshu et al., 2006). While effective, these strains cannot be used for tick studies unless the bptA gene can be added back. This limitation is addressed in the strain containing the bbe02::kanR mutation since it exhibits the high transformation frequency seen for the strains lacking lp25 and contains the other lp25 genes (Kawabata et al., 2004). Another alternative is to complement back the entire lp25 containing resistance to gentamicin or streptomycin (Gilbert et al., 2007; Grimm et al., 2004) after making a mutant in the lp25 deficient background and complementing the inactivating gene either in cis or in trans. In addition to the use of the aforementioned strains, other investigators have described additional infectious strains from strains B31, 297, and N40 that are competent for B. burgdorferi transformation (Rosa et al., 2010). Although transformable, it is not clear how these strains differ from wild type strain B31 in regard to their restriction/modification activity. Taken together, investigators have numerous antibiotic resistance markers and strains to utilize in order to mutagenize borrelial genes of interest.
There are several different ways that one can genetically alter B. burgdorferi. They include insertional disruption, randomized or “targeted” transposition, site-specific mutagenesis, and genetic deletion (Figure 2). A less widely used but potential effective way to inactivate a gene is to clone an incomplete and internal fragment of a gene of interest into a suicide vector containing a selectable and functional antibiotic resistance determinant in B. burgdorferi and to select for a single crossover event (Figure 2, panel A). Although single crossovers have been used successfully in B. burgdorferi (Stevenson et al., 1998), this approach is presumed to be less efficient than double crossover based modalities probably due to the smaller size of the homologous fragment required for the single crossover event (see below).
Figure 2.

Genetic Mutagenesis Schematic. In all of the panels, the white arrow represents the antibiotic resistance marker used for selection in B. burgdorferi. A. Single crossover mutants. A single crossover in the gene of interest occurs by homologous recombination, resulting in the incorporation of the entire suicide vector (shown above) including the antibiotic resistance determinant. Here, the suicide vector contains a truncated segment of the gene to be mutagenized (the middle gene) that mediates the single crossover event. B. Insertional inactivation. Generation of an insertional disruption mutation whereby recombination occurs between homologous sequences carried on the suicide vector (above) and the genetic target (again the middle gene). C. Deletion mutant. Double crossover requires homologous DNA regions flanking the gene of interest and generates a deletion mutation whereby allelic exchange replaces the target gene (middle gene) with an antibiotic resistant cassette. D. Site directed mutagenesis. In this case, a specific mutation is introduced in the gene of interest (middle gene), as indicated by a “gray box”, and is genetically linked to a selectable marker. As with the insertional inactivation and deletion mutagenesis, the flanking regions provided in the suicide vector (above) facilitates allelic exchange via double crossover recombination.
More commonly employed are insertional mutations that exploit pre-existing (if present) or engineered restriction enzyme sites within a gene of interest whereby one inserts an antibiotic resistance determinant that is expressed in B. burgdorferi (Figure 2, panel B). In this case, homologous sequences that are upstream and downstream of the disrupted gene (between 1–2 kb in length) are included to promote the desired double crossover event. A disadvantage of this experimental design is the potential (albeit rare) for the synthesis of truncated protein that exhibits reduced but detectable activity. Also, if the targeted gene is within an operon, polar effects may occur that could contribute to the phenotype(s) being tracked.
Similar to insertional mutants, transposon mutagenesis inserts an antibiotic cassette into DNA randomly and, when done in the conventional “global” manner, requires extensive characterization to map the insertions to an individual locus (Botkin et al., 2006; Lin et al., 2009; Rosa et al., 2010). Mutant libraries in both non-infectious and infectious B. burgdorferi have been constructed using this type of transposon-based mutagenesis (Botkin et al., 2006; Morozova et al., 2005; Stewart et al., 2004). Alternatively, one can target discrete regions for in vitro-based transposon mutagenesis to specifically target a specific genetic region (Akerley et al., 2002; Seshu et al., 2004). In the latter example, a fragment containing the region to be mutagenized is amplified by PCR and incubated with the transposon and transposable antibiotic resistance marker. Various transposon insertions are then mapped using molecular based techniques. As with insertional inactivation, this method can also result in truncated proteins and be subject to polarity issues.
