

Abstract
The E3 ubiquitin ligase NEDD4-2 (encoded by the Nedd4L gene) regulates the amiloride-sensitive epithelial Na+ channel (ENaC/SCNN1) to mediate Na+ homeostasis. Mutations in the human β/γENaC subunits that block NEDD4-2 binding or constitutive ablation of exons 6–8 of Nedd4L in mice both result in salt-sensitive hypertension and elevated ENaC activity (Liddle syndrome). To determine the role of renal tubular NEDD4-2 in adult mice, we generated tetracycline-inducible, nephron-specific Nedd4L KO mice. Under standard and high-Na+ diets, conditional KO mice displayed decreased plasma aldosterone but normal Na+/K+ balance. Under a high-Na+ diet, KO mice exhibited hypercalciuria and increased blood pressure, which were reversed by thiazide treatment. Protein expression of βENaC, γENaC, the renal outer medullary K+ channel (ROMK), and total and phosphorylated thiazide-sensitive Na+Cl– cotransporter (NCC) levels were increased in KO kidneys. Unexpectedly, Scnn1a mRNA, which encodes the αENaC subunit, was reduced and proteolytic cleavage of αENaC decreased. Taken together, these results demonstrate that loss of NEDD4-2 in adult renal tubules causes a new form of mild, salt-sensitive hypertension without hyperkalemia that is characterized by upregulation of NCC, elevation of β/γENaC, but not αENaC, and a normal Na+/K+ balance maintained by downregulation of ENaC activity and upregulation of ROMK.
Introduction
Hypertension is a major risk factor for stroke, myocardial infarction, and heart and kidney failure. The postmacula densa segments of the nephron, namely the distal convoluted tubule (DCT), the connecting tube (CNT) and the collecting duct (CD), control Na+ and K+ balance and thus extracellular volume and blood pressure. Na+ reabsorption occurs in the DCT by electroneutral cotransport via the thiazide-sensitive Na+Cl– cotransporter (NCC) and in the late portion of the DCT (DCT2), CNT, and CD by electrogenic Na+ reabsorption through the amiloride-sensitive epithelial Na+ channel (ENaC) (1). ENaC provides the driving force for K+ secretion via the renal outer medullary K+ channel (ROMK) (2).
The significance of these 3 renal segments for controlling Na+ balance and blood pressure is underscored by genetic diseases affecting ENaC and NCC. Gain-of-function mutations within SCNN1 cause Liddle syndrome, which is characterized by increased ENaC activity, and result in salt retention and hypertension (3). Such mutations in β- and γENaC lead to inactivation of the PY motif that interacts with WW domains of the ubiquitin-protein ligase NEDD4-2 (encoded by the Nedd4L gene), which ubiquitylates and degrades the channel (4–8). With increased aldosterone secretion, serum- and glucocorticoid-regulated kinase 1 (SGK1) interferes with ENaC ubiquitylation by NEDD4-2 (9). In addition, ENaC has been shown to be activated by proteolytic cleavage of the α and γ subunits that reflects ENaC activation and leads to channel with a higher open probability (10–12). Moreover, the α subunit is needed to bring β- and γENaC to the apical membrane, making functional channels (13–15).
The activity of NCC plays a major role in Na+ balance and blood pressure regulation. Thiazide diuretics, which inhibit NCC and lower blood pressure, and genetic mutations that suppress or heighten NCC function cause hypotension or hypertension, respectively (16). NCC is also a downstream molecular target of the with-no-lysine WNK1/WNK4 kinases (17, 18), the kelch-like KLHL3/Cullin 3 (CUL3) ubiquitin-protein ligase complex (19, 20), and the SGK1 kinase (21). Mutations in human proteins lead to overactive NCC and cause pseudohypoaldosteronism type II (PHAII) (19, 20, 22), characterized by hyperkalemia, hypertension, hypercalciuria, and metabolic acidosis that can be treated by thiazides (23). Very recently, we have shown both in vitro and in vivo that Nedd4-2 is involved in NCC regulation (24).
Generation of constitutive Nedd4L KO mice, by removing exons 6 to 8 of the Nedd4L gene (Nedd4L-Δ6-8 KO mice), demonstrated the importance of NEDD4-2 in the control of ENaC activity and blood pressure (25). Indeed, these mice displayed impaired Na+ excretion and hypertension, apparently mediated by ENaC overactivity (25). No effect on NCC was observed. To determine the role of renal NEDD4-2 in controlling Na+ balance and blood pressure during adulthood and to dissect the mechanisms behind this regulation, we developed inducible renal tubule–specific Nedd4Lflox/flox/Pax8-rTA/LC1 KO mice (Nedd4LPax8/LC1). Here, we show that loss of Nedd4L in adult renal tubules leads to some features of salt-sensitive PHAII with overactive NCC, leading to increased blood pressure and hypercalciuria. Interestingly, compensation by increased ROMK abundance and downregulation of ENaC activity allows the maintenance of a normal Na+/K+ balance.
Results
Generation of inducible renal tubule–specific Nedd4L KO mice.
