

Abstract
Prolactin is essential for normal mammary gland development and differentiation, and has been shown to promote tumor cell proliferation and chemotherapeutic resistance. Soluble isoforms of the prolactin receptor have been reported to regulate prolactin bioavailability by functioning as “prolactin binding proteins.” Included in this category is Δ7/11, a product of alternate splicing of the prolactin receptor primary transcript. However, the direct interactions of prolactin withΔ7/11, and the resulting effect on cell behavior, have not been investigated. Herein, we demonstrate the ability of Δ7/11 to bind prolactin using a novel proximity ligation assay and traditional immunoprecipitation techniques. Biochemical analyses demonstrated that Δ7/11 was heavily glycosylated, similar to the extracellular domain of the primary prolactin receptor, and that glycosylation regulated the cellular localization and secretion of Δ7/11. Low levels of Δ7/11 were detected in serum samples of healthy volunteers, but were undetectable in human milk samples. Expression of Δ7/11 was also detected in six of the 62 primary breast tumor biopsies analyzed; however, no correlation was found with Δ7/11 expression and tumor histotype or other patient demographics. Functional analysis demonstrated the ability of Δ7/11 to inhibit prolactin-induced cell proliferation as well as alter prolactin-induced rescue of cell cycle arrest/early senescence events in breast epithelial cells. Collectively, these data demonstrate that Δ7/11 is a novel regulatory mechanism of prolactin bioavailability and signaling.
Keywords: Prolactin, prolactin receptor, mammary, alternative splicing, prolactin-binding protein
Introduction
Prolactin is a ubiquitous and pleiotropic polypeptide hormone that plays a critical role in breast development, differentiation, and lactation (Das and Vonderhaar 1997; Trott, et al. 2008). It is produced by lactotrophic cells within the anterior pituitary, as well as by many extrapituitary tissues including the mammary epithelium, skin, placenta, uterus, brain, and immune cells (Oakes, et al. 2008). Through the use of diverse animal models, over 300 functions have been identified for prolactin, including an influence on behavior, electrolyte balance, regulation of metabolism and immune response (Bernichtein, et al. 2010; Bole-Feysot, et al. 1998). The co-expression of the prolactin receptor in these tissues suggests that an autocrine-paracrine loop of action exists (Hovey, et al. 2002; Oakes, et al. 2008; Trott, et al. 2008). However, due to the inability to distinguish between pituitary and extrapituitary prolactin (Bernichtein, et al. 2010) and the difficulty in detecting the protein in tissue culture models (Ginsburg and Vonderhaar 1995), our comprehension of the physiological role of extrapituitary prolactin, as well as its regulation, remains poorly understood.
The prolactin receptor is a member of the type-1 cytokine receptor superfamily, which includes the receptors for growth hormone, colony stimulating factor, and the interleukins (Bole-Feysot, et al. 1998). These receptors are single chain transmembrane proteins that consist of an extracellular, a transmembrane, and an intracellular domain (Trott, et al. 2004; Trott, et al. 2008; Vonderhaar, et al. 1985). Upon ligand binding, receptor homodimerization occurs and cell signaling is induced through activation of the receptor-associated kinases. The canonical prolactin signaling pathways include the Janus kinase 2 (Jak2)/STAT5, c-Src/Fyn, PI3K/AKT, Nek3-Rac1, and Grb2-MAPK pathways (Harris, et al. 2004; Hennighausen, et al. 1997; Nguyen, et al. 2008). Adding to the complexity of this multifaceted role of prolactin, the primary transcript of the prolactin receptor (PrlR) has alternative splice products that yield different lengths of the cytoplasmic tails, each of which are reported to have distinctive signaling properties (Clevenger, et al. 2009).
Autocrine/paracrine regulation of bioavailable extrapituitary prolactin to its receptor has been proposed to be regulated by soluble forms of the PrlR (Dannies 2001; Kline and Clevenger 2001; Trott, et al. 2003). This mechanism of regulation has been demonstrated for many proteins including tumor necrosis factor-α, ciliary neutrophic factor, growth hormone, leptin, interleukins 1, 2, and 6, transferrin, and nerve growth factor (Dannies 2001; Rose-John and Heinrich 1994). These soluble receptor “binding proteins” are generated either by alternate splicing of mRNA for the receptor or through post-translational proteolytic cleavage of the extracellular domain of the receptor. To date, two prolactin-binding proteins (PrlBP) have been described in human serum, milk, and cell lysates (Kline and Clevenger 2001; Trott, et al. 2003). Kline and Clevenger (Kline and Clevenger 2001) first characterized a PrlBP in human serum and milk by co-precipitating the PrlBP with antibodies generated against human prolactin and the extracellular domain (ECD) of the human PrlR, as well as by demonstrating the binding affinity of PrlBP for prolactin and growth hormone. The structural properties of PrlBP were confirmed using limited proteolysis and mass spectrometry, and the ability of PrlBP to antagonize the growth promoting function of prolactin was demonstrated in a cell culture model (Kline and Clevenger 2001).
A second soluble form of the PrlR was subsequently described (Trott, et al. 2003). This PrlR isoform results from alternative splicing of the mRNA from exon 7 to exon 11, thus giving its name Δ7/11. Expression of Δ7/11 was reported in human colon and breast tumor samples, as well as in tumor-associated histologically normal breast tissue. Fluorescent immunohistochemistry illustrated vesiculated intracellular distribution, consistent with its fate as a secreted protein. Similar to the previously identified human PrlBP, Δ7/11 was shown to bind to growth hormone. However, its binding affinity for prolactin and the physiological function is yet to be described.
Herein, we demonstrate the ability of Δ7/11 to directly bind prolactin using novel proximity ligation assays and traditional immunoprecipitation techniques. The biochemical properties and relative binding affinities of Δ7/11 for prolactin were investigated. Δ7/11 was found to be glycosylated and was not detectable in human milk, two properties that distinguish it from PrlBP. Furthermore, we demonstrate the ability of Δ7/11 to alter prolactin-induced cell proliferation and signaling of breast cancer cells.
Materials and Methods
Cell culture
T47D, MDA MB231, MCF7, Hs578t, and Chinese Hamster Ovary K1 (CHO) cell lines were obtained from American Type Culture Collection (Manassas, VA) and cultured as recommended. Cells were passaged using trypsinization (0.05% trypsin-EDTA, Invitrogen) and counted on a hemocytometer using trypan blue exclusion.
Production of recombinant Δ7/11
Recombinant protein was produced based on published data (Trott, et al. 2003), isolated and purified by ProteinOne (Rockville, MD) in their E. coli expression system. Protein was validated via sequencing and western blot analysis.
Generation of human prolactin receptor isoform overexpressing CHO cells and collection of conditioned media
Design of the flag-tagged human PrlR isoform constructs was previously described (Trott, et al. 2003). CHO cells were plated in normal growth medium. Twenty-four h post-plating, cells were transfected with either pEF6C (Empty Vector) or pEF6C-Δ7/11DNA using Fugene HD Transfection Reagent (Roche Applied Science, Indianapolis, IN). After two days of growth, stably transfected cells were selected in the presence of blasticidin (2 μg/ml; Invitrogen). The presence of the protein was confirmed via dot blot analysis using FLAG (Sigma) and PrlR isoform specific antibodies (Antibody Production and Purification Unit, National Cancer Institute, National Institutes of Health, Bethesda, MD). For collection of conditioned media, near-confluent stably transfected CHO cells were washed and placed in serum-free media for 24 h. Media were collected, centrifuged to remove cells, and then directly used to treat cells. When indicated, conditioned media were concentrated using Amicon Ultra centrifugation filter (MWCO 10K; Millipore, Billerica, MA).
Generation of human prolactin antibody
The prolactin antibody was produced by PRIMM (IMGEN Technologies, Cambridge, MA) via immunization of rabbits with a recombinant His-tagged prolactin protein (residues 23–220, GI: 531103) and affinity column purified by the Antibody Production and Purification Unit (APPU; National Cancer Institute, Bethesda, MD). Initial affinity column purification was followed by an additional purification using a GE Superdex 200 2.6/60 on an Akta Purifier (GE Healthcare Bio-Sciences Corp., Piscataway NJ) in PBS containing 0.1% sodium azide. The resulting antibody was validated by western blot analysis against the immunizing protein.