To isolate a gene deletion, the gene in question is replaced with an antibiotic cassette. As with insertional inactivation, 1–2 kb of the upstream and downstream regions of the deleted gene are amplified by PCR and a antibiotic resistance determinant is cloned to replace the gene of interest (Figure 2, panel C). If the construct is designed such that orientation is defined, the antibiotic cassette can be placed in the same orientation of the deleted gene, generating a non-polar mutant (if relevant to the gene of interest). One potential limitation is the loss of sequences via the deletion event that unintentionally affect the expression of adjacent genes. This can be evaluated by transcript quantification of the linked genes, whereby the deletion mutant is compared to the parental strain.
Site-directed mutagenesis of the coding sequence in the gene of interest can be used to characterize specific functional domains within a given protein. Specifically, this method can be used to inactivate one function of a multi-function protein particularly when one function is absolutely required for viability. A specific example of this is the rrp2 locus that could not be deleted in B. burgdorferi (Yang et al., 2003). However, if the ATP binding domain was mutated (a single residue was mutagenized) the resulting Rrp2 protein could no longer direct the synthesis of B. burgdorferi RpoS, indicating that this domain is required for rrp2 activation (Yang et al., 2003). The complication of this approach is that a selectable marker must be genetically linked to the mutagenized gene, which may hinder expression of adjacent genes. Also, the crossover event must be carefully evaluated since it is possible that recombination events other than those needed to obtain the desired mutation may occur, thereby retaining the wild type allele (Figure 2, panel D).
Equally important to mutagenesis and, in some circumstances, more challenging is the complementation of mutant borrelial strains with the wild type allele. Complementation is absolutely essential to ensure that that the phenotype being tracked is due to the genetically modified gene and not the result of a second site mutation or lack of a borrelial plasmid. As with other bacteria, genetic complementation is commonly accomplished in trans on one of several available shuttle vectors containing an E. coli origin of replication and the region required for autonomous replication and cell partitioning from B. burgdorferi (Table 2) (Elias et al., 2003; Frank et al., 2003; Stewart et al., 2001). Specifically, shuttle vectors have been developed using the autonomous replication region from cp9, cp26, cp32, lp17, lp25, and lp28-1 (Table 2 and (Beaurepaire and Chaconas, 2005; Byram et al., 2004; Eggers et al., 2002; Elias et al., 2003; Frank et al., 2003; Stewart et al., 2003, 2001) and, in some studies, have been used to promote the loss of a targeted plasmid due to plasmid incompatibility (Grimm et al., 2004; Eggers et al., 2002). The recovery rate of positive transformants with an autonomous replicating plasmid is greater than of those requiring allelic exchange, but if there is no selective pressure to maintain the complemented gene, it is possible that the plasmid could be lost during the infectious process. An example of this type of selective pressure is seen for the pBBE22 shuttle vector, which contains the bbe22/pncA locus from lp25 (Table 2) (Purser et al., 2003). The pBBE22 plasmid is maintained in B. burgdorferi clones missing lp25 during the infection of mice since the nicotinamidase encoded by the bbe22/pncA gene is absolutely required for survival in the mammalian host.
An alternative method to generating trans complementation constructs is a recombinase-based system that does not require traditional restriction enzyme digestion and ligation. Currently, the most commonly used system is the Gateway™ system from Invitrogen. Our laboratory has modified existing borrelial shuttle vectors to contain attachment (att) sites to generate a new collection of borrelial shuttle/destination vectors (Figure 1 and Table 2). The cloned att sites promote homologous recombination with an entry vector that contains any gene(s) of interest when combined in the presence of the LR recombinase. Following transformation of E. coli with the resulting in vitro recombination mixture only cells containing the recombined fragment will survive. Gateway™-based complementation of a B. burgdorferi dbpBA deletion strain was recently reported, whereby intact dbpBA was delivered into a customized borrelial shuttle/destination vector, designated pBBE22gate (Table 2), confirming the utility of this approach (Weening et al., 2008). Since the Gateway™ recombinase-based approach requires no restriction digestion, the isolation of the desired recombinant construct is sequence independent (past the addition of att sites during the construction of the destination vector). As such, the isolation of the desired recombinant is not hindered by the absence of restriction sites, the presence of multiple restriction sites in the borrelial genomic fragment being cloned, or the lack of restriction sites available for cloning into preexisting borrelial shuttle vectors.