Germline deletion of exons 6 to 8 of the Nedd4L gene in mice leads to ENaC overactivation and hypertension (25). To determine the role of renal NEDD4-2 in regulating Na+ balance and blood pressure in adult mice, we generated inducible renal tubule–specific Nedd4L KO mice using the same floxed allele used previously (25) and a combination of the inducible Tet-On and Cre-loxP systems, as previously described (24). Double-transgenic Nedd4Lflox/flox/Pax8-rTA/LC1 (Nedd4LPax8/LC1) conditional KO mice and single-transgenic Nedd4Lflox/flox/Pax8-rTA (Nedd4LPax8) or Nedd4Lflox/flox/LC1 (Nedd4LLC1) control littermates were treated with doxycycline as described previously (24). Immunofluorescence analysis showed that Cre recombinase is expressed in every tubular cell along the renal tubules of doxycycline-treated KO, but not in the blood vessels (Figure 1A). Consistent with previous report (26), Nedd4L mRNA could be detected in the proximal tubule (PT), DCT, CNT, and CD in microdissected tubules of control mice, but not in KO (Figure 1B). Western blot on total kidney lysates revealed a decrease of the 130-kDa NEDD4-2 protein in doxycycline-treated KO mice (Figure 1C). Using several antibodies against NEDD4-2, we observed that the remaining band at 130 kDa was consistently present at low levels in the KO mice (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI61110DS1). However, this band was completely lost when Western blots were performed on microdissected tubules (Figure 1D). These data indicate that the lower expression of NEDD4-2 observed on whole kidney extracts of KO mice corresponds to NEDD4-2 expressed in nontubular cells that were not targeted using the Pax8-rTA/LC1 system. Moreover, no additional truncated NEDD4-2 protein could be detected with the existing NEDD4-2 antibodies in the KO. As it was previously reported that the Pax8-rTA/LC1-mediated recombination system was leaky in some hepatocytes (27), we performed PCR to check the presence of recombination in liver and observed a weak band corresponding to the Nedd4L null allele in the KO (Supplemental Figure 2A). However, immunoblotting on whole KO liver showed normal NEDD4-2 protein expression in the KO (Supplemental Figure 2B). NEDD4-1 protein, closely related to NEDD4-2 (28), was not upregulated in the Nedd4LPax8/LC1 KO mice, indicating that there is no compensation by this paralogue (Supplemental Figure 3).
Figure 1. Generation of inducible renal tubule–specific Nedd4L KO mice.

Nedd4Lflox/flox/Pax8-rTA/LC1 KO mice and control littermates (Nedd4LPax8 or Nedd4LLC1) were obtained and treated with doxycycline as described previously (24) to induce Nedd4L ablation in renal tubular cells. (A) Immunofluorescence for CRE and calbindin CaBP28 on kidney sections from control (left panel) and induced Nedd4LPax8/LC1 KO (right panel). CRE recombinase is expressed in all renal tubules in induced KO mice, including CaBP28-positive DCT/CNT cells (CRE: red; CaBP28: green; DAPI: blue). Scale bars: ∼50 μm. C, CNT; D, DCT. (B) Quantitative real-time PCR analysis for Nedd4L, Slc12a3 (encoding NCC), and aquaporin 2 (Aqp2) mRNA on microdissected renal tubules normalized to Gapdh: Nedd4L mRNA is not detected in Nedd4LPax8/LC1 KO, and Slc12a3 mRNA levels are unchanged (n = 4 per group, 8 days of high-Na+ diet). Slc12a3 was used as DCT marker and Aqp2 as CNT/CD marker. (C and D) Analysis of NEDD4-2 protein expression by Western blot in whole kidney lysates (C) and microdissected tubules (D). The lower NEDD4-2 expression observed in KO whole-kidney lysates is absent in microdissected tubules (n = 3 mice per genotype). Load: unspecific band used as loading control. C, CNT/CD; P, PT; T, TAL.
Nedd4LPax8/LC1 KO mice show normal urine and plasma Na+ and K+ levels.
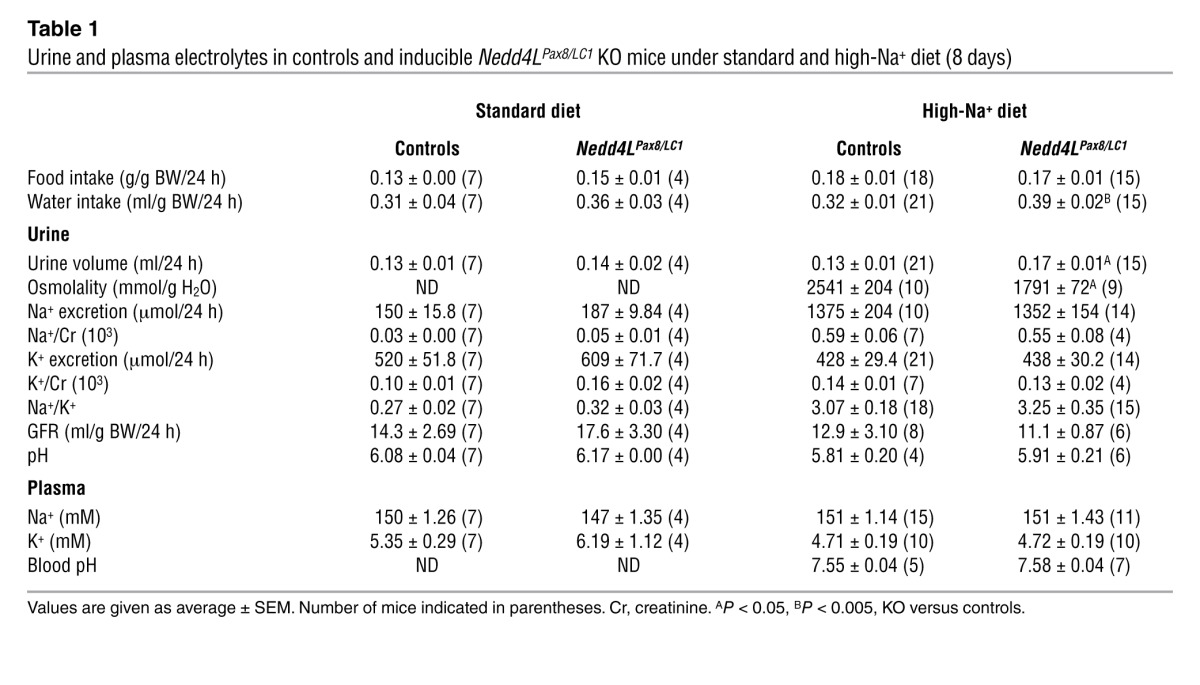
Control and KO mice were fed with a standard or high-Na+ diet and placed in metabolic cages to analyze Na+/K+ balance. Plasma aldosterone levels were strongly decreased in the KO mice under both diets, suggesting increased Na+ retention (Figure 2A). However, KO did not show any change in plasma Na+ and K+ concentrations (Table 1). Moreover, after a change from standard to high-Na+ diet, KO mice showed a tendency to retain Na+ compared with controls, but this was not statistically significant (Supplemental Figure 4). These data indicate either that the Nedd4LPax8/LC1 KO mice are able to maintain a normal Na+/K+ balance or that they have a modest Na+ retention that could not be detected by measuring urinary Na+ excretion. Interestingly, when fed with a high-Na+ diet, KO mice displayed increased water consumption, elevated urine output, and diluted urine (Table 1), as previously observed in constitutive Nedd4L-Δ6-8 KO mice (25).
