

Abstract
Members of the epidermal growth factor receptor family (EGFR/Erb1, Erb2/HER2, ErbB3/HER3 and ErbB4/HER4) are key targets for inhibition in cancer therapy1. Critical for activation is the formation of an asymmetric dimer by the intracellular kinase domains, in which the C-terminal lobe (C-lobe) of one kinase domain induces an active conformation in the other2. The cytoplasmic protein Mig6 (Mitogen-induced gene 6) interacts with and inhibits the kinase domains of EGFR and ErbB23–5. Crystal structures of complexes between the EGFR kinase domain and a fragment of Mig6 show that a ~25-residue epitope (segment 1) from Mig6 binds to the distal surface of the C-lobe of the kinase domain. Biochemical and cell-based analyses confirm that this interaction contributes to EGFR inhibition by blocking the formation of the activating dimer interface. A longer Mig6 peptide that is extended C-terminal to segment 1 has increased potency as an inhibitor of the activated EGFR kinase domain, while retaining a critical dependence on segment 1. We show that signaling by EGFR molecules that contain constitutively active kinase domains still requires formation of the asymmetric dimer, underscoring the importance of dimer interface blockage in Mig6-mediated inhibition.
Prior to activation, the EGFR kinase domain is in an autoinhibited conformation which resembles that of inactive cyclin-dependent kinases (CDKs) and the Src family kinases2,6. Conversion to the active form requires interactions between the distal surface of the C-lobe of one kinase domain and the N-terminal lobe (N-lobe) of the other in the asymmetric activating dimer2. This conformational change resembles closely the activation switch induced in CDKs by cyclins7, even though the C-lobe of the EGFR kinase domain is structurally unrelated to cyclins.
If the cyclin/CDK-like asymmetric dimer is indeed critical for EGFR activation, then the modulation of this interaction might underlie naturally occurring mechanisms of EGFR regulation. We looked for protein inhibitors of EGFR that are known to function by interacting with the intracellular portions of the receptor. One such protein is Mig6 (or receptor associated late transducer, RALT, the gene for which is also named gene 33), which is a feedback inhibitor of both EGFR and ErbB23,5. Mig6 inhibits EGFR-mediated signals in mouse skin8, and deletion of the Mig6 gene leads to hyper-activation of EGFR 9,10.
The N-terminal region of Mig6 is not implicated in EGFR inhibition (Fig. 1a). The C-terminal region shows sequence similarity only to a non-catalytic region of the ACK1 tyrosine kinase (Fig 1a), which also binds to the EGFR cytoplasmic domain11. A segment within this region of Mig6 (residues 323–372) is critical for ErbB2 and EGFR binding (Fig. 1a)12,13. We determined the crystal structure of a 60-residue fragment spanning this segment (residues 315–374) bound to the EGFR kinase domain (Supplemental Material). This structure and structures of EGFR complexed to two overlapping 40- and 25-residue fragments (residues 325–364 and 340–364) define a 25-residue epitope of Mig6 that is sufficient for binding to the EGFR kinase domain (residues 337–361, denoted Mig6segment 1). The structure of the 40-residue peptide complex has been determined at 2.9 Å resolution.
Figure 1. Structure of the EGFR kinase domain/Mig6segment 1.

a, Schematic diagram of human Mig6 primary structure. Regions of interest, including the previously defined EGFR/ErbB2 binding region4,5,12, are boxed and labeled. b, Two orthogonal views of the EGFR kinase domain/Mig6segment 1 complex. A channel which peptide inhibitors of some other kinases are docked is indicated15,16. The electron density around Mig6segment 1 in the right panel is contoured at 3σ and is from a simulated annealing omit map with coefficients (|Fo|-|Fc|)eiαC, where the calculated structure factors are generated from a model that does not contain Mig6. c, Detailed view of the interface between the EGFR kinase domain and Mig6segment 1. Hydrogen bonds are represented by dashed lines. d, Comparison of the Mig6segment 1 binding interface and the kinase domain asymmetric dimer interface on the distal surface of the kinase C-lobe. A large portion of the surface is shared by the two interfaces (outlined), and it is clear that binding of the EGFR kinase domain by Mig6segment 1 would block the formation of the asymmetric activating dimer. (c) and (d) are in similar orientations as that in the right panel of (b).