Recent publications suggest that shuttle vectors such as pBSV2 are present in multiple copies in B. burgdorferi. This may result in altered regulation or the overexpression of genes relative to their native genomic arrangement, which may contain important cis-acting elements. For these reasons, cis complementation might be preferred since the same upstream regulatory sequences would be retained. The limitation of this type of construct is the requirement for a linked antibiotic resistance marker (and distinct from the one used to initially mutagenize the gene of interest) that might alter the expression of genetically linked genes, particularly if the mutated gene is an internal gene in an operon. Alternatively, the intact complement gene can be introduced at a heterologous site within the genome. Examples of this include integration between the bb0445 and bb0446 genes in the borrelial chromosome (Li et al., 2007) and within a non-coding region in cp26 (Dunham-Ems et al., 2009; Kenedy et al., 2009).
Critical Parameters and Troubleshooting
Transformation
Improved efficiency of borrelial transformation over the years stems from the refinement of existing protocols, the isolation of strains with decreased ability to eliminate introduced DNA, and the development of antibiotic resistance determinants that function in B. burgdorferi (Bono et al., 2000; Elias et al., 2003; Frank et al., 2003; Lawrenz et al., 2002; Sartakova et al., 2000, 2003). The protocol for the electrotransformation of B. burgdorferi by Samuels is effectively unchanged and is widely used for the past 15 years (Samuels, 1995). The only difference in the protocol listed here is the substitution of a HEPES/sucrose buffer relative to the initially suggested phosphate buffered and glycerol solutions (Seshu et al., 2004). The electroporation conditions imposed for B. burgdorferi are similar to other bacterial organisms with the exception that extremely large amounts of DNA (micrograms) are used. This requirement can complicate transformation because the increase in DNA concentration may result in the unintended concomitant concentration of salt and subsequent arcing of the transformation mix. To avoid this, it is imperative that the DNA used be as pure (devoid of salt) and concentrated as is possible. Linearization of suicide vectors increases the likelihood of allelic exchange by double crossover in the genome. In vitro methylation of DNA can also improve transformation efficiency due to protection against the restriction of the transformed DNA (Chen et al., 2008).
Maintenance of Segmented Genome
The segmented genome of B. burgdorferi (21 plasmids; 12 linear and 9 circular, and a single linear chromosome for the prototypical strain B31) can be difficult to maintain in its entirety during in vitro cultivation in complete BSK media. BSK media (either BSK-II or commercially available BSK-H) is an undefined, nutrient rich media that supports in vitro growth. The rich nature of the BSK media often results in unintentional contamination; as such, careful sterile technique is critical. Since B. burgdorferi is a BSL-2 organism, work with this agent requires a type II A biosafety cabinet (BSC). In addition to maintaining proper BSL-2 practices, this level of containment is also important in reducing the exposure to unwanted contaminants.
Another important issue experienced by most borrelial researchers is the loss of plasmids following in vitro cultivation. To address this concern, plasmid content should be vigilantly monitored following long-term cultivation (passage) in vitro and genetic manipulation (after electrotransformation and selection in vitro). This issue is a by-product of the segmented borrelial genome, whereby daughter cells must receive the full complement of plasmids to retain their entire genetic compendium. It is likely that the loss of plasmids observed in some labs, and the increased rate of plasmid loss, is again due to subtle differences in cultivation methods between research groups. In summary, it is clear that many research labs are successful in cultivating and maintaining B. burgdorferi even though different practices are used between groups, as indicated in a previous Current Protocols by Zuckert (Zuckert, 2007). This is due, in all probability, to the fact that most of these differences have negligible effects. The intent here is to provide a perspective so that new investigators are cognizant of issues relevant to borrelial cultivation and transformation and can account for them accordingly.