Figure 2. Inducible Nedd4LPax8/LC1 KO mice show decreased plasma aldosterone and salt-sensitive blood pressure increase.

(A) Plasma aldosterone levels were decreased in induced Nedd4LPax8/LC1 KO (n = 7–10) compared with controls (n = 6–9) under both standard and high-Na+ diets. (B) Graphs represent SBP and DBP 12-hour night averages measured by telemetry in controls (n = 8) and Nedd4LPax8/LC1 KO (n = 6) under standard diet and after 3 and 12 days of high-Na+ diet. (C) SBP and DBP 24-hour profiles (day, white zone; night, gray zone) in controls (gray curve) and Nedd4LPax8/LC1 KO (black curve) after 12 days of high-Na+ diet. ZT, Zeitgeber time (ZT0 or ZT24, light on; ZT12, light off). *P < 0.05, †P < 0.01, ‡P < 0.005, KO versus controls.
Table 1.
Urine and plasma electrolytes in controls and inducible Nedd4LPax8/LC1 KO mice under standard and high-Na+ diet (8 days)
Loss of renal tubule NEDD4-2 in adult mice leads to salt-sensitive blood pressure increase.
Systolic and diastolic arterial blood pressures (SBP and DBP) were measured by telemetry for 3 consecutive 24-hour periods upon standard and high-Na+ diets. No significant difference between Nedd4LPax8/LC1 KO and control littermates was observed under standard diet (Figure 2B). However, following 12 days of high-Na+ diet, the difference between controls and KO reached 10 mmHg for the SBP during the active night period (Figure 2, B and C). These data indicate that renal tubule NEDD4-2 in adult mice is important for maintaining normal blood pressure under high-salt intake.
Nedd4L inactivation in adult renal tubules leads to increased β- and γENaC protein expression, but reduced αENaC proteolysis.
Because NEDD4-2 has been shown to participate in the degradation of ENaC, it was expected that Nedd4L inactivation would result in more ENaC expression. Immunofluorescence showed that both β- and γENaC are strongly increased in Nedd4LPax8/LC1 KO renal tubules (Figure 3, B and C, and Supplemental Figure 5). However, the cellular localization of both subunits was mainly cytoplasmic. Interestingly, αENaC expression was not increased in KO mice and seen predominantly at intracellular sites (Figure 3A and Supplemental Figure 5). Consistently, Scnn1a mRNA levels were lower in microdissected tubules of KO mice that paralleled the decreased plasma aldosterone levels observed in these mice (Figure 3D). On the other hand, Scnn1b and Scnn1g mRNA expression was unchanged (Figure 3, E and F). It is known that proteolytic cleavage of the α and γ subunits is involved in ENaC activation (10, 12, 29). We therefore analyzed by immunoblotting whether loss of NEDD4-2 would affect ENaC cleavage. Nedd4LPax8/LC1 KO mice showed a strong increase in the 90-kDa band for βENaC (Figure 3H). The same was observed for γENaC, but no 70-kDa cleaved form could be detected (Figure 3H). For αENaC, there was no change in the expression of the 90-kDa uncleaved band (quantification of blots from 3 independent experiments: controls: 1.00 ± 0.22, n = 16; KO: 0.94 ± 0.18, n = 17; P = 0.818), but the 30-kDa cleaved band was decreased in the KO (controls: 1.00 ± 0.20, n = 16; KO: 0.48 ± 0.13, n = 17; P = 0.044) (Figure 3G). When aldosterone was administered to the Nedd4LPax8/LC1 KO mice using osmotic minipumps, both uncleaved and cleaved forms of αENaC expression increased and achieved comparable levels to those in the controls (Figure 3I). These results indicate that the downregulation of the αENaC proteolytic cleavage is due to decreased aldosterone. Taken together, our data suggest that ENaC activity is downregulated in Nedd4LPax8/LC1 KO mice.
Figure 3. Nedd4LPax8/LC1 KO mice have increased β- and γENaC abundance, but not αENaC.

(A–C) Immunostaining for α- (A), β- (B), and γENaC (C) on kidney cryosections of control and KO mice under high-Na+ diet. Cytoplasmic β- and γENaC abundance is increased in CD of KO, whereas αENaC is decreased. Scale bars: ∼20 μm. (D) TaqMan analysis of Scnn1a mRNA on microdissected renal tubules shows downregulation in KO (n = 4 per group). (E and F) TaqMan analysis for Scnn1b (E) and Scnn1g (F) mRNA on whole kidney (n = 6 per genotype). mRNA expression was normalized to Gapdh mRNA levels and expressed relative to control values. (G and H) Representative Western blots for α-, β-, and γENaC: the cleaved 30-kDa αENaC band was decreased in Nedd4LPax8/LC1 KO mice (G), whereas β- and γENaC expression were elevated (H). For γENaC, lanes were run on the same gel but were noncontiguous, as indicated by the vertical black line. (I) Aldosterone infusion in Nedd4LPax8/LC1 KO mice results in increased αENaC protein (uncleaved and cleaved form) expression, suggesting that the reduced expression observed in the KO is due to the decreased plasma aldosterone. Kidney extract from Scnn1a KO mice was used as negative control. Lanes that were run on the same gel but were noncontiguous are indicated with the vertical black lines. *P < 0.05, KO versus controls. Ctrl, control mice.