The EGFR kinase domain bound to Mig6segment 1 adopts the Src/CDK-like inactive conformation, and not the active conformation normally seen in crystals of the kinase domain (Fig. 1b)2,6. The interface, which buries 1800 Å2 of surface area, involves an extended conformation of the Mig6 peptide and disparate binding elements on the kinase domain (Fig. 1b and c; Supplemental Material). Mig6segment 1 lies within a shallow depression on the distal surface of the C-lobe of the kinase domain, formed by helices αG and αH and the loops connecting helices αF-αG, αG-αH and αH-αI. The interations are mainly polar, although a few hydrophobic residues from helix αH contribute to the interface.
The footprint of Mig6segment 1 on the kinase domain overlaps the cyclin-like face of the kinase domain in the asymmetric kinase domain dimer and so binding of Mig6 to an EGFR kinase domain will prevent it from acting as a cyclin-like activator for other kinase domains (Fig. 1 and 4d). Residues in EGFR located at the Mig6segment 1 binding interface are conserved2, suggesting that Mig6 will also bind to other EGFR family members.
Figure 4. A double-headed mechanism for EGFR inhibition by Mig6.

a, Co-transfection experiment showing that EGFRactivator can activate EGFRactivatable, and that Mig6 can inhibit this activation. b, Co-transfection experiments showing that full-length EGFR containing the L834R/V924R double mutation only shows autophosphorylation when co-transfected with EGFRactivator. Co-transfection combinations in (a) and (b) are represented by the cartoons in the respective lower panels for clarity. The I682Q, D813N, L834R and V924R mutations are denoted in the cartoons by a circle, diamond, star and triangle, respectively. c, a schematic diagram showing the double-headed mechanism for EGFR inhibition by Mig6 involving both segment 1 and segment 2.
Mig6segment 1 binds to the EGFR kinase domain with micromolar affinity. The dissociation constant for a 30-residue fluorescein-labeled Mig6 peptide (residues 334–363, spanning the entire binding epitope of segment 1) is 13.0+/−1.3 μM (Fig. 2a and Supplemental Table 1). Val924 in the C-lobe of the kinase domain is located in the center of the asymmetric kinase domain dimer interface and also participates in the interaction between the kinase domain and Mig6segment 1 (Fig. 1b and c)2. A V924R mutation in the kinase domain abolishes peptide binding (Fig. 2a). Met346, Phe352 and Tyr358 in Mig6 are within the kinase/Mig6segment 1 interface (Fig. 1c), and mutation of any of these residues also abrogates binding (Fig. 2b).
Figure 2. Binding and inhibition of EGFR by Mig6segment 1.

a, Titrations of the wild type EGFR kinase domain and the V924R and I682Q mutants to the 30-residue (residues 334–363) fluorescein-labeled Mig6 peptide. b, Titrations of the wild type EGFR kinase domain to the wild type and three mutant 30-residue fluorescein-labeled peptides. n.d. denotes “not determined” (KD values cannot be determined reliably). c, Inhibition of the activity of the EGFR kinase domain by peptides spanning Mig6segment 1 in the vesicle-based kinase assay. The 60-, 40- and 30-residue peptides contain the entire binding epitope of segment 1, while the 25-residue peptide lacks the N-terminal 3 residues. The mutations were introduced in the 30-residue peptide. See Supplemental Table 1 for the residue boundaries. Fl: fluorescein. d, Cell-based assay showing that Mig6 inhibits full-length EGFR autophosphorylation, whereas mutations in segment 1 abolishes the inhibition.
The EGFR kinase domain has very low activity in solution, but is activated upon increasing its local concentration by tethering it to lipid vesicles, which promotes the formation of the asymmetric dimer2. Various Mig6 peptides which contain segment 1 inhibit the activity of the kinase domain attached to lipid vesicles, with IC50 values of ~10 μM (Fig. 2c). A 25-residue peptide (residues 340–364), which lacks 3 residues in the N-terminal portion of Mig6segment 1, is much less potent (Fig. 2c and Supplemental Material). Peptides that contain mutations which disrupt the binding interface (M346A, F352A and Y358A) do not inhibit kinase activity significantly. An EGFR kinase domain bearing an I682Q mutation is not stimulated by concentration at the membrane because it is unable to form the asymmetric dimer2. The basal activity of this mutant in solution is not inhibited by Mig6segment 1, which has the same binding affinity for this mutation as for the wild type kinase domain (Fig. 2a and Supplemental Figure 3). Thus, Mig6segment 1 is only able to inhibit the kinase domain in the context of asymmetric dimer formation.