Solution phase proximity ligation assay (spPLA)
All assays probes and reagents were purchased from Applied Biosystems (Berkeley, CA) and used as directed. Two sets of probes were used. The first set of probes was designed to detect Δ7/11 using the SF1a rabbit monoclonal antibody and the commercially available mouse monoclonal antibody that detect the extracellular domain of the prolactin receptor (Invitrogen Cat.# 35-9200). The second set of probes was designed to detect human prolactin and used the prolactin antibody (PRIMM) described above. Applied Biosystems linked the short DNA strands to the primary antibodies for detection. Each set of probes (primary antibodies linked to the DNA probe) was used for dual recognition of the target protein complex in situ, i.e. in their physiological context. If the DNA strands are in close proximity they interact to form a unique DNA amplicon, which is subsequently amplified and detected by quantitative real-time PCR. The procedure is as follows: Briefly, diluted samples and the spPLA probes (62.5 pM) were incubated overnight at 4°C. The following day, ligation buffer mix was added to each well, incubated for 10 min at 37 °C, followed by the addition of protease to each well, incubation for 10 min at 37 °C, and then inactivated by a five min incubation at 95 °C. Samples were then mixed with TaqMan Fast PCR Master Mix (Life Technologies, Carlsberg, CA) and products measured using the StepOnePlus Real Time PCR System (Life Technologies). Data were analyzed via theΔ Ct method. Controls were reaction mixture alone and reaction mixture containing the probe pairs only (no protein added). For detection of Δ7/11 in human serum, a panel of normal and matched breast cancer serum samples was purchased from SeraCare Life Sciences, Inc., (Oceanside, CA).
Proliferation assays
Breast cancer cells, proliferating in log phase, were placed in RPMI1640 containing 0.01% charcoal stripped serum (control media) for 24 h followed by three days of treatment. For treatment with conditioned media: cells were treated with fresh conditioned media from stably transfected CHO cells expressing either human Δ7/11 or the empty vector control in the presence or absence of 100 ng/ml recombinant human prolactin. For treatment with recombinant protein: cells were treated with control media, control media with 100 ng/ml prolactin, control media with 250 ng/ml recombinantΔ7/11, or the combination. For all proliferation assays, cells were trypsinized and counted on a hemocytometer using trypan blue exclusion. Data represent three independent experiments +/− SD.
Cell cycle analysis
Two × 104 cells/cm2 were plated in growth medium for 24 h, then washed and incubated in media containing 0.01% charcoal stripped serum overnight. Cells were then treated with prolactin, Δ7/11 or the combination as indicated, in media containing 0.01% charcoal stripped serum for three days. Following treatment, cells were washed in DPBS and fixed in 70% ethanol for 24 h to three days. On the day of analysis, cells were treated with RNase (100 U, Sigma) for 20 min at 37°C, followed by incubation with propidium iodide (50 μg/ml in DPBS, Invitrogen) for 60 min at 4°C. Prior to analysis on the FACSCalibur (BD Biosciences, Franklin Lakes, NJ), cells were filtered though a 40-micron nylon mesh filter. Side and forward scatter were used to exclude cell debris/dead cells/clumps, and a second dot plot was employed to gate propidium iodide positive cells. A minimum of 10,000 cells was acquired. The percentage of cells in G0/G1, S and G2/M phase, and CV was calculated using the MODFIT-LT software (Verity Software House, Topsham, ME).
De-glycosylation assays
Human milk (a kind gift from Dr. Gilbert Smith; National Cancer Institute, Bethesda MD), human serum, recombinant Δ7/11 protein and conditioned media collected from CHO cells stably expressing Δ7/11 were subjected to de-glycosylation using the PNGase kit from New England BioLabs (Ipswich, MA) as directed. Briefly, samples were denatured for 10 min at 100°C, followed by incubation with the PNGase enzyme cocktail for 1 h at 37 °C. Reducing sample buffer (BioRad) was added, samples were incubated for 10 min at 90 °C, and then separated by SDS-PAGE. Proteins were transferred to PVDF membranes using the iBlot (Invitrogen) at P3, for 7 min. Membranes were blocked for 1 h in 5% milk in TBS buffer with 0.1% Tween (TBST) at room temperature, and then incubated with primary antibody (5 μg/mL SF1a rabbit monoclonal antibody) overnight at 4°C, washed, incubated with the appropriate secondary antibody conjugated to Alexa680 (Invitrogen) in TBST with 5% milk for 1 h at room temperature, and then imaged using the Li-COR Odyssey (Lincoln, NE). Membranes were then stripped with 1M NaOH for 5 min, washed, probed with a PrlR antibody that recognized the extracellular domain of all PrlR isoforms (5 μg/ml; Invitrogen), and imaged as described above.
For cell culture de-glycosylation studies, T47D cells were transiently transfected with a FLAG-tagged human Δ7/11 construct (previously described in (Trott, et al. 2003). Three days post-transfection, cells were treated with 500 ng/ml of tunicamycin (Sigma) for 48 h. This concentration was previously determined to affect glycosylation of proteins, and not protein synthesis (Banerjee, et al. 1993). Cells were treated for two days; conditioned media collected and concentrated using Amicon Ultra-10 filtration system (EMD Millipore, Billerica, MA). Cell lysates were collected and separated into membrane and cytosol fractions using the BioVision FractionPREP kit (Milpitas, CA) as directed, then analyzed via western blot as described below. For immunofluorescence detection of Δ7/11 localization, cells were briefly fixed in 100% methanol, washed, and then blocked in 1× PBS containing 5.0% goat serum and 1.0 % BSA (blocking buffer) for 30 minutes at room temperature. Cells were then incubated with a 1:50 dilution of the primary antibody (anti-FLAG, Sigma) in 2% BSA and 2% goat serum overnight at 4°C, washed with 1× PBS and then incubated with appropriate secondary antibody (anti-mouse Alexa Fluor 488; Invitrogen) at a 1:1,000 dilution in blocking buffer for 30 minutes at room temperature. Coverslips were mounted using Prolong Gold antifade reagent with DAPI (Invitrogen). Imaging was performed using the Carl Zeiss LSM510 confocal imaging system (Carl Zeiss MicroImaging) at ×20 magnification.
PCR analysis of breast tumor biopsies
Collection of patient samples was performed in accordance with the guidelines of the National Cancer Institute’s Institutional Review Board, protocol OH99-C-NO57. Total RNA was isolated from tumor biopsies using the RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. RNA was reverse transcribed using MMLV reverse transcriptase (Invitrogen) and primed with oligo-dT and random hexamers (Invitrogen). The cDNA was amplified using gene primers that recognize all three isoforms of the prolactin receptor as described in (Trott, et al. 2003) and PCR Mastermix (Roche Applied Science, Mannheim Germany). PCR data were analyzed via gel electrophoresis.
Western blotting
Equal concentrations of protein, as determined by the Bradford assay, were separated by SDS-PAGE under reducingconditions. Membraneswere blocked in 5% non-fat milk in TBST for 1 h at room temperature, then incubated with primary antibody (P38, P-P38 (Tyr 182) and PERK, Santa Cruz; FLAG, Sigma; PSTAT5 and PAKT, Cell Signaling Technology) overnight at 4 °C in TBST + 5% BSA, washed,and incubated with the appropriate secondary antibody conjugatedto horseradish peroxidase (GE Healthcare, Piscataway, NJ) in TBST with 5% milk for 1 h at room temperature. Peroxidaseactivity was detected using the enhanced chemiluminescence detectionsystem (ECL Plus, GE Healthcare) as directed. GAPDH was used as a control to show equal loading (Santa Cruz). Western blots were quantified using NIH ImageJ 64.