Complex BSK media
It is possible that the reproducibility of experimentation between laboratories may be affected by a number of parameters that are associated with laboratory-specific differences in the cultivation of B. burgdorferi. One issue of note involves the source and composition of the BSK media used. As indicated herein, BSK media can be made on site, as either BSK-II or BSK-H media, or can be purchased commercially as BSK-H media. BSK media contains several undefined components, including bovine serum albumin (BSA), rabbit serum, neopeptone, and yeastolate. While these compounds are likely to be similar from different vendors, it is possible that compositional differences may exist that may affect the growth of an identical strain of B. burgdorferi between different research labs. For example, it is well established that B. burgdorferi does not synthesize its own fatty acids and is dependent on the media (or infected host) to provide these essential components so the borrelial cell can generate lipids. Because of this requirement, B. burgdorferi incorporates both saturated and polyunsaturated fatty acids present with the BSA and rabbit serum, as well as other components, provided in the BSK-II or BSK-H media. When making ones own media it is important to screen lots of BSA to find one that supports the growth of infectious B. burgdorferi. The exact reason for this screening step is not clear but may be due to different lots being rich in fatty acids that support the growth of the laboratory strain being tested.
It is also possible to culture adapt B. burgdorferi to different BSK media that does not initially support the growth of a particular strain. However one should use caution with this approach since it is possible that unintended mutations and/or plasmid loss (see below for more on plasmid composition) may occur during this process resulting in a strain with an altered phenotype. If adaptation is attempted, investigators are advised to test the strain for plasmid content and altered infectivity potential to determine if any overt (and unwanted) changes have been acquired during this process.
Time Considerations
The process of transforming B. burgdorferi is a time intensive process. First, it takes ample time to grow the large volume of cells needed for transformation. Next, following transformation, it takes the mutants between 2–3 weeks before putative transformants appear. Confirmation of the desired mutant via molecular techniques can take an additional 1–2 weeks depending on the approach being used. Once the mutant is confirmed, the process of transforming the genetic complement ensues, which, from start to finish, can take an additional 3–4 weeks. As such, although the ability to make mutants in B. burgdorferi represents an important advance, the process itself is still cumbersome. Regardless of these temporal limitations, transformants must be carefully screened and validated to confirm that antibiotic resistance (and desired mutation) are present and not the result of spontaneous secondary mutations that can afford antibiotic resistance to B. burgdorferi. It is ideal to verify PCR screening results by Southern or Western analysis (assuming in the latter case that an antibody reagent exists for the targeted gene product). Finally, once a mutation is confirmed, the plasmid profile should be determined to ensure that plasmids critical for tick and mammalian infections are present.
Supplementary Material
Acknowledgement
Work in the Skare laboratory is supported by the Public Health Service grants R01-AI042345 and R01-AI058086 from the National Institute of Allergy and Infectious Diseases, NIH.
Cited Literature
- Akerley BJ, Rubin EJ, Novick VL, Amaya K, Judson N, Mekalanos JJ. A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. PNAS U.S.A. 2002;99:966–971. doi: 10.1073/pnas.012602299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour AG. Isolation and cultivation of Lyme disease spirochetes. Yale J Biol Med. 1984;57:521–525. [PMC free article] [PubMed] [Google Scholar]