Loss of renal tubule NEDD4-2 in adult mice leads to overactive NCC.
We have recently showed that NEDD4-2 is involved in the regulation of NCC at the posttranslational level (24). Here, we confirm, by using immunofluorescence, that loss of NEDD4-2 leads to increased NCC protein abundance (Figure 4A) without any change of Slc12a3 mRNA expression in microdissected renal tubules (Figure 1B). NCC immunostaining was hardly visible in control mice fed a high-Na+ diet, while an immunofluorescent signal was easily detectable in the KO mice. Immunoblotting data indicated that the elevated NCC phosphorylation at T53, T58, and S71, known to be involved in the activation of the cotransporter, followed the increased NCC protein abundance in KO (ref. 30 and Figure 4, B and C). To confirm our observation, we looked at NCC expression in kidneys from P19 Nedd4L-Δ15-16 KO mice generated by Boase et al. (31) and observed an increase of expression and phosphorylation of the cotransporter also in these mice (Supplemental Figure 6). Thus, we confirm that loss of NEDD4-2 leads to increased NCC. PHAII patients and mouse models with overactive NCC display hypercalciuria that can be treated by thiazides, whereas hypocalciuria is observed in Slc12a3 KO mice (32). NCC activity is thus correlated with renal Ca2+ excretion. Based on this observation, we measured the urinary Ca2+ and found that the Nedd4LPax8/LC1 KO excrete more Ca2+ under a high-Na+ diet. Interestingly, thiazide treatment markedly reduced the elevated Ca2+ excretion in the KO, similar to what occurs in PHAII patients (Figure 4D). In addition, thiazide, but not amiloride, decreased the difference in blood pressure between controls and KO during the active night period (Figure 4E). Taken together, these data suggest that loss of NEDD4-2 in renal tubules in adult mice leads to increased NCC activity.
Figure 4. NCC is overactivated in Nedd4LPax8/LC1 KO mice.

(A) Immunofluorescence for NCC on kidney sections of control and KO mice under high-Na+ diet. NCC expression is increased in DCT of KO. Scale bars: ∼20 μm. (B) Western blot analysis for total NCC and phosphorylated pT53-, pT58-, pS71-, and pT43/53/58-NCC. A representative blot on 5 controls and 5 KO is shown. For pS71-NCC and pT43/53/58-NCC, lanes that were run on the same gel but noncontiguous are indicated with a vertical black line. (C) Graphs show quantification of Western blots for phosphorylated NCC and the corresponding total NCC from 2 independent experiments (pT53-NCC and pT58-NCC: n = 5 per genotype; pS71-NCC and p43/53/58-NCC: n = 11 per genotype). Protein expression was normalized to the amount of β-actin or β-tubulin and expressed relative to control values. (D) Urine Ca2+ measurement in control and KO mice under high-Na+ diet (10 days). Nedd4LPax8/LC1 KO are hypercalciuric as shown by the 3-fold increased urine Ca2+/creatinine (Cr) ratio, which can be corrected by thiazide (n = 4 per group). Vh, vehicle; Tz, thiazide. (E) Plot showing telemetric measurement on controls versus KO under high-Na+ diet. The increased SBP and DBP observed in KO after 5 weeks of high-Na+ diet can be prevented by thiazide, but not by amiloride (n = 4 per group). *P < 0.05, **P < 0.01, KO versus controls. †P < 0.05, ††P < 0.01, thiazide versus vehicle.
Nedd4LPax8/LC1 KO mice have increased ROMK abundance.
Increased NCC activity has been shown to be linked with K+ retention (33, 34). Therefore, the absence of hyperkalemia in Nedd4LPax8/LC1 KO mice was unexpected, as they do also show decreased ENaC activity and plasma aldosterone that should decrease the electrochemical driving force for K+ excretion via apical K+ channels such as ROMK in the ENaC-expressing cells. We therefore analyzed the abundance and subcellular localization of ROMK in control and KO mice. Interestingly, immunofluorescence showed a strong increase in ROMK protein abundance and apical localization in the DCT and CNT of Nedd4LPax8/LC1 KO (Figure 5A) and in CD and thick ascending limb (TAL) (Figure 5A). Kcnj1 mRNA expression levels (Figure 5B) were not different between controls and KO, indicating that loss of NEDD4-2 affects ROMK expression at the posttranslational level.
Figure 5. Nedd4LPax8/LC1 KO mice have increased ROMK abundance.

(A) Immunofluorescence for ROMK on kidney sections from control and KO mice fed with high-Na+ diet (10 days) shows increase in the channel in DCT2 (D2), CNT (CN), CD and TAL (T) in KO. Scale bars: ∼40 μm. (B) TaqMan analysis showed that Kcnj1 mRNA levels are not changed between controls and KO mice on whole kidney.
Nedd4L ablation in renal tubules during embryonic development results in a phenotype similar to that seen when the deletion occurs at adult age.