Immunoprecipitation
Recombinant human prolactin (500 ng) and recombinant human Δ7/11 (1 μg) were incubated alone or in combination with either SF1a rabbit monoclonal antibody (5 μg/ml) or a commercially available antibody directed against human prolactin (5 μg/ml, PRIMM) in PBS, overnight at 4°C. The next day, protein A/G PLUS beads (Invitrogen) were added; samples were incubated for 1.5 h at 4°C, and then washed in PBS. The immunobead complexes were pelleted, resuspended in reducing sample buffer (BioRad), and the eluted proteins were subjected to SDS-PAGE and western blot analysis as described above. Primary antibodies: PrlR ECD antibody (1:500; Invitrogen) or a polyclonal antibody directed against human prolactin (2 μg/ml). Secondary antibodies: goat anti-mouse FITC (1:3000; Invitrogen), or Clean-blot secondary anti-rabbit (1:100; Pierce Biotechnology, Rockledge, IL). Controls included beads incubated with the proteins but no antibody and beads incubated with normal rabbit serum. For conditioned media immunoprecipitation, the same procedure was performed with the following modifications: (1) 800 ng of recombinant human prolactin, (2) 1.0 ml of concentrated conditioned media from Δ7/11-expressing CHO cells was the source of Δ7/11 and concentrated conditioned media from CHO cells expressing the empty vector were used as the control.
Statistical analysis
Data was evaluated for significance via t-tests or one-way analysis of variance (ANOVA) with the appropriate post hoc analysis (Tukey, Bonferroni) using GraphPad InStat Software version 3.0b (San Diego, CA). Data was considered significant at P < 0.05.
Results
Evaluation of recombinant Δ7/11 and prolactin binding interactions
Δ7/11 was identified as an alternative splice variant of the primary human PrlR transcript using RT-PCR of breast tumor tissue RNA (Trott, et al. 2003). Figure 1A illustrates the alternate splicing of the exons found within the discussed prolactin receptor isoforms, and Fig. 1B shows the extracellular and intracellular domains present in the membrane bound and soluble prolactin receptor variants (adapted from (Trott, et al. 2003). Bioinformatic analysis suggested that Δ7/11 was a secreted binding protein and the ability of Δ7/11 to bind ligand was examined via immunoprecipitation of FLAG-tagged Δ7/11 with 125I labeled human growth hormone (hGH). Results confirmed the ability of Δ7/11 to bind ligand, and the ability of Δ7/11 to bind 125I-hGH was competitively inhibited by the addition of ovine prolactin, suggesting that Δ7/11 was also binding prolactin (Trott, et al. 2003). However, the direct interaction of Δ7/11 with prolactin was not investigated. Therefore, to directly assess the ability of Δ7/11 to bind prolactin, a novel in-solution proximity ligation assay (spPLA) was used.
Fig. 1. Schematic representation of the human prolactin receptor variants.
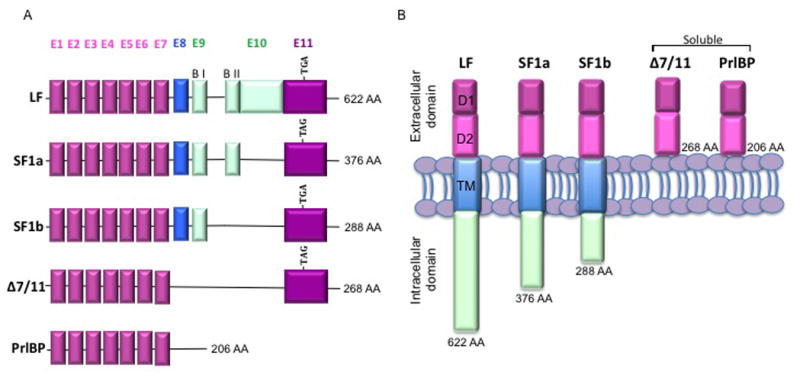
(A) Major human prolactin receptor isoforms generated by alternate splicing. Pink = extracellular domain; blue = transmembrane domain; green = intracellular domain; E = exon; B I = proline-rich JAK2 docking site, B II= hydrophobic region; TGA/TAG = stop codon. (B) Illustration of the major human prolactin receptor variants. LF = long form; SF1a = short form 1a; SF1b = short form 1b, PrlBP = prolactin binding protein; TM = transmembrane domain; D1 and D2 = N terminal subdomain; AA = amino acid number including signal peptide. Adapted from Trott, et al. 2003.
A monoclonal antibody directed against the short form 1a isoform of the PrlR (SF1a) and a PrlR extracellular domain antibody (ECD) were each linked to DNA-strands to form the detection probes. Upon proximal binding of the probe pairs to their respective target proteins, the complimentary DNA strands hybridize and are ligated to form a unique DNA amplicon, which is subsequently detected by quantitative real-time PCR. Based on the structure of the PrlR isoforms (Trott, et al. 2004), the SF1a and ECD set of probes will detect only the SF1a and Δ7/11 proteins. Initial optimization experiments demonstrated the detectable range of Δ7/11 protein; 2 pg - 4 ng (Fig. 2A). Additional probes to detect human prolactin were developed using a human specific antibody (design/production described in Materials and Methods). The specificity of the prolactin probes to human prolactin compared to ovine prolactin was also evaluated, and results show a preferential detection of human prolactin (Fig. 2B). To further establish the selectivity of this assay, we next examined whether a similarly sized and relevant peptide hormone, hGH, could be detected in the assay. Results confirmed the specificity of the assay as hGH was below detectable limits of the assay (Fig. 2C).
Fig. 2. Optimization of solution-phase proximity ligation assays.
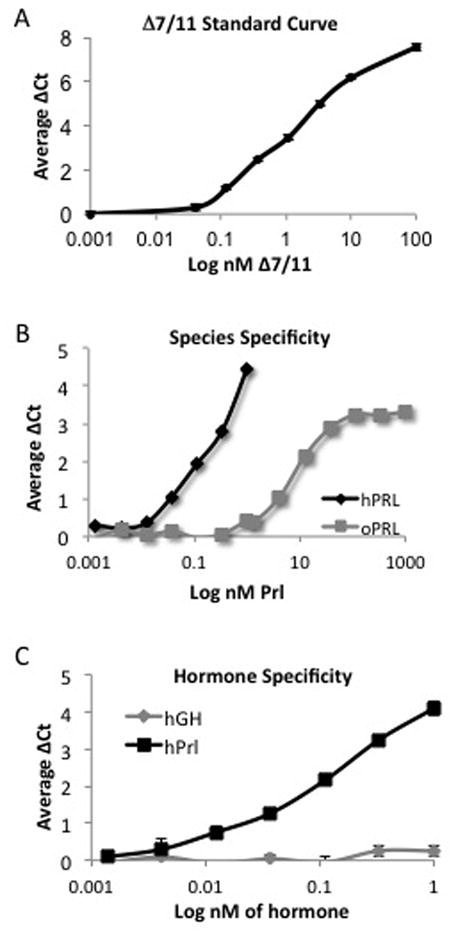
To generate detection probes, SF1a and prolactin receptor antibodies were linked to DNA strands, and upon proximal binding of the antibodies to their respective target proteins, the DNA strands ligate and the PCR amplicon is detected using quantitative real-time PCR. (A) Representative data of Δ7/11 detection using a monoclonal antibody directed against the extracellular domain and the SF1a isoform of the prolactin receptor. (B) Representative data of ovine and human prolactin (oPRL and hPrl, respectively) detection using human prolactin-specific probes. (C) Representative data of growth hormone (hGH) and human prolactin detection using human prolactin-specific probes. Data represent average ΔCt of a minimum of three replicates +/− SD.
Following optimization of the probes, we tested the ability of recombinant human Δ7/11 to directly interact with prolactin. As shown in Fig 3A, in vitro incubation of Δ7/11 with prolactin resulted in a dose dependent increase in detection of protein interactions using SF1a and prolactin probe pairings. The maximal detection of protein binding with 10 nM of prolactin occurred at 500 nM of Δ7/11. When using only prolactin-specific probes, a dose dependent inhibition of prolactin detection was identified (Fig. 3B). Collectively, these results demonstrate direct protein interactions of Δ7/11 and prolactin.
Fig. 3. Detection of Δ7/11 and prolactin interactions.