- Barthold SW, Beck DS, Hansen GM, Terwilliger GA, Moody KD. Lyme borreliosis in selected strains and ages of laboratory mice. J Infect Dis. 1990;162:133–138. doi: 10.1093/infdis/162.1.133. [DOI] [PubMed] [Google Scholar]
- Barthold SW, Moody KD, Terwilliger GA, Duray PH, Jacoby RO, Steere AC. Experimental Lyme arthritis in rats infected with Borrelia burgdorferi. J Infect Dis. 1988;157:842–846. doi: 10.1093/infdis/157.4.842. [DOI] [PubMed] [Google Scholar]
- Barthold Stephen W, Cadavid D, Philipp MT. Animal Models of Borreliosis. In: Samuels DS, Radolf JD, editors. Borrelia: Molecular Biology, Host Interaction and Pathogenesis. Norfolk, UK: Caister Academic Press; 2010. pp. 359–412. [Google Scholar]
- Beaurepaire C, Chaconas G. Mapping of essential replication functions of the linear plasmid lp17 of B. burgdorferi by targeted deletion walking. Molecular Microbiology. 2005;57:132–142. doi: 10.1111/j.1365-2958.2005.04688.x. [DOI] [PubMed] [Google Scholar]
- Blevins JS, Revel AT, Smith AH, Bachlani GN, Norgard MV. Adaptation of a luciferase gene reporter and lac expression system to Borrelia burgdorferi. Appl Environ Microbiol. 2007;73:1501–1513. doi: 10.1128/AEM.02454-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bono JL, Elias AF, Kupko JJ, Stevenson B, Tilly K, Rosa P. Efficient Targeted Mutagenesis in Borrelia burgdorferi. J Bacteriol. 2000;182:2445–2452. doi: 10.1128/jb.182.9.2445-2452.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botkin DJ, Abbott AN, Stewart PE, Rosa PA, Kawabata H, Watanabe H, Norris SJ. Identification of Potential Virulence Determinants by Himar1 Transposition of Infectious Borrelia burgdorferi B31. Infect. Immun. 2006;74:6690–6699. doi: 10.1128/IAI.00993-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylan JA, Lawrence KA, Downey JS, Gherardini FC. Borrelia burgdorferi membranes are the primary targets of reactive oxygen species. Molecular Microbiology. 2008;68:786–799. doi: 10.1111/j.1365-2958.2008.06204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, Davis JP. Lyme disease-a tick-borne spirochetosis? Science. 1982;216:1317–1319. doi: 10.1126/science.7043737. [DOI] [PubMed] [Google Scholar]
- Byram R, Stewart PE, Rosa P. The Essential Nature of the Ubiquitous 26-Kilobase Circular Replicon of Borrelia burgdorferi. J. Bacteriol. 2004;186:3561–3569. doi: 10.1128/JB.186.11.3561-3569.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caimano MJ, Eggers CH, Hazlett KR, Radolf JD. RpoS is not central to the general stress response in Borrelia burgdorferi but does control expression of one or more essential virulence determinants. Infect Immun. 2004;72:6433–6445. doi: 10.1128/IAI.72.11.6433-6445.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens S, van Vugt R, Tilly K, Rosa PA, Stevenson B. Homology throughout the multiple 32-kilobase circular plasmids present in Lyme disease spirochetes. J Bacteriol. 1997;179:217–227. doi: 10.1128/jb.179.1.217-227.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens S, Palmer N, van Vugt R, Huang WM, Stevenson B, Rosa P, Lathigra R, Sutton G, Peterson J, Dodson RJ, et al. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol. 2000;35:490–516. doi: 10.1046/j.1365-2958.2000.01698.x. [DOI] [PubMed] [Google Scholar]
- Chen Q, Fischer JR, Benoit VM, Dufour NP, Youderian P, Leong JM. In Vitro CpG Methylation Increases the Transformation Efficiency of Borrelia burgdorferi Strains Harboring the Endogenous Linear Plasmid lp56. J Bacteriol. 2008;190:7885–7891. doi: 10.1128/JB.00324-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham-Ems SM, Caimano MJ, Pal U, Wolgemuth CW, Eggers CH, Balic A, Radolf JD. Live imaging reveals a biphasic mode of dissemination of Borrelia burgdorferi within ticks. J Clin Invest. 2009;119:3652–3665. doi: 10.1172/JCI39401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers CH, Caimano MJ, Clawson ML, Miller WG, Samuels DS, Radolf JD. Identification of loci critical for replication and compatibility of a Borrelia burgdorferi cp32 plasmid and use of a cp32-based shuttle vector for the expression of fluorescent reporters in the Lyme disease spirochaete. Mol Microbiol. 2002;43:281–295. doi: 10.1046/j.1365-2958.2002.02758.x. [DOI] [PubMed] [Google Scholar]