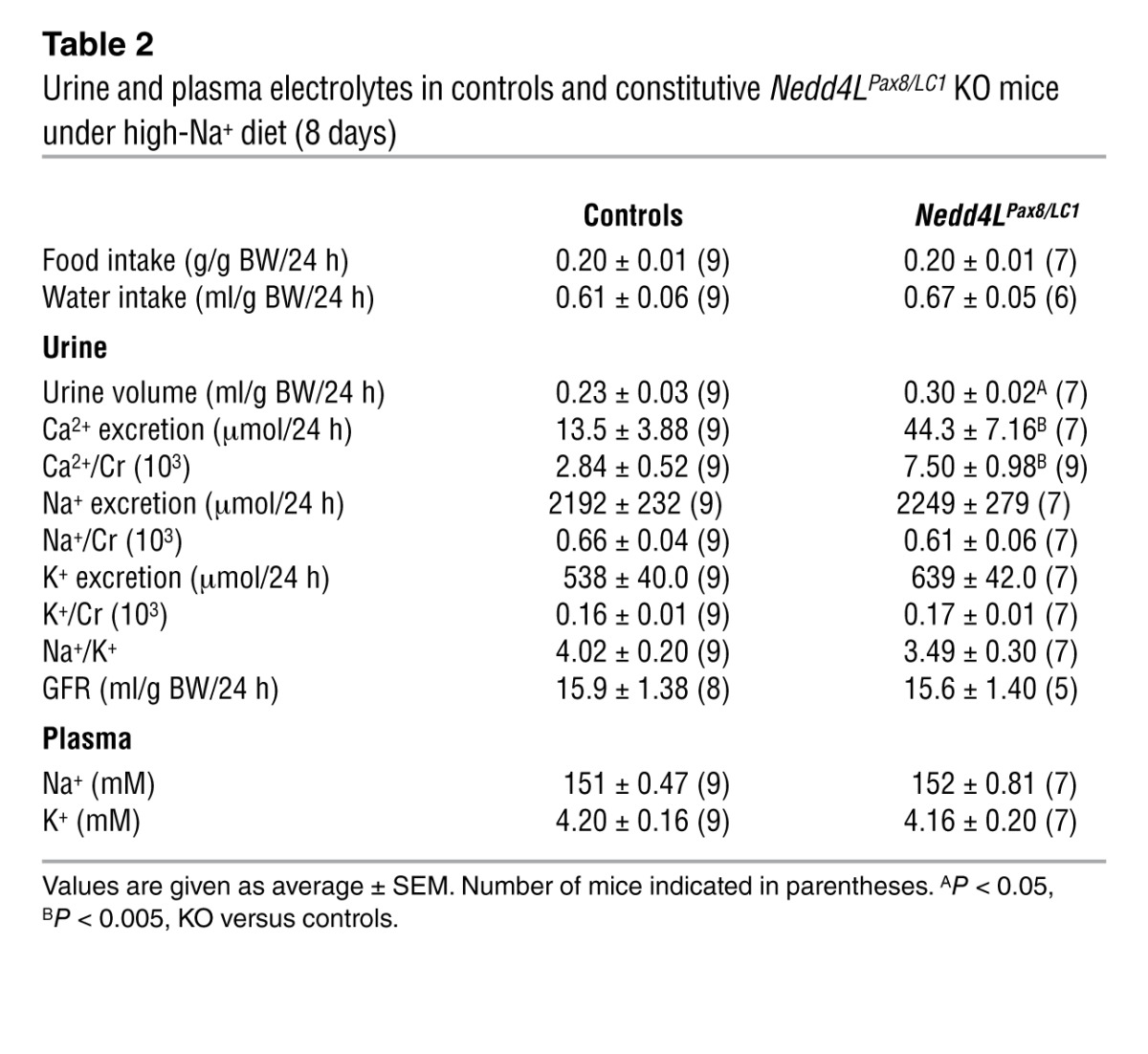
Because the total Nedd4L-Δ6-8 KO mice showed no change in plasma aldosterone under standard and high-Na+ diets, increase in αENaC expression, and no alteration of NCC abundance (25), we hypothesized that the time of Nedd4L deletion may account for the differences with the Nedd4LPax8/LC1 KO mice. We treated the pregnant mothers of Nedd4LPax8/LC1 KO mice and control littermates with doxycycline during gestation to induce deletion during embryonic development, as previously reported (27). We observed that the constitutive Nedd4L ablation in renal tubules had effects on NCC and α-, β- and γENaC expression (Figure 6, A–C) similar to those seen when the deletion was induced during adulthood. Moreover, the constitutive Nedd4LPax8/LC1 KO mice showed decreased plasma aldosterone (Figure 6D) and hypercalciuria (Table 2). These data suggest that the differences in phenotype between the Nedd4LPax8/LC1 KO mice and the total Nedd4L-Δ6-8 KO mice are most likely not due to early developmental adaptation, but rather to nonrenal NEDD4-2 or to the differences in genetic background between these mouse models.
Figure 6. Nedd4L ablation in renal tubules during embryonic development results in phenotype similar to that seen when the deletion occurs at adult age.

Mice were treated with doxycycline during embryonic development to induce Nedd4L ablation early on. (A–C) Western blot analysis on 5-week-old control and KO mice showed increased NCC (A), β-, and γENaC (C) expression, and decreased cleaved αENaC under high-Na+ diet (10 days) (B). (D) Plasma aldosterone levels were decreased in KO (n = 7) compared with control mice (n = 7) under standard diet. *P < 0.05, KO versus controls.
Table 2.