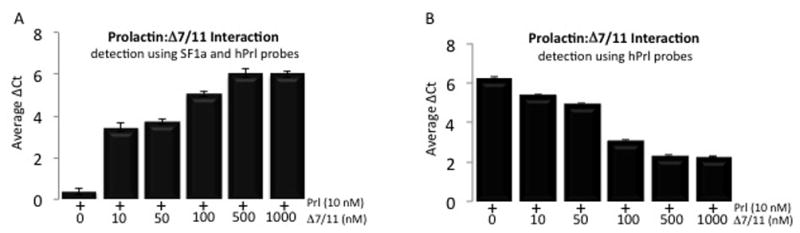
The direct interaction of Δ7/11 and recombinant human prolactin measured by solution-phase proximity ligation assays; SF1a and prolactin antibodies were linked to DNA-strands, upon proximal binding of the antibodies to their respective target proteins, the DNA strands ligate and the PCR amplicon is detected using quantitative real-time PCR. (A) Representative data of Δ7/11 and prolactin interactions using a Δ7/11 specific probe. (B) Representative data of Δ7/11 and prolactin interactions using a hPrl-specific probe. Data represent average ΔCt of a minimum of three replicates +/− SD.
To confirm results obtained from the spPLA assays, standard immunoprecipitation experiments were performed. Both recombinant Δ7/11 and Δ7/11-expressing CHO cell conditioned media were tested for their ability to bind human prolactin. The two sources of Δ7/11 were incubated with prolactin and immunoprecipitated with either a prolactin antibody or the SF1a antibody, followed by western blot analysis using a PrlR ECD or human prolactin antibody. Results confirmed our observations from the spPLA assay; both sources of Δ7/11 bound to prolactin (Fig. 4). Similar to the spPLA assays, we were unable to detect Δ7/11 when incubated with prolactin and immunoprecipitated with the prolactin antibody (Fig. 4, bottom panel). The inability to detect Δ7/11 may be due to variations in the potency and form of Δ7/11 produced in mammals compared to the recombinant Δ7/11 produced by bacteria. The different sources ofΔ7/11 may have altered the binding affinity or strength of the protein-protein interactions, which resulted in the loss of the protein complex during the co-immunoprecipitation procedure. Alternatively, additional post-translational modifications of the Δ7/11 by mammalian cells might have altered the binding affinity or physically blocked the binding site for the prolactin antibody to bind to its epitope on prolactin. For example, glycosylation (i.e. the addition of a carbohydrate to the Δ7/11 molecule) may cause a steric hindrance in the ability of the prolactin antibody to reach the epitope on prolactin when bound to glycosylated Δ7/11; therefore, only the free prolactin, not bound to Δ7/11, was able to be immunoprecipitated when prolactin antibody was used to detect prolactin and Δ7/11 interactions.
Fig. 4. Endogenous and recombinant Δ7/11 binds prolactin.
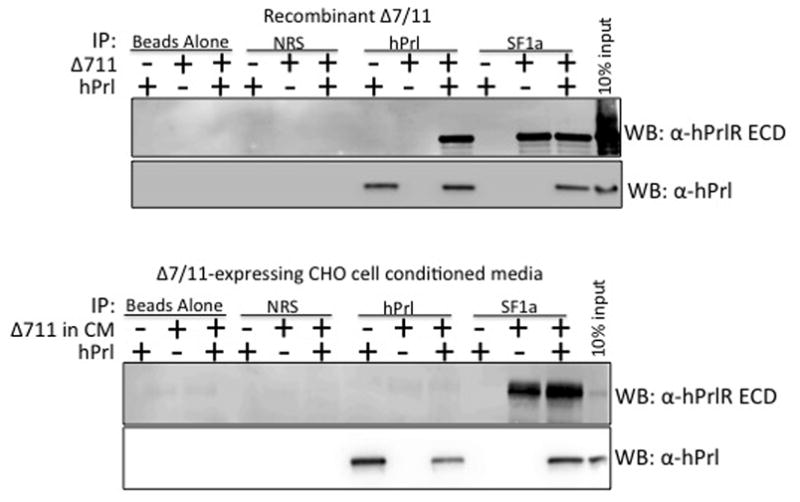
Recombinant human prolactin (500 ng) and recombinant Δ7/11 (1 μg) alone or in combination, were immunoprecipitated with either a prolactin antibody (hPrl; lanes 7–9) or a prolactin receptor isoform antibody that recognizes Δ7/11 (SF1a; lanes 10–12). Immunobead complexes were dissociated, and proteins detected via western blot analysis using an antibody directed against the extracellular domain (ECD) of the prolactin receptor (PrlR) or a prolactin antibody. Top Panels: representative western blots of recombinant proteins immunoprecipitated and analyzed with the indicated antibodies. Bottom Panels: representative western blots of concentrated conditioned media (CM) from CHO-expressing cells as the source of Δ7/11. Controls: lanes 1–3, beads incubated with the proteins but no antibody; lanes 4 – 6, beads incubated with normal rabbit serum (NRS).
Glycosylation of Δ7/11
Previous studies describe a prolactin binding protein (PrlBP) that is found in human serum and milk, and is not glycosylated despite the high level of glycosylation on the asparagine residues of the PrlR ECD (Buteau, et al. 1998; Kline and Clevenger 2001). Therefore, the glycosylation status of Δ7/11 was investigated. Δ7/11-expressing CHO cell lysates and recombinant Δ7/11 were incubated in the presence or absence of Peptide N-glycosidase F, and then evaluated for a shift in electrophoretic mobility by western blot analysis. Δ7/11 was detected using either a SF1a or a PrlR ECD monoclonal antibody, both of which recognize the lower molecular weight Δ7/11 in addition to other PrlR isoforms (SF1a; 57 kDa, Δ7/11; 39.1 – 40.6 kDa depending on glycosylation (Trott, et al. 2004)). As shown in Fig. 5, CHO cell lysates repeatedly demonstrated a distinct shift in electrophoretic mobility when treated with the glycosidase, suggesting glycosylation of Δ7/11. The recombinant Δ7/11, produced in bacteria, lacked glycosylation. In addition to the cell lysates and recombinant Δ7/11, human serum and milk were treated with glycosidase and tested for the presence of Δ7/11. Δ7/11 could not be detected in serum or milk via western blot analysis using either the SF1a or PrlR ECD monoclonal antibody. However, consistent with previous reports (Kline and Clevenger 2001) the prolactin binding protein was detected in serum and milk, and did not exhibit any shift in electrophoretic mobility (Fig. 5B).
Fig. 5. Endogenously produced Δ7/11 is glycosylated.