- Elias AF, Bono JL, Kupko JJ, Stewart PE, Krum JG, Rosa PA. New antibiotic resistance cassettes suitable for genetic studies in Borrelia burgdorferi. J Mol Microbiol Biotechnol. 2003;6:29–40. doi: 10.1159/000073406. [DOI] [PubMed] [Google Scholar]
- Elias AF, Stewart PE, Grimm D, Caimano MJ, Eggers CH, Tilly K, Bono JL, Akins DR, Radolf JD, Schwan TG, et al. Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect Immun. 2002;70:2139–2150. doi: 10.1128/IAI.70.4.2139-2150.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KL, Bundle SF, Kresge ME, Eggers CH, Samuels DS. aadA confers streptomycin resistance in Borrelia burgdorferi. J Bacteriol. 2003;185:6723–6727. doi: 10.1128/JB.185.22.6723-6727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- Gherardini FC, Boylan JA, Lawrence K, Skare JT. Metabolism and Physiology of Borrelia. In: Samuels DS, Radolf JD, editors. Borrelia: Molecular Biology, Host Interaction and Pathogenesis. Norfolk, UK: Caister Academic Press; 2010. pp. 103–138. [Google Scholar]
- Gilbert MA, Morton EA, Bundle SF, Samuels DS. Artificial regulation of ospC expression in Borrelia burgdorferi. Mol Microbiol. 2007;63:1259–1273. doi: 10.1111/j.1365-2958.2007.05593.x. [DOI] [PubMed] [Google Scholar]
- Grimm D, Eggers CH, Caimano MJ, Tilly K, Stewart PE, Elias AF, Radolf JD, Rosa PA. Experimental assessment of the roles of linear plasmids lp25 and lp28-1 of Borrelia burgdorferi throughout the infectious cycle. Infect Immun. 2004;72:5938–5946. doi: 10.1128/IAI.72.10.5938-5946.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde JA, Shaw DK, III, R S, Trzeciakowski JP, Skare JT. The BosR regulatory protein of Borrelia burgdorferi interfaces with the RpoS regulatory pathway and modulates both the oxidative stress response and pathogenic properties of the Lyme disease spirochete. Molecular Microbiology. 2009;74:1344–1355. doi: 10.1111/j.1365-2958.2009.06951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata H, Norris SJ, Watanabe H. BBE02 disruption mutants of Borrelia burgdorferi B31 have a highly transformable, infectious phenotype. Infect Immun. 2004;72:7147–7154. doi: 10.1128/IAI.72.12.7147-7154.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenedy MR, Vuppala SR, Siegel C, Kraiczy P, Akins DR. CspA-Mediated Binding of Human Factor H Inhibits Complement Deposition and Confers Serum Resistance in Borrelia burgdorferi. Infect Immun. 2009;77:2773–2782. doi: 10.1128/IAI.00318-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labandeira-Rey M, Seshu J, Skare JT. The absence of linear plasmid 25 or 28-1 of Borrelia burgdorferi dramatically alters the kinetics of experimental infection via distinct mechanisms. Infect Immun. 2003;71:4608–4613. doi: 10.1128/IAI.71.8.4608-4613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labandeira-Rey M, Skare JT. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect Immun. 2001;69:446–455. doi: 10.1128/IAI.69.1.446-455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrenz MB, Kawabata H, Purser JE, Norris SJ. Decreased electroporation efficiency in Borrelia burgdorferi containing linear plasmids lp25 and lp56: impact on transformation of infectious B. burgdorferi. Infect Immun. 2002;70:4798–4804. doi: 10.1128/IAI.70.9.4798-4804.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Pal U, Ramamoorthi N, Liu X, Desrosiers DC, Eggers CH, Anderson JF, Radolf JD, Fikrig E. The Lyme disease agent Borrelia burgdorferi requires BB0690, a Dps homologue, to persist within ticks. Mol Microbiol. 2007;63:694–710. doi: 10.1111/j.1365-2958.2006.05550.x. [DOI] [PubMed] [Google Scholar]