Urine and plasma electrolytes in controls and constitutive Nedd4LPax8/LC1 KO mice under high-Na+ diet (8 days)
Discussion
Mutations in human β- and γENaC leading to inactivation of the PY motif that interacts with NEDD4-2 WW domains cause Liddle syndrome, characterized by increased ENaC activity, salt retention, and hypertension (3). Surprisingly, constitutive deletion of exons 6 to 8 of the Nedd4L gene in the mouse results in amiloride-sensitive salt-induced hypertension, but no sign of volume expansion or impaired Na+/K+ balance (25). These observations raise the question on the role of renal NEDD4-2 in controlling ENaC and blood pressure. To address this issue, we generated inducible renal tubule–specific Nedd4L KO (Nedd4LPax8/LC1) mice. No Nedd4L mRNA corresponding to the deleted region as well as no protein product could be detected in microdissected renal tubules of Nedd4LPax8/LC1 KO mice, suggesting that Nedd4L is efficiently deleted in the targeted renal tubular cells. Recently, another constitutive Nedd4L KO mouse model was generated by constitutively deleting exons 15 and 16 of the Nedd4L gene (Nedd4L-Δ15-16 KO mice) (31). Such deletion led to a perinatal lethal lung phenotype in contrast to the Nedd4L-Δ6-8 KO model. Boase et al. provided evidence that production of a partial NEDD4-2 protein product in the lungs of the Nedd4L-Δ6-8 KO may be the reason for the differences between the 2 models (31). Such truncated NEDD4-2 protein product was not detected in kidneys of our Nedd4LPax8/LC1 KO mice.
Surprisingly, despite the increase in NCC, and β- and γENaC protein levels and the decreased circulating aldosterone, suggesting Na+ retention and confirming our previous report (24), Nedd4LPax8/LC1 KO mice showed normal urine and plasma Na+ and K+ levels. NCC has been reported to be activated by increasing its cell-surface expression and phosphorylation state, mainly via the WNK-SPAK/OSR1 pathway (17, 35). We found that NCC abundance was increased in Nedd4LPax8/LC1 KO mice and is accompanied by a proportional increase in phosphorylation of T53, T58, and S71 (30, 35). We also observed that Nedd4LPax8/LC1 KO mice have salt-sensitive blood pressure increase and hypercalciuria that could both be corrected by thiazides, corroborating the assumption that the increased NCC is functional. Of interest, thiazides did not completely correct the high blood pressure, suggesting that mechanisms other than NCC activation are likely to contribute. Interestingly, β- and γENaC abundance were increased in the Nedd4LPax8/LC1 KO mice, but their cellular localization was mainly intracellular, and γENaC appeared to be in its uncleaved form. Unexpectedly, Scnn1a mRNA expression and proteolytic cleavage of the subunit were decreased in the Nedd4LPax8/LC1 KO mice, suggesting ENaC downregulation. Moreover, amiloride treatment of Nedd4LPax8/LC1 KO mice did not decrease the high blood pressure, indicating that ENaC is not involved in the hypertensive phenotype. Interestingly, the ENaC downregulation observed in the Nedd4LPax8/LC1 KO mice paralleled the decreased plasma aldosterone and could be rescued by administrating aldosterone to the KO mice. These data confirm (a) the crucial role of aldosterone in controlling αENaC expression in the kidney (36), and (b) the importance of αENaC for apical trafficking of the 2 other ENaC subunits (37). To our knowledge, this is the first in vivo study showing differential regulation between the upregulation of β/γENaC (a likely result of the lack of ubiquitylation) and the downregulation of proteolytic activation of αENaC due to decreased aldosterone. Interestingly, no human mutation in the gene encoding αENaC (Scnn1a) has been reported to cause Liddle syndrome. Taken together, these results give new insights about how the different mechanisms involved in ENaC regulation can interact and compensate each other if one is impaired. However, our results stand in contrast with the phenotype of Liddle syndrome, where ENaC activity is increased despite the low circulating aldosterone levels (38). It is likely that deletion of Nedd4L affects other factors, such as NCC upregulation and consequent compensatory pathways, or other factors involved in controlling ENaC activity or trafficking (39). Because modifications in the abundance of individual renal Na+ transporters often result in compensatory changes in the expression levels of other Na+ transporters to maintain Na+ balance (40), we propose that NEDD4-2 deficiency leads to increased NCC-mediated Na+ reabsorption, which is compensated by decreased ENaC activity. Consistent with our hypothesis, KO mice for the kidney-specific KS-Wnk1 isoform, in which NCC is strongly upregulated, present a very similar phenotype with downregulation of the 3 ENaC subunits (41). Of interest, the phenotype of the Nedd4LPax8/LC1 KO mice is the mirror image of the Slc12a3 KO, which show increased plasma aldosterone, decreased calciuria, and upregulation of ENaC activity in CD (28).
Our results in the Nedd4LPax8/LC1 KO mice are however different to what was observed in the total Nedd4L-Δ6-8 KO mice, in which NCC was unchanged and all 3 ENaC subunits were increased (25). One substantial difference between the 2 models is the plasma aldosterone that is much higher in the total Nedd4L-Δ6-8 KO mice and could be attributed to the systemic effects. This difference in plasma aldosterone could explain many of the changes, especially with regard to ENaC. Compensatory mechanisms that could occur during development in the total Nedd4L-Δ6-8 KO mice may also explain the difference with our inducible system. However, when we induced the renal Nedd4L deletion during embryonic development, we obtained a phenotype similar to that in the postnatal deletion of renal Nedd4L, with decreased plasma aldosterone, increased NCC, and decreased αENaC proteolysis. Therefore, the differences with the total Nedd4L-Δ6-8 KO model could rather be due to variations in genetic background or to loss of nontubular NEDD4-2 function. Indeed, Van Huysse et al. recently demonstrated that the salt-induced elevated blood pressure in the Nedd4L-Δ6-8 KO mouse model depends critically on ENaC overexpression in the brain (42).