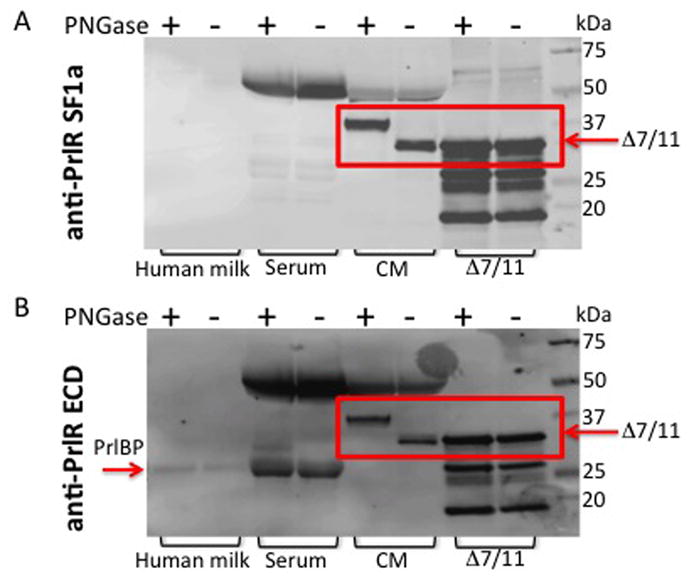
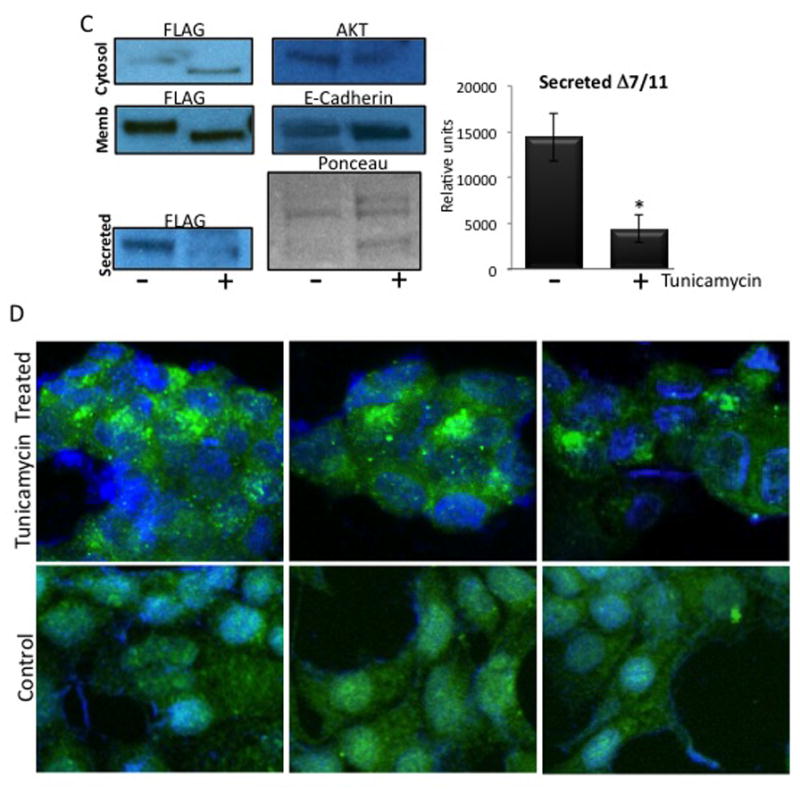
(A) Samples were de-glycosylated with PNGase F treatment, and then subjected to western blot analysis using an antibody directed against the prolactin receptor short form 1a (SF1a), which recognizes Δ7/11. (B) Membranes were stripped and re-probed using an antibody that recognizes the extracellular domain common to the prolactin receptor isoforms (anti-PrlR ECD). Serum = human serum. CM = concentrated conditioned media from CHO cells stably expressing Δ7/11. Δ7/11 = recombinant Δ7/11 produced in E. coli. Approximate molecular weights: hPRLBP, 25 kDa; glycosylated Δ7/11, 39.1 kDa; glycosylated Δ7/11, 40.6 kDa. (C) T47D cells transiently transfected with FLAG-tagged Δ7/11 were treated +/− tunicamycin (500 ng/ml) for 48 hrs. Conditioned media were collected and concentrated, cell lysates were separated into membrane and cytosolic fractions. Representative western blots of cytosolic, membrane, and secreted proteins are shown. AKT, E-Cadherin and Ponceau S stained membrane are shown as loading controls for cytosolic, membrane, and conditioned media, respectively. Histogram shows the quantitation of the amount of Δ7/11 secreted from cells from three independent experiments (*P<0.05). (D) Representative images of T47D cells transiently transfected with FLAG-tagged Δ7/11, treated +/− tunicamycin for 48 hrs, and then analyzed via immunocytofluorescence. FLAG-tagged Δ7/11 stained with Alexa488 (green); DNA stained with DAPI (blue).
To analyze the functional significance of Δ7/11 glycosylation, enzymatic de- glycosylation studies were performed in cultured cells. T47D cells were transiently transfected with FLAG-tagged Δ7/11 and grown in the presence or absence of 500 ng/ml of tunicamycin (Sigma) for 48 h. This concentration was previously determined to affect glycosylation of proteins, and not protein synthesis (Banerjee, et al. 1993). Similar to previous studies showing that glycosylation is required for PrlR long form membrane localization (Banerjee, et al. 1993), we show that the secretion and localization of δ7/11 is regulated by glycosylation. Both western and immunofluorescence analyses show altered Δ7/11 localization upon treatment. Specifically, cells treated with tunicamycin had increased levels of Δ7/11 in the cytoplasm, and significantly decreased levels of secreted Δ7/11 (P<0.05, Fig. 5C). The shift in the molecular weight of Δ7/11 indicates de-glycosylation. Immunocytofluorescence showed an increase in punctate perinuclear localization of Δ7/11compared to control cells, suggesting that non-glycosylated Δ7/11 accumulates in the Golgi area. Collectively, this data suggest that the functional mechanism of Δ7/11 glycosylation involves proper localization and secretion of the protein, and has no effect on the ability of Δ7/11 to bind prolactin.
We further examined human serum for the presence of Δ7/11 using the spPLA for enhanced detection. Ten serum samples from breast cancer patients and thirteen healthy volunteer serum samples were analyzed. A control sample of serum supplemented with recombinant Δ7/11 was used as a positive control. Three of the healthy volunteer serum samples had low but detectable levels of Δ7/11 (Fig. 6), suggesting that in contrast to the prolactin binding protein, Δ7/11 is not abundantly secreted into circulation. Δ7/11 has also been identified in both breast tumors and the patient-matched, associated normal breast tissue (Trott, et al. 2003). To extend upon this initial observation and investigate Δ7/11 expression with clinical variables, we analyzed an additional 62 primary breast tumor biopsies for Δ7/11 expression. Consistent with previous results, approximately ten percent (6 of 62) of the tumors were positive for Δ7/11 expression (Supplementary Fig 1). No correlation was found with histotype (lobular vs. ductal carcinomas), estrogen receptor positivity of the tumor cells, or with patient age.
Fig. 6. Δ7/11 in healthy human serum samples.
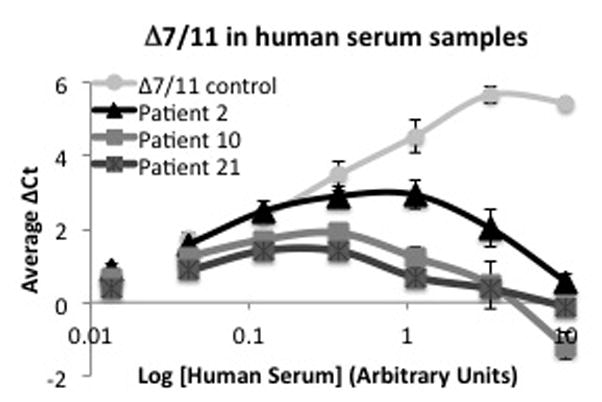
Representative data illustrating the detection of in human serum samples via solution-phase proximity ligation assays. Δ7/11 control = human serum supplemented with recombinant Δ7/11 as a positive control.
Δ7/11 inhibits prolactin-stimulated proliferation
To begin to elucidate the functional role of Δ7/11, proliferation assays were performed. T47D breast cancer cells were chosen as they are reported to readily respond to prolactin-stimulated proliferation (Das and Vonderhaar 1996; Perks, et al. 2004). All cell lines used in the current study have been reported to express levels of prolactin mRNA or secrete low levels of autocrine prolactin (Clevenger, et al. 1995; Ginsburg and Vonderhaar 1995; Perks, et al. 2004; Shaw-Bruha, et al. 1997; Vonderhaar 1999). These studies have shown that in the short time our cells were treated and the media collected (overnight under minimal media conditions), that autocrine prolactin secretion is negligible at best. Thus, the negligible amounts of prolactin secreted by the cells is not enough to demonstrate significant differences in cell proliferation between the control and Δ7/11 treated cells during the 3-day period observed. To observe maximal effects of prolactin response, cells were incubated in 0.01% charcoal stripped serum overnight prior to treatment for 3 days. As shown in Figure 7, recombinant Δ7/11 had no effect on cell proliferation alone; however, Δ7/11 was able to completely inhibit prolactin-induced cell proliferation at all concentrations tested (25 and 250 ng/ml, P<0.05). As Δ7/11 was found to be glycosylated (Fig. 5), we investigated whether the glycosylation would alter the observed inhibitory effect of Δ7/11 on prolactin-induced proliferation. Δ7/11-expressing CHO cell conditioned media was used as the source of Δ7/11, and both T47D and MDA MB231 breast cancer cells were treated with Δ7/11, prolactin, or the combination. Similar to results obtained with the recombinant protein, Δ7/11 was found to significantly inhibit prolactin-induced cell proliferation in both cell lines (Fig. 7B). The breast cancer cell response to prolactin was reduced overall; potentially due to other cytokines and growth factors secreted into the CHO cell conditioned media Lastly, to confirm our findings from the proliferation assays, cell cycle analysis was performed. T47D cells were treated similarly to the proliferation assays; however, the cells were fixed, stained with propidium iodide, and analyzed via flow cytometry 24 h post treatment to observe initial cell cycle events. In agreement with the proliferation assays, prolactin stimulated an increase in the percent of cells in S phase, and Δ7/11 inhibited this response (Fig. 7C).