- Lin T, Gao L, Edmondson DG, Jacobs MB, Philipp MT, Norris SJ. Central Role of the Holliday Junction Helicase RuvAB in vlsE Recombination and Infectivity of Borrelia burgdorferi. PLoS Pathog. 2009;5 doi: 10.1371/journal.ppat.1000679. e1000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova OV, Dubytska LP, Ivanova LB, Moreno CX, Bryksin AV, Sartakova ML, Dobrikova EY, Godfrey HP, Cabello FC. Genetic and physiological characterization of 23S rRNA and ftsJ mutants of Borrelia burgdorferi isolated by mariner transposition. Gene. 2005;357:63–72. doi: 10.1016/j.gene.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Nadelman RB, Wormser GP. Lyme borreliosis. Lancet. 1998;352:557–565. doi: 10.1016/S0140-6736(98)01146-5. [DOI] [PubMed] [Google Scholar]
- Norris SJ, Coburn J, Leong JM, Hu LT, Hook M. Pathobiology of Lyme Disease Borrelia. In: Samuels DS, Radolf JD, editors. Borrelia: Molecular Biology, Host Interaction and Pathogenesis. Norfolk, UK: Caister Academic Press; 2010. pp. 299–332. [Google Scholar]
- Pal U, Fikrig Erol. Tick Interactions. In: Samuels DS, Radolf JD, editors. Borrelia: Molecular Biology, Host Interaction and Pathogenesis. Norfolk, UK: Caister Academic Press; 2010. pp. 279–298. [Google Scholar]
- Parveen N, Cornell KA, Bono JL, Chamberland C, Rosa P, Leong JM. Bgp, a secreted glycosaminoglycan-binding protein of Borrelia burgdorferi strain N40, displays nucleosidase activity and is not essential for infection of immunodeficient mice. Infect Immun. 2006;74:3016–3020. doi: 10.1128/IAI.74.5.3016-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piesman J, Mather TN, Sinsky RJ, Spielman A. Duration of tick attachment and Borrelia burgdorferi transmission. J. Clin. Microbiol. 1987;25:557–558. doi: 10.1128/jcm.25.3.557-558.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack RJ, Telford SR, Spielman A. Standardization of medium for culturing Lyme disease spirochetes. J Clin Microbiol. 1993;31:1251–1255. doi: 10.1128/jcm.31.5.1251-1255.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purser JE, Lawrenz MB, Caimano MJ, Howell JK, Radolf JD, Norris SJ. A plasmid-encoded nicotinamidase (PncA) is essential for infectivity of Borrelia burgdorferi in a mammalian host. Mol Microbiol. 2003;48:753–764. doi: 10.1046/j.1365-2958.2003.03452.x. [DOI] [PubMed] [Google Scholar]
- Purser JE, Norris SJ. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2000;97:13865–13870. doi: 10.1073/pnas.97.25.13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revel AT, Blevins JS, Almazan C, Neil L, Kocan KM, de la Fuente J, Hagman KE, Norgard MV. bptA (bbe16) is essential for the persistence of the Lyme disease spirochete, Borrelia burgdorferi, in its natural tick vector. Proc Natl Acad Sci U S A. 2005;102:6972–6977. doi: 10.1073/pnas.0502565102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa PA, Tilly K, Stewart PE. The burgeoning molecular genetics of the Lyme disease spirochaete. Nat Rev Microbiol. 2005;3:129–143. doi: 10.1038/nrmicro1086. [DOI] [PubMed] [Google Scholar]
- Rosa PA, Cabello FC, Samuels DS. Genetic Manipulation of Borrelia burgdorferi. In: Samuels DS, Radolf JD, editors. Borrelia: Molecular Biology, Host Interaction and Pathogenesis. Norfolk, UK: Caister Academic Press; 2010. pp. 189–220. [Google Scholar]
- Samuels DS. Electrotransformation of the spirochete Borrelia burgdorferi. 1995 doi: 10.1385/0-89603-310-4:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartakova M, Dobrikova E, Cabello FC. Development of an extrachromosomal cloning vector system for use in Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2000;97:4850–4855. doi: 10.1073/pnas.080068797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartakova ML, Dobrikova EY, Terekhova DA, Devis R, Bugrysheva JV, Morozova OV, Godfrey HP, Cabello FC. Novel antibiotic-resistance markers in pGK12-derived vectors for Borrelia burgdorferi. Gene. 2003;303:131–137. doi: 10.1016/s0378-1119(02)01146-0. [DOI] [PubMed] [Google Scholar]