Interestingly, despite the strong increase in renal NCC expression levels, the Nedd4LPax8/LC1 KO mice displayed some but not all the features characteristic of PHAII. The role of NCC overactivation in developing PHAII has been highlighted by human mutations and animal models. Wilson et al. first identified mutations in the WNK1 and WNK4 genes in some patients with PHAII (22). The PHAII-Wnk4Q562E transgenic mouse model developed by Lalioti et al. exhibits increased NCC and PHAII features that could be abolished by crossing these mice with Slc12a3 KO mice (33). Yang et al. generated mice with 1 normal copy of Wnk4 and 1 mutant Wnk4D561A (34). These mice displayed increased phosphorylation and expression of NCC as well as hypertension and hyperkalemia that could be treated with thiazide. However, there is now increasing evidence that overactive NCC alone is not sufficient to lead to PHAII and that the deregulation of other channels or transporters might be required. As mentioned above, KS-Wnk1 KO mice, with strong increase in NCC and decreased ENaC, showed only few of the PHAII features (41). In addition, a recent study of NCC overexpression in mice showed no effect on blood pressure, plasma, or urine electrolyte and urinary Ca2+ (43). One of the PHAII features that is missing in the Nedd4LPax8/LC1 KO mice is the hyperkalemia. Several lines of evidence have shown that NCC is important for K+ conservation, as Gitelman patients (44) and Slc12a3 KO mice exhibit hypokalemia (45) and hyperkalemia is observed in PHAII patients (33, 34). The increased ROMK abundance observed in the Nedd4LPax8/LC1 KO mice, which could result from the lack of NEDD4-2–mediated ubiquitylation (46), may lead to higher K+ excretion, as it has been reported for the KS-Wnk1 KO mice (41). Another possibility could be that the Nedd4LPax8/LC1 KO mice have some residual ENaC activity sufficient for excreting enough K+ to maintain a normal kalemia. Whether some ENaC-independent K+ secretion occurs in the Nedd4LPax8/LC1 KO mice remains to be addressed.
In conclusion, we provide evidence that inactivation of Nedd4L exons 6 to 8 in renal tubules of adult mice does not lead to a Liddle syndrome phenotype associated with elevated ENaC activity, but rather causes a salt-sensitive PHAII-like syndrome with NCC upregulation, increased blood pressure, and hypercalciuria. Based on these results, NEDD4-2 appears to primarily target NCC and might thus be an attractive target to treat hypertension, avoiding the most severe side effect of thiazides, namely hypokalemia. Moreover, there is an interesting clinical association of hypercalciuria with hypertension and an increased risk of nephrolithiasis in hypertension (47–50). Defects in NEDD4-2 could therefore underlie these associations, and its study could lead to important mechanistic insights. The recent discoveries that mutations in KLHL3 and CUL3 (part of a ubiquitin-protein ligase complex) lead to PHAII demonstrate the importance of the ubiquitylation process in the control of NCC activation. In addition, Khan et al. very recently demonstrated that NCC ubiquitylation is regulated by phosphorylation of the cotransporter and may contribute to the control of NCC surface expression (51). Finally, our data suggest that under high-Na+ intake, NEDD4-2 is not crucial for regulating renal ENaC, as the low plasma aldosterone leading to decreased αENaC proteolytic cleavage is probably sufficient to counterbalance the increased β- and γENaC abundance in the Nedd4LPax8/LC1 KO mice and thus to compensate for the increased NCC activity and maintain normal Na+/K+ balance. Why other pathways known to regulate NCC, such WNK-SPAK/OSR1, do not compensate for the increased NCC activity resulting from the lack of NEDD4-2 represents an important question for further investigation.
Methods
Generation and induction of renal tubule–specific Nedd4L KO mice.
Inducible renal tubule–specific Nedd4Lflox/flox/Pax8-rTA/LC1 KO (Nedd4LPax8/LC1) mice were generated as described previously (24). Genotyping and recombination of PCR were performed as described in Supplemental Methods. Three- to 4-week-old KO and single transgenic homozygous Nedd4LPax8 or Nedd4LLC1 littermates (controls) were treated with doxycycline (2 mg/ml in 2% sucrose in drinking water) for 11 days. All experiments were started 1 day after the end of the induction, if not stated otherwise. To delete Nedd4L during embryonic development, pregnant females were treated with doxycycline as described above during the breeding and gestation periods, and resulting pups were treated until weaning. Induced mice were fed a standard (0.18% Na+; Sniff) or high-Na+ diet (>3.2% Na+) for 8 days and placed in metabolic cages to measure BW, water, and food consumption and to collect urine for 24 hours. For thiazide treatment, mice were injected i.p. at Zeitgeber time ZT2 (2 hours after light on) with vehicle or thiazide (20 mg/kg BW) and urine was collected 6, 12, 24, and 30 hours after injection. Creatinine and Ca2+ were measured by the Laboratory of Clinical Chemistry at the Lausanne Hospital (CHUV) using a Modular Analytics System (Roche Diagnostics). Mice were anesthetized by isoflurane inhalation for blood collection by retroorbital plexus puncture and sacrificed by cervical dislocation for tissue collection.
Inducible renal tubule–specific Scnn1a KO mice.
Scnn1aflox/flox/Pax8-rTA/LC1 KO mice were generated and induced as described above for the Nedd4LPax8/LC1 KO mice, but using Scnn1aflox/flox mice (52).
Microdissection of mouse renal tubules.
Kidneys were perfused and microdissected as described previously (53). For each tubular segment (PT, TAL, DCT, or CNT/CD), tubules microdissected from 3 mice were pooled together with equal tubular length (controls: 6–8 cm; KO: 12 cm to be sure not to miss any weak signal in comparison with controls).
Real-time quantitative PCR.
Total RNA was isolated from kidney using the RNAquous Kit (Ambion) (1 μg) or from renal tubules microdissected as described in (53) using the RNeasy MicroKit (QIAGEN). RNA was reverse-transcribed and used for real-time quantitative PCR as described (24). Information about TaqMan Gene Expression Assays (Applied Biosystems) and primer/probe sequences are given in Supplemental Table 1. Regarding Nedd4L, the probe and primers were chosen to be located in the exon 6 to 8 region that was deleted in the KO mice after doxycycline treatment.
Measurement of urine and plasma metabolites and aldosterone.