We tested the inhibition of EGFR autophosphorylation by full length Mig6 in a cell-based assay. Co-expression of the wild type Mig6 with EGFR decreases the EGF-induced autophosphorylation of EGFR, whereas individual introduction of mutations in Mig6segment 1 (M346A, F352A or Y358A) abolishes this effect (Fig. 2d), confirming that segment 1 is important for inhibition of EGFR by full length Mig6.
An intriguing property of Mig6 is its ability to bind more tightly to activated EGFR than to the unliganded receptor3,5,12. Mig6segment 1 alone cannot confer this property, because the kinase residues that interact with it do not change conformation upon activation2,6,14. The C-terminus of Mig6segment 1 is located within a channel leading into the kinase active site (Fig. 1b), utilized by peptidic inhibitors of protein kinases that interact directly with the active sites15,16. The region of Mig6 that is C-terminal to segment 1 (segment 2, Fig. 1a) contains a region of strong homology to ACK13,5,11. Since Mig6 and ACK1 are both sensitive to the activation state of EGFR3,5,11,12, there may be specific interactions between segment 2 and the activation loop and/or the N-lobe of the kinase domain.
To test the role of segment 2, we produced a longer peptide (residues 336–412, Mig6segment 1–2), and analyzed its effect on a variant of the EGFR kinase domain that contains a mutation (L834R) that renders it constitutively active in the absence of concentration on vesicles2. Mig6segment 1–2 inhibits this mutant kinase domain with an IC50 value of ~200 nM (Fig. 3a). Mig6segment 1–2 bearing a mutation within segment 1 (Y358A) inhibited L834R much less efficiently (IC50 ~5 μM). Mig6segment 1 (the 30-residue peptide) did not inhibit this mutant kinase, consistent with its dimerization-independent activity. Interestingly, Mig6segment 1–2 appears to be much less potent in inhibiting the basal activity of the wild type kinase domain in solution, and Mig6segment 1–2 bearing a mutation in segment 1 (Y358A) does not show any inhibition under the same conditions (Fig. 3b). These results suggest that segment 2 is responsible for the inhibition of the activated EGFR kinase domain, and that both segments 1 and 2 are important for the high potency of inhibition.
Figure 3. Inhibition of the EGFR kinase activity by Mig6segment 1–2.
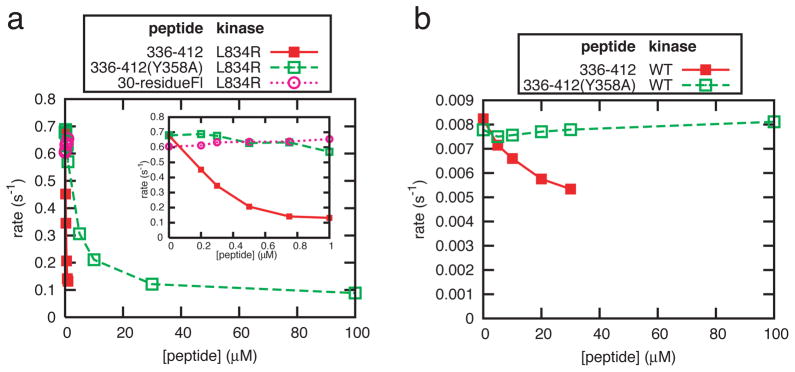
a, Inhibition of the L834R mutant kinase in solution by peptides 336–412, 336–412(Y358A) (containing both segment 1 and 2). The 30-residue peptide (containing segment 1 only) is used as a control. The insert shows an expanded view at low peptide concentrations. b, Inhibition of the wild type kinase in solution by peptides 336–412 and 336–412(Y358A). Titration of peptide 336–412 beyond 20 μM leads to unreliable results due to precipitation of the protein and peptide (See Method).
Could Mig6 function by binding primarily to the activated kinase in an asymmetric kinase domain dimer, and not to the cyclin-like activator kinase? The Mig6segment 1 interaction would then be important for anchorage of Mig6 to EGFR, but not directly relevant for shutting down kinase activity. Such a role may be operative in autoinhibition of ACK1, the kinase domain of which has a conserved segment 1 binding surface, with the Mig6 homologous segments present within the same protein (Supplemental Material). We expect, however, that the asymmetric EGFR dimer will dissociate, and that activated kinase molecules can subsequently serve as cyclin-like activators. This may facilitate the lateral propagation of EGFR activation, which can spread across the cell surface even when EGF is localized to a small region17,18. The interaction between Mig6segment 1 and the kinase domain would block further transmission of the activating signal.