- Schwan TG, Burgdorfer W, Schrumpf ME, Karstens RH. The urinary bladder, a consistent source of Borrelia burgdorferi in experimentally infected white-footed mice (Peromyscus leucopus) J Clin Microbiol. 1988;26:893–895. doi: 10.1128/jcm.26.5.893-895.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshu J, Boylan JA, Hyde JA, Swingle KL, Gherardini FC, Skare JT. A conservative amino acid change alters the function of BosR, the redox regulator of Borrelia burgdorferi. Mol Microbiol. 2004;54:1352–1363. doi: 10.1111/j.1365-2958.2004.04352.x. [DOI] [PubMed] [Google Scholar]
- Seshu J, Esteve-Gassent MD, Labandeira-Rey M, Kim JH, Trzeciakowski JP, Hook M, Skare JT. Inactivation of the fibronectin-binding adhesin gene bbk32 significantly attenuates the infectivity potential of Borrelia burgdorferi. Mol Microbiol. 2006;59:1591–1601. doi: 10.1111/j.1365-2958.2005.05042.x. [DOI] [PubMed] [Google Scholar]
- Skare JT, Carroll JA, Yang XF, Samuels DS, Akins DR. Gene Regulation, Transcriptomics and Proteomics. In: Samuels DS, Radolf JD, editors. Borrelia: Molecular Biology, Host Interaction and Pathogenesis. Norfolk, UK: Caister Academic Press; 2010. pp. 67–102. [Google Scholar]
- Steere AC, Coburn J, Glickstein L. The emergence of Lyme disease. J Clin Invest. 2004;113:1093–1101. doi: 10.1172/JCI21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AC, Grodzicki RL, Kornblatt AN, Craft JE, Barbour AG, Burgdorfer W, Schmid GP, Johnson E, Malawista SE. The spirochetal etiology of Lyme disease. New England Journal of Medicine. 1983;308:733–740. doi: 10.1056/NEJM198303313081301. [DOI] [PubMed] [Google Scholar]
- Stevenson B, Bono JL, Elias A, Tilly K, Rosa P. Transformation of the Lyme disease spirochete Borrelia burgdorferi with heterologous DNA. J Bacteriol. 1998;180:4850–4855. doi: 10.1128/jb.180.18.4850-4855.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PE, Chaconas G, Rosa P. Conservation of plasmid maintenance functions between linear and circular plasmids in Borrelia burgdorferi. J Bacteriol. 2003;185:3202–3209. doi: 10.1128/JB.185.10.3202-3209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PE, Hoff J, Fischer E, Krum JG, Rosa PA. Genome-wide transposon mutagenesis of Borrelia burgdorferi for identification of phenotypic mutants. Appl Environ Microbiol. 2004;70:5973–5979. doi: 10.1128/AEM.70.10.5973-5979.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PE, Thalken R, Bono JL, Rosa P. Isolation of a circular plasmid region sufficient for autonomous replication and transformation of infectious Borrelia burgdorferi. Mol Microbiol. 2001;39:714–721. doi: 10.1046/j.1365-2958.2001.02256.x. [DOI] [PubMed] [Google Scholar]
- Weening EH, Parveen N, Trzeciakowski JP, Leong JM, Hook M, Skare JT. Borrelia burgdorferi lacking DbpBA exhibits an early survival defect during experimental infection. Infection and immunity. 2008;76:5694–5705. doi: 10.1128/IAI.00690-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XF, Alani SM, Norgard MV. The response regulator Rrp2 is essential for the expression of major membrane lipoproteins in Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2003;100:11001–11006. doi: 10.1073/pnas.1834315100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XF, Pal U, Alani SM, Fikrig E, Norgard MV. Essential Role for OspA/B in the Life Cycle of the Lyme Disease Spirochete. J Exp Med. 2004;199:641–648. doi: 10.1084/jem.20031960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckert WR. Current Protocols in Microbiology. John Wiley & Sons, Inc.; 2007. Laboratory Maintenance of Borrelia burgdorferi; pp. 12C.1.1–12C.1.10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.