Fig. 7. Δ7/11 inhibits prolactin-induced breast epithelial cell proliferation.
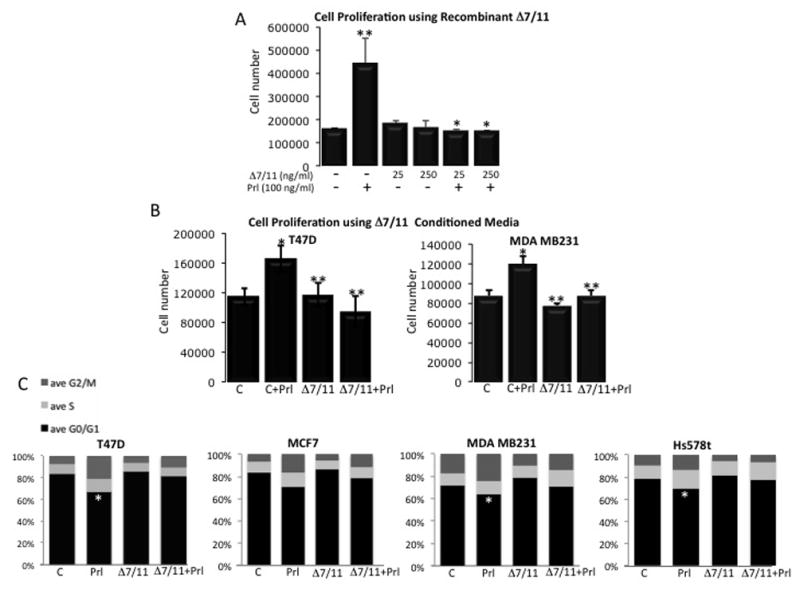
(A) T47D breast cancer cells were plated at 2×104 cells/cm2 in growth media. Twenty four h post-plating cells were washed, incubated overnight in media containing 0.01% charcoal stripped serum, and then treated for three days as indicated. Cell number was determined via trypsinization and counting on a hemocytometer using trypan blue exclusion. Data represent mean +/− SD of three independent experiments. * Indicates significance between Prl and Δ7/11+Prl: P<0.05, ** indicates significance between control and Prl P<0.05. (B) 24-h conditioned media were collected from CHO cells expressing either the empty vector (C = control) or Δ7/11 expression vector (Δ7/11) and used to treat cells. T47D and MDA MB231 breast cancer cells were plated at 2×104 cells/cm2 in growth media for 24 h, washed, incubated in median containing 0.01% charcoal stripped serum overnight, and then treated for three days as indicated. Cell number was determined as described in (A). Data represent mean +/− SD of three independent experiments. * Indicates significance between C and C+Prl, ** indicates significance between C+Prl and Δ7/11 or Δ7/11+Prl: P<0.05. Prl = 100 ng/ml prolactin. (C) The indicated breast cancer cell lines were cultured as described in (A) and treated as indicated for 24 h (C = serum free media, Prl = 100 ng/ml prolactin, Δ7/11 = 250 ng/ml recombinant Δ7/11). Following treatment, cells were fixed, stained with propidium iodide, and analyzed via flow cytometry. Percentage of cells in S phase was calculated using MODFIT-LT software. Data represent mean +/− SD of three independent experiments. *P<0.05 between Prl and all other treatments.
Δ7/11 alters breast epithelial cell signaling
To investigate the mechanisms by which Δ7/11 influenced prolactin-stimulated cell behavior, the pathways reported to be activated by prolactin were examined. T47D cells were incubated in media containing 0.01% charcoal stripped serum for 24 h prior to treatment for 5 and 60 min with prolactin, Δ7/11, or the combination. As shown in Fig. 8, 24 h culture in low serum containing media induced early onset senescence events in the cells, as indicated by phosphorylation of P38 in the control treatment groups. Treatment with prolactin was able to rescue a significant portion of cells from P38 induced early senescence/cell cycle arrest as indicated by the significant reduction of P38 phosphorylation in the cell lysates. The addition of Δ7/11 had no significant effect on the cells compared to controls. The combined treatment ofΔ7/11 and prolactin resulted in levels of P38 phosphorylation similar to controls or cells treated with Δ7/11 alone, suggesting that the addition of Δ7/11 was able to inhibit the prolactin-induced rescue of early onset senescence/cell cycle arrest events. This observation was the most prominent and consistent signaling mechanism observed across all cell lines tested. The basal activation/phosphorylation and prolactin-stimulated activation/phosphorylation of other common signaling pathways varied among the breast cell lines tested (AKT, STAT5, STAT3, and ERK1/2, Supplementary Fig. 2). This is potentially due to autocrine secretion of prolactin as well as the expression levels of the various prolactin receptor isoforms (Trott, et al. 2004; Vonderhaar 1999).
Fig. 8. Δ7/11 inhibits prolactin-induced rescue of early senescence events in breast epithelial cells.
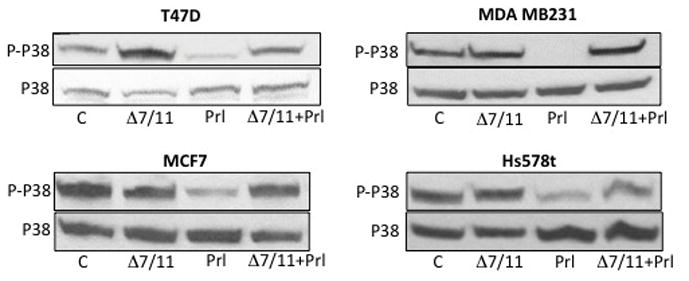
Cells were plated at 2×104 cells/cm2 in growth media. Twenty four h post-plating cells were washed, incubated overnight in 0.01% charcoal stripped serum, and then treated with serum free media (C), serum-free media + 100 ng/ml prolactin (Prl), serum-free media + 250 ng/ml recombinant Δ7/11, or the combination of Prl and Δ7/11 for 5 min. A. Cell lysates (50 μg) were resolved by SDS-PAGE and immunoblotted with antibodies specific for phosphorylated (P) P38 or total P38. Figure shows representative immunoblots of two separate experiments.
Discussion
The data presented in this report show direct interactions between prolactin and Δ7/11, a soluble isoform of the PrlR. We also demonstrate the glycosylation of Δ7/11 and its ability to inhibit prolactin-stimulated cell proliferation. Collectively, these data demonstrate a novel regulatory mechanism of prolactin bioavailability and signaling.
Similar to the PrlBP, the precise source of Δ7/11 generation in breast and colon tissues is unknown. In addition to the nine cell lines analyzed for Δ7/11 message in the initial report (Trott, et al. 2003), we screened an additional 12 breast cancer cell lines and two primary normal breast epithelial cell preparations from reduction mammoplasty tissue. Similar to the original report, we were unable to detect Δ7/11 via RT-PCR in these cells grown in vitro on tissue culture dishes (data not shown). This presents the possibility that the source of Δ7/11 in breast tissue originates from stromal or immune cells. Alternatively, as the polarity and 3D organization of epithelial cells is known to directly regulate gene expression (Rooney and Streuli 2011; Schedin and Keely 2011), the possibility remains that breast epithelial cells in a 3D organization or the appropriate extracellular matrix will produce Δ7/11. It is noteworthy that Δ7/11 has only been detected in organized tissues, and not in monolayers of cells in culture. It is well recognized that prolactin signaling is altered when cells are cultured in 3D (Katz and Streuli 2007; Prince, et al. 2002; Rooney and Streuli 2011), therefore, future studies exploring the role of Δ7/11 on prolactin regulation in polarized cells or on different extracellular matrices would be of interest to the field.