To exam this potential, we co-transfected cells with two variants of EGFR. One form (EGFRactivator) resembles ErbB3 in that it is catalytically inactive (the catalytic base, Asp 813, is mutated to Asn) but can serve as a cyclin-like activator. To promote its interaction with Mig6 we introduced the L834R mutation, which destabilizes the inactive conformation, into EGFRactivator. To prevent EGFRactivator from assuming the “activated” position in the asymmetric dimer we also introduced the I682Q mutation2. The second EGFR variant (EGFRactivatable) is catalytically active, but has the V924R mutation, which prevents it from serving as an activator2. We tested the effects of Mig6 on EGFR phosphorylation in co-transfections with these two variants. The results show that EGFRactivator can activate EGFRactivatable in the presence of EGF (Fig. 4a), consistent with our previous findings2. Co-transfection of Mig6 with EGFRactivator and EGFRactivatable suppresses this activation (Fig. 4a). Mig6segment 1 does not bind to the kinase domain bearing the V924R mutation, and an intact Mig6segment 1 is required for inhibition of EGFR in cellular assays (Fig. 2). We therefore interpret the results of the triple transfection experiment (Fig. 4a) to mean that Mig6 binds to EGFRactivator and prevents the activation of EGFRactivatable.
Full-length EGFR bearing the activating L834R mutation is not fully phosphorylated in cells19,20, suggesting that the formation of the asymmetric dimer is still required for robust autophosphorylation even when the kinase domain is rendered constitutively active. We confirmed this by introducing the V924R mutation, which prevents the kinase domain from serving as the cyclin-like activator, into EGFR with a constitutively active kinase domain (L834R/V924R). EGFR(L834R/V924R) fails to undergo autophosphorylation (Fig. 4b), although the kinase activity of this double mutant is comparable to that of the kinase domain bearing the single activating mutation (L834R) (Supplemental Figure 6). EGF-stimulated autophosphorylation is restored when this double mutant is co-transfected with the kinase-dead EGFRactivator (Fig. 4b). These results further underscore the importance of blockage of the asymmetric dimer interface by Mig6, as it can prevent both the activation of kinase domains and downstream signaling by activated kinase domains.
Our results demonstrate that Mig6 uses a double-headed mechanism for inhibiting EGFR, with the blockage of the asymmetric cyclin/CDK-like dimer being a particularly interesting aspect of the inhibition (Fig. 4c). This mechanism provides direct confirmation for the critical role of the asymmetric kinase domain dimer in the activation of EGFR family receptors. In addition, our results suggest an approach for the development of a new class of inhibitors that act by binding to the cyclin-like face of the C-lobe of the kinase domains of this family. This region is not conserved in other protein kinases, and so such inhibitors may enable the development of cancer therapies with a high degree of specificity towards EGFR family members.
Methods
Peptide preparation
All the Mig6 peptides used in this study are listed in Supplemental Table 1. The 60-residue peptide was expressed as a GST-fusion in Escherichia coli BL21 (DE3) by using pGEX6p1 (Amersham) (BamHI/XhoI) and purified using a glutathione sepharose column. The protein was treated with the PreScission protease to release the Mig6 peptide, which was further purified using a Hitrap SP column (Amersham). The longer peptides (336–412 and 336–412(Y358A)) were cloned as Trp ΔLE fusions and expressed as inclusion bodies as described previously21. To prevent cleavage of the Mig6 peptides by subsequent cyanogen bromide treatment the single methionine in these peptides (M346) was mutated to leucine. This mutation does not affect the binding to the EGFR kinase domain significantly (Supplemental Fig. 4). The fusion proteins were cleaved with cyanogen bromide and the released Mig6 peptides were purified. All other Mig6 peptides were synthesized using solid phase peptide synthesis via the Fmoc strategy with Wang resin on a Protein Technologies PS3 synthesizer. The peptide identities were confirmed by mass spectrometry.