Multiple isoforms of prolactin have been identified in circulation, with the 23 kDa identified as the most prominent in humans (Fonseca, et al. 1991). Previous studies have shown that the 23 kDa protein is involved in classic prolactin functions, including exhibiting proliferative and pro-angiogenic properties. Conversely, the 16 kDa form of prolactin was demonstrated as a potent anti-proliferative and anti-angiogenic factor in various cell types both in vitro and in vivo (Bentzien, et al. 2001; Clapp, et al. 2006; Kim, et al. 2003; Nguyen, et al. 2007). It would be of interest to investigate the binding affinity for Δ7/11 on these two functionally opposing prolactin isoforms. Our data suggests that Δ7/11 may have a higher affinity for the 23 kDa prolactin as we observed an inhibition of prolactin-stimulated proliferation in all cell types investigated. Furthermore, 16 kDa prolactin is a cleavage product of the 23 kDa protein, proposed to be formed by the action of the protease cathepsin D (Piwnica, et al. 2004). Whether the binding of Δ7/11 to prolactin alters the enzymatic action of cathepsin D is yet to be investigated. These studies may help to elucidate the production and function of these two functionally opposing prolactins.
It is of note that Δ7/11 is glycosylated, though the function of this biochemical modification remains unknown. The PrlBP is not glycosylated and has the ability to bind both prolactin and growth hormone, as well as inhibit prolactin-stimulated proliferation in a cell culture model (Kline, et al. 2002). In the present study, both the glycosylated and un-glycosylated forms of Δ7/11 were able to bind and inhibit prolactin. In attempt to understand the functional role of Δ7/11 glycosylation, we observed the cellular localization of Δ7/11 in the presence of tunicamycin, an inhibitor of N-glycosylation. Previous studies showed that glycosylation of the asparagyl residues of the extracellular domain of the prolactin receptor is crucial for its cell surface localization, but has no effect on PrlR signaling (Buteau, et al. 1998;Cahoreau, et al. 1994). Similarly, we found that treatment of cells overexpressing Δ7/11 with tunicamycin resulted in accumulation of protein in the Golgi area. This suggests that the levels of Δ7/11 secreted into the extracellular space can be controlled cell-autonomously by post-translational modifications. In addition to Δ7/11 glycosylation regulating cellular localization, one can hypothesize that the glycosylation may influence the binding affinities for the different forms of prolactin (16 or 23 kDa) or bind the extracellular domains of the different PrlR isoforms. Given the sequence similarity of Δ7/11 to the PrlR, and the ability of the various PrlR isoforms to homo- and hetero-dimerize, the direct interaction of Δ7/11 and the PrlR is yet another potential mechanism for enhancement or inhibition of prolactin signaling.
Prolactin induces differentiation in mammary epithelial cells through activation of the JAK/STAT5 signaling pathway (Vonderhaar and Ziska 1989), and has been shown to activate other common proliferative signal transduction pathways in breast epithelial cells (AKT, ERK1/2) (Brisken and O’Malley 2010; LaPensee and Ben-Jonathan 2010). In our studies, we found that alterations in the STAT signaling pathways varied across the cell lines (both basal activation and activation by prolactin). We hypothesize that that this is due to the varying amounts of endogenous prolactin secreted by each cell type, as well as the different levels of the prolactin receptor isoforms produced in each cell (Trott, et al. 2004; Vonderhaar 1999). Nevertheless, one conclusive pattern was consistently observed in all cell lines tested: we found that the underlying mechanism of Δ7/11-induced inhibition of proliferation involved the regulation of P38 activation. Whether prolactin was rescuing the cells from cell cycle arrest or early senescence was not determined. However, our data suggest that prolactin was consistently able to rescue the cells from either cell cycle arrest/early senescence events while simultaneously inactivating P38 (i.e. de-phosphorylation), thus leading to the observed increase in cell proliferation compared to control cells or cells treated with Δ7/11 alone. Conversely, Δ7/11 blocked the ability of prolactin to rescue the cells in all cells lines tested. The role of prolactin as an inducible survival factor in mammary cells is well known, however, our observation that prolactin may rescue cells from early senescent events via inactivation of P38 presents a unique and understudied mechanisms of prolactin signaling. More studies investigating this novel signaling mechanism are warranted.
In conclusion, we present data detailing the ability of Δ7/11, a soluble secreted form of the PrlR, to directly bind to prolactin and inhibit prolactin-induced cell proliferation. Unlike the previously reported PrlBP, Δ7/11 was found to be glycosylated. Lastly, the inability to detectΔ7/11 in human serum samples suggests that Δ7/11 is a tissue-specific factor responsible for local regulation of prolactin function. These data highlight the role of Δ7/11 as a novel regulatory factor of prolactin bioavailability.
Supplementary Material
Acknowledgments
Funding
This research was supported by the Center for Cancer Research, an Intramural Research Program of the National Cancer Institute and by the NCCU-BBRI-Lineberger Partnership in Cancer Research U54 CA156735.
Footnotes
Declaration of Interests.
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Banerjee R, Ginsburg E, Vonderhaar BK. Characterization of a monoclonal antibody against human prolactin receptors. Int J Cancer. 1993;55:712–721. doi: 10.1002/ijc.2910550503. [DOI] [PubMed] [Google Scholar]
- Bentzien F, Struman I, Martini JF, Martial J, Weiner R. Expression of the antiangiogenic factor 16K hPRL in human HCT116 colon cancer cells inhibits tumor growth in Rag1(−/−) mice. Cancer Res. 2001;61:7356–7362. [PubMed] [Google Scholar]
- Bernichtein S, Touraine P, Goffin V. New concepts in prolactin biology. J Endocrinol. 2010;206:1–11. doi: 10.1677/JOE-10-0069. [DOI] [PubMed] [Google Scholar]
- Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr Rev. 1998;19:225–268. doi: 10.1210/edrv.19.3.0334. [DOI] [PubMed] [Google Scholar]
- Brisken C, O’Malley B. Hormone action in the mammary gland. Cold Spring Harb Perspect Biol. 2010;2:a003178. doi: 10.1101/cshperspect.a003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buteau H, Pezet A, Ferrag F, Perrot-Applanat M, Kelly PA, Edery M. N-glycosylation of the prolactin receptor is not required for activation of gene transcription but is crucial for its cell surface targeting. Mol Endocrinol. 1998;12:544–555. doi: 10.1210/mend.12.4.0085. [DOI] [PubMed] [Google Scholar]
- Cahoreau C, Garnier L, Djiane J, Devauchelle G, Cerutti M. Evidence for N-glycosylation and ubiquitination of the prolactin receptor expressed in a baculovirus-insect cell system. FEBS Lett. 1994;350:230–234. doi: 10.1016/0014-5793(94)00772-1. [DOI] [PubMed] [Google Scholar]
- Clapp C, Gonzalez C, Macotela Y, Aranda J, Rivera JC, Garcia C, Guzman J, Zamorano M, Vega C, Martin C, Jeziorski MC, de la Escalera GM. Vasoinhibins: a family of N-terminal prolactin fragments that inhibit angiogenesis and vascular function. Front Horm Res. 2006;35:64–73. doi: 10.1159/000094309. [DOI] [PubMed] [Google Scholar]