Structure determination
The EGFR kinase domain constructs used are identical to those used previously2, except for the K799E mutant, which includes six fewer residues at the N-terminus (spanning residues 678–998 of EGFR), and proteins were purified as described2. The wild type kinase domain was first co-crystallized with the 60-residue Mig6 peptide and the structure was determined at 3.5 Å resolution. This revealed that a ~25-residue segment of the peptide is bound to the distal surface of the C-lobe of the EGFR kinase domain and that the rest of the peptide is disordered (Supplemental Fig. 1a). A 25-residue peptide (residues 340–364 in Mig6) was designed based on the initial structure and cocrystallized with both the wild type and a mutant (K799E) form of the EGFR kinase domain. The K799E mutation does not affect the conformation of the kinase domain or its interaction with Mig6segment 1 (Supplemental Figure 5), but crystals of this mutant kinase domain in complex with the peptide diffracted X-rays to higher resolution. The structure shows that this 25-residue peptide lacks the N-terminal part of the kinase binding epitope (Supplemental Figure 1b). This peptide was then extended to include residues 325–364 in Mig6 (the 40-residue peptide) and co-crystallized with the EGFR (K799E) kinase domain. The structure of this peptide/kinase domain complex was determined at 2.9 Å. There are four kinase domains in the asymmetric unit, all of which adopt the same conformation. Two of the four kinase domains are bound to the Mig6 peptide, and the Mig6 binding surfaces of the other two are occupied by crystal contacts. Crystallization conditions, data collection and structural refinement statistics are summarized in Supplemental Table 2.
Binding assays
Fluorescein-labeled 30-residue wild type, M346L, M346A, F352A and Y358A Mig-6 peptides were diluted to final concentrations of 5, 8, 3.1, 3.5 and 2.7 μM in a buffer containing 10 mM Tris, 50 mM NaCl and 2 mM DTT, pH 7.5. These peptides in the cuvette were then titrated with the wild type or mutant forms of the EGFR kinase domain at 20 °C. For the competition assays, the labeled 30mer wild type peptide (5 μM) and kinase domain (60 μM) were mixed and titrated with unlabeled competitor peptides. The fluorescence anisotropy at each titration step was monitored. The I682Q and K799E mutant kinases used in the binding assays contain the N-terminal 6xHis tag and linker fragment before the kinase domain, whereas this N-terminal fragment in the wild type and V924R mutant kinases was removed by Tobacco Etch Virus protease treatment.
Kinase assays
The assays were conducted as described previously2. The substrate peptide was kept at 1 mM in all the experiments. The reported rates are the initial velocities normalized by the kinase concentrations. The wild type kinase concentrations in the vesicle-based and solution-based assays were 3.5 and 14 μM respectively. Preliminary experiments showed that peptide 336–412 (Mig6segment 1–2) inhibited the L834R mutant kinase much more strongly and also caused precipitation when both the kinase and the peptide were at high concentrations. We therefore reduced the concentration of L834R in the assays to 200 nM. The higher intrinsic activity of this mutant and usage of MnCl2 at 10 mM instead of MgCl2 allowed us to measure kinase activity at such a low kinase concentration22.
Cell-based assays
Cos-7 cells were co-transfected using Fugene 6 (Roche) with the N-terminal FLAG-tagged EGFR-pcDNA3.1 constructs (as used before2) and the wild type or mutants of the Mig6 genes with a C-terminal Myc tag (also in pcDNA3.1). Cells were cultured for 36 hours after transfection and serum-starved for 12 hours. Cells were treated with EGF (50 ng/ml) for ~5 minutes at 37 °C, lysed and subjected to Western blot analyses. The levels of total EGFR, EGFR autophosphorylation and Mig6 were probed using the anti-EGFR antibody SC03 (Santa Cruz), anti-phosphotyrosine antibody 4G10 (Upstate) and an anti-Myc antibody (Cell Signaling), respectively.
Acknowledgments
We thank Xiaoxian Cao and Ann Fisher for cell culture support; David King for mass spectrometry; the staff at beamlines 8.2.1 and 12.3.1 of the Advanced Light Source for technical support; Markus Seeliger, Sebastian Deindl, Patricia Pellicena, Jodi Gureasco, Steven Jacques and other members in the Kuriyan and Cole groups for technical help and discussions. We thank Todd Miller for discussions on ACK1 and one of the reviewers for suggesting the co-transfection experiments. This work is supported in part by grants from the NCI to J.K. and NIH to P.A.C.. R.B. is supported by the Susan G. Komen Breast Cancer Foundation.