- Clevenger CV, Chang WP, Ngo W, Pasha TL, Montone KT, Tomaszewski JE. Expression of prolactin and prolactin receptor in human breast carcinoma. Evidence for an autocrine/paracrine loop. Am J Pathol. 1995;146:695–705. [PMC free article] [PubMed] [Google Scholar]
- Clevenger CV, Gadd SL, Zheng J. New mechanisms for PRLr action in breast cancer. Trends Endocrinol Metab. 2009;20:223–229. doi: 10.1016/j.tem.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Dannies PS. A serum prolactin-binding protein: implications for growth hormone. Trends Endocrinol Metab. 2001;12:427–428. doi: 10.1016/s1043-2760(01)00497-0. [DOI] [PubMed] [Google Scholar]
- Das R, Vonderhaar BK. Activation of raf-1, MEK, and MAP kinase in prolactin responsive mammary cells. Breast Cancer Res Treat. 1996;40:141–149. doi: 10.1007/BF01806209. [DOI] [PubMed] [Google Scholar]
- Das R, Vonderhaar BK. Prolactin as a mitogen in mammary cells. J Mammary Gland Biol Neoplasia. 1997;2:29–39. doi: 10.1023/a:1026369412612. [DOI] [PubMed] [Google Scholar]
- Fonseca ME, Ochoa R, Moran C, Zarate A. Variations in the molecular forms of prolactin during the menstrual cycle, pregnancy and lactation. J Endocrinol Invest. 1991;14:907–912. doi: 10.1007/BF03347114. [DOI] [PubMed] [Google Scholar]
- Ginsburg E, Vonderhaar BK. Prolactin synthesis and secretion by human breast cancer cells. Cancer Res. 1995;55:2591–2595. [PubMed] [Google Scholar]
- Harris J, Stanford PM, Oakes SR, Ormandy CJ. Prolactin and the prolactin receptor: new targets of an old hormone. Ann Med. 2004;36:414–425. doi: 10.1080/07853890410033892. [DOI] [PubMed] [Google Scholar]
- Hennighausen L, Robinson GW, Wagner KU, Liu W. Prolactin signaling in mammary gland development. J Biol Chem. 1997;272:7567–7569. doi: 10.1074/jbc.272.12.7567. [DOI] [PubMed] [Google Scholar]
- Hovey RC, Trott JF, Vonderhaar BK. Establishing a framework for the functional mammary gland: from endocrinology to morphology. J Mammary Gland Biol Neoplasia. 2002;7:17–38. doi: 10.1023/a:1015766322258. [DOI] [PubMed] [Google Scholar]
- Katz E, Streuli CH. The extracellular matrix as an adhesion checkpoint for mammary epithelial function. Int J Biochem Cell Biol. 2007;39:715–726. doi: 10.1016/j.biocel.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Luo W, Chen DT, Earley K, Tunstead J, Yu-Lee LY, Lin SH. Antitumor activity of the 16-kDa prolactin fragment in prostate cancer. Cancer Res. 2003;63:386–393. [PubMed] [Google Scholar]
- Kline JB, Clevenger CV. Identification and characterization of the prolactin-binding protein in human serum and milk. J Biol Chem. 2001;276:24760–24766. doi: 10.1074/jbc.M011786200. [DOI] [PubMed] [Google Scholar]
- Kline JB, Rycyzyn MA, Clevenger CV. Characterization of a novel and functional human prolactin receptor isoform (deltaS1PRLr) containing only one extracellular fibronectin-like domain. Mol Endocrinol. 2002;16:2310–2322. doi: 10.1210/me.2001-0033. [DOI] [PubMed] [Google Scholar]
- LaPensee EW, Ben-Jonathan N. Novel roles of prolactin and estrogens in breast cancer: resistance to chemotherapy. Endocr Relat Cancer. 2010;17:R91–107. doi: 10.1677/ERC-09-0253. [DOI] [PubMed] [Google Scholar]
- Nguyen N, Stellwag EJ, Zhu Y. Prolactin-dependent modulation of organogenesis in the vertebrate: Recent discoveries in zebrafish. Comp Biochem Physiol C Toxicol Pharmacol. 2008;148:370–380. doi: 10.1016/j.cbpc.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Nguyen NQ, Cornet A, Blacher S, Tabruyn SP, Foidart JM, Noel A, Martial JA, Struman I. Inhibition of tumor growth and metastasis establishment by adenovirus-mediated gene transfer delivery of the antiangiogenic factor 16K hPRL. Mol Ther. 2007;15:2094–2100. doi: 10.1038/sj.mt.6300294. [DOI] [PubMed] [Google Scholar]
- Oakes SR, Rogers RL, Naylor MJ, Ormandy CJ. Prolactin regulation of mammary gland development. J Mammary Gland Biol Neoplasia. 2008;13:13–28. doi: 10.1007/s10911-008-9069-5. [DOI] [PubMed] [Google Scholar]
- Perks CM, Keith AJ, Goodhew KL, Savage PB, Winters ZE, Holly JM. Prolactin acts as a potent survival factor for human breast cancer cell lines. Br J Cancer. 2004;91:305–311. doi: 10.1038/sj.bjc.6601947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piwnica D, Touraine P, Struman I, Tabruyn S, Bolbach G, Clapp C, Martial JA, Kelly PA, Goffin V. Cathepsin D processes human prolactin into multiple 16K-like N-terminal fragments: study of their antiangiogenic properties and physiological relevance. Mol Endocrinol. 2004;18:2522–2542. doi: 10.1210/me.2004-0200. [DOI] [PubMed] [Google Scholar]
- Prince JM, Klinowska TC, Marshman E, Lowe ET, Mayer U, Miner J, Aberdam D, Vestweber D, Gusterson B, Streuli CH. Cell-matrix interactions during development and apoptosis of the mouse mammary gland in vivo. Dev Dyn. 2002;223:497–516. doi: 10.1002/dvdy.10070. [DOI] [PubMed] [Google Scholar]
- Rooney N, Streuli CH. How integrins control mammary epithelial differentiation: a possible role for the ILK-PINCH-Parvin complex. FEBS Lett. 2011;585:1663–1672. doi: 10.1016/j.febslet.2011.05.014. [DOI] [PubMed] [Google Scholar]
- Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem J. 1994;300(Pt 2):281–290. doi: 10.1042/bj3000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schedin P, Keely PJ. Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb Perspect Biol. 2011;3:a003228. doi: 10.1101/cshperspect.a003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw-Bruha CM, Pirrucello SJ, Shull JD. Expression of the prolactin gene in normal and neoplastic human breast tissues and human mammary cell lines: promoter usage and alternative mRNA splicing. Breast Cancer Res Treat. 1997;44:243–253. doi: 10.1023/a:1005879103367. [DOI] [PubMed] [Google Scholar]
- Trott JF, Hovey RC, Koduri S, Vonderhaar BK. Alternative splicing to exon 11 of human prolactin receptor gene results in multiple isoforms including a secreted prolactin-binding protein. J Mol Endocrinol. 2003;30:31–47. doi: 10.1677/jme.0.0300031. [DOI] [PubMed] [Google Scholar]
- Trott JF, Hovey RC, Koduri S, Vonderhaar BK. Multiple new isoforms of the human prolactin receptor gene. Adv Exp Med Biol. 2004;554:495–499. doi: 10.1007/978-1-4757-4242-8_71. [DOI] [PubMed] [Google Scholar]
- Trott JF, Vonderhaar BK, Hovey RC. Historical perspectives of prolactin and growth hormone as mammogens, lactogens and galactagogues--agog for the future! J Mammary Gland Biol Neoplasia. 2008;13:3–11. doi: 10.1007/s10911-008-9064-x. [DOI] [PubMed] [Google Scholar]
- Vonderhaar BK. Prolactin involvement in breast cancer. Endocr Relat Cancer. 1999;6:389–404. doi: 10.1677/erc.0.0060389. [DOI] [PubMed] [Google Scholar]
- Vonderhaar BK, Bhattacharya A, Alhadi T, Liscia DS, Andrew EM, Young JK, Ginsburg E, Bhattacharjee M, Horn TM. Isolation, characterization, and regulation of the prolactin receptor. J Dairy Sci. 1985;68:466–488. doi: 10.3168/jds.S0022-0302(85)80847-X. [DOI] [PubMed] [Google Scholar]
- Vonderhaar BK, Ziska SE. Hormonal regulation of milk protein gene expression. Annu Rev Physiol. 1989;51:641–652. doi: 10.1146/annurev.ph.51.030189.003233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.