Footnotes
The atomic coordinates are deposited at the Protein Data Bank under accession codes 2RF9, 2RFD and 2RFE.
References
- 1.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5 (5):341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 2.Zhang X, et al. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125 (6):1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 3.Hackel PO, Gishizky M, Ullrich A. Mig-6 is a negative regulator of the epidermal growth factor receptor signal. Biol Chem. 2001;382 (12):1649–1662. doi: 10.1515/BC.2001.200. [DOI] [PubMed] [Google Scholar]
- 4.Xu Dazhong, Makkinje Anthony, Kyriakis John M. Gene 33 Is an Endogenous Inhibitor of Epidermal Growth Factor (EGF) Receptor Signaling and Mediates Dexamethasone-induced Suppression of EGF Function. J Biol Chem. 2005;280 (4):2924–2933. doi: 10.1074/jbc.M408907200. [DOI] [PubMed] [Google Scholar]
- 5.Fiorentino L, et al. Inhibition of ErbB-2 mitogenic and transforming activity by RALT, a mitogen-induced signal transducer which binds to the ErbB-2 kinase domain. Mol Cell Biol. 2000;20 (20):7735–7750. doi: 10.1128/mcb.20.20.7735-7750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wood ER, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64 (18):6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 7.Jeffrey PD, et al. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376 (6538):313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 8.Ballaro C, et al. Targeted expression of RALT in mouse skin inhibits epidermal growth factor receptor signalling and generates a Waved-like phenotype. EMBO Rep. 2005;6 (8):755–761. doi: 10.1038/sj.embor.7400458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferby I, et al. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat Med. 2006;12 (5):568–573. doi: 10.1038/nm1401. [DOI] [PubMed] [Google Scholar]
- 10.Zhang YW, et al. Evidence that MIG-6 is a tumor-suppressor gene. Oncogene. 2007;26 (2):269–276. doi: 10.1038/sj.onc.1209790. [DOI] [PubMed] [Google Scholar]
- 11.Shen F, et al. Activated Cdc42-associated Kinase 1 Is a Component of EGF Receptor Signaling Complex and Regulates EGF Receptor Degradation. Mol Biol Cell. 2007;18 (3):732–742. doi: 10.1091/mbc.E06-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anastasi S, et al. Feedback inhibition by RALT controls signal output by the ErbB network. Oncogene. 2003;22 (27):4221–4234. doi: 10.1038/sj.onc.1206516. [DOI] [PubMed] [Google Scholar]
- 13.Anastasi S, et al. The evolutionarily conserved EBR module of RALT/MIG6 mediates suppression of the EGFR catalytic activity. Oncogene. 2007 doi: 10.1038/sj.onc.1210590. [DOI] [PubMed] [Google Scholar]
- 14.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277 (48):46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 15.Depetris RS, et al. Structural basis for inhibition of the insulin receptor by the adaptor protein Grb14. Mol Cell. 2005;20 (2):325–333. doi: 10.1016/j.molcel.2005.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lei M, Robinson MA, Harrison SC. The active conformation of the PAK1 kinase domain. Structure. 2005;13 (5):769–778. doi: 10.1016/j.str.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Reynolds AR, et al. EGFR activation coupled to inhibition of tyrosine phosphatases causes lateral signal propagation. Nat Cell Biol. 2003;5 (5):447–453. doi: 10.1038/ncb981. [DOI] [PubMed] [Google Scholar]
- 18.Ichinose J, Murata M, Yanagida T, Sako Y. EGF signalling amplification induced by dynamic clustering of EGFR. Biochem Biophys Res Commun. 2004;324 (3):1143–1149. doi: 10.1016/j.bbrc.2004.09.173. [DOI] [PubMed] [Google Scholar]
- 19.Lynch, Thomas J, et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. N Engl J Med. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 20.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305 (5687):1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 21.Conti E, Kuriyan J. Crystallographic analysis of the specific yet versatile recognition of distinct nuclear localization signals by karyopherin alpha. Structure. 2000;8 (3):329–338. doi: 10.1016/s0969-2126(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 22.Wedegaertner PB, Gill GN. Activation of the purified protein tyrosine kinase domain of the epidermal growth factor receptor. J Biol Chem. 1989;264 (19):11346–11353. [PubMed] [Google Scholar]