Urinary and plasma Na+ and K+ were measured using a flame photometer (Cole-Palmer Instrument), and urine osmolality with an Advanced 2020 osmometer (Advanced Instruments). Glomerular filtration rate (GFR) was estimated based on creatinine clearance. Plasma aldosterone levels were measured as described (24).
Telemetry.
Experiments were performed on 1.5-month-old male controls and induced KO as described (41). Mice were fed a standard diet for 7 days, a low-Na+ diet (<0.01% Na+; Sniff) for 7 days, and then a high-Na+ diet for up to 5 weeks. After a 10-day recovery period, cardiovascular parameters were recorded 9 seconds every minute for 24 hours in a light/dark-cycle (ZT0-ZT12/ZT12-ZT24). For thiazide and amiloride experiments, mice were injected i.p. with either vehicle, thiazide (20 mg/kg BW), or amiloride (5 mg/kg BW) at ZT11 and blood pressure was measured during the active night period between 2 hours (ZT13) and 9 hours (ZT20) after injection.
Aldosterone treatment.
The ALZET 1003D osmotic mini-pumps (preloaded with vehicle or aldosterone to get a dose of 150 μg/kg BW/d for 3 days) were implanted s.c. Mice were kept on a low-Na+ diet, from the implantation day until sacrifice, to avoid aldosterone escape.
Immunoblotting.
Frozen tissues were homogenized and protein extracted as described (24). Anti–α-, –β-, and –γENaC were used as described (14). Kidneys from inducible renal tubule–specific Scnn1a KO mice were used as controls for the ENaC-specific bands. For analysis of protein expression in Nedd4L total KO mice, kidneys from P19 Nedd4L-Δ15-16 total KO mice (31) were used. NEDD4-2 was detected using an anti–NEDD4-2 generated in the laboratory of S. Kumar and diluted 1/1000 (31) or with other antibodies directed against different regions of NEDD4-2 and described in Supplemental Methods. NEDD4-1 was detected as described in Supplemental Methods. NCC was detected with an anti-NCC antibody (Chemicon). For anti-phosphorylated pT53-NCC and pT58-NCC, peptide-synthesis, immunizations of rabbits, and antibody purifications were custom-made by Pineda Antibody Services. Phospho-antibodies were preincubated with the corresponding non–phospho-peptides and diluted 1/5000. Anti-phosphorylated pS71-NCC and anti-pT43/53/58-NCC were provided by D. Alessi (MRC Protein Phosphorylation Unit, University of Dundee, Dundee, United Kingdom) and preincubated with the corresponding non–phospho-peptide (35, 54). Anti–β-actin or anti–β-tubulin antibody (Sigma-Aldrich) was used as loading control. For protein analysis on microdissected renal tubules, Western blots were performed as described (53) using the NEDD4-2 antibody previously described (31).
Immunofluorescence.
Kidneys were processed and cryosections incubated with antibodies against CRE, α-, β-, γENaC, NCC, and ROMK1,2 as described (15, 55).
Statistics.
All values are presented as mean ± SEM. The data were analyzed using unpaired 2-tailed Student’s t test, KO versus controls. For thiazide and amiloride injection, a paired t test was used, diuretic injected versus vehicle injected. A P value of less than 0.05 was considered significant.
Study approval.
All experimental procedures were approved by the Swiss Federal Veterinary Office and carried out in accordance with the local animal welfare act.
Supplementary Material
Acknowledgments
We thank J.P. Arroyo, O. Bonny, N. Faresse, G. Gamba, D. Pouly, R.D. Rajuram, B. Rossier, and L. Schild for critically reading the manuscript. We are very thankful to R. Garcia Barros and M. Stifanelli for their technical assistance. This work was supported by the Leducq Foundation Transatlantic Network on Hypertension (to O. Staub and E. Hummler), the Swiss National Science Foundation 310030-141013 (to O. Staub), 310030-122243 (to J. Loffing) and 3100A0-102125/1 (to E. Hummler), the Swiss NCCR Kidney.ch (to O. Staub, J. Loffing, and E. Hummler), the Swiss Kidney Foundation (to C. Ronzaud), and National Health and Medical Research Council grant APP1020755 and fellowship 1002863 (to N.A. Boase and S. Kumar).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(2):657–665. doi:10.1172/JCI61110.
See the related Commentary beginning on page 546.
References
- 1.Reilly RF, Ellison DH. Mammalian distal tubule: physiology, pathophysiology, and molecular anatomy. Physiol Rev. 2000;80(1):277–313. doi: 10.1152/physrev.2000.80.1.277. [DOI] [PubMed] [Google Scholar]
- 2.Giebisch G. Renal potassium transport: mechanisms and regulation. Am J Physiol. 1998;274(5 pt 2):F817–F833. doi: 10.1152/ajprenal.1998.274.5.F817. [DOI] [PubMed] [Google Scholar]
- 3.Shimkets RA, et al. Liddle’s syndrome: heritable human hypertension caused by mutations in the β subunit of the epithelial sodium channel. Cell. 1994;79(3):407–414. doi: 10.1016/0092-8674(94)90250-X. [DOI] [PubMed] [Google Scholar]
- 4.Snyder PM, Steines JC, Olson DR. Relative contribution of Nedd4 and Nedd4-2 to ENaC regulation in epithelia determined by RNA interference. J Biol Chem. 2004;279(6):5042–5046. doi: 10.1074/jbc.M312477200. [DOI] [PubMed] [Google Scholar]
- 5.Staub O, et al. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. . EMBO J. 1996;15(10):2371–2380. [PMC free article] [PubMed] [Google Scholar]
- 6.Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O. A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. . FASEB J. 2001;15(1):204–214. doi: 10.1096/fj.00-0191com. [DOI] [PubMed] [Google Scholar]
- 7.Abriel H, et al. Defective regulation of the epithelial Na+ channel (ENaC) by Nedd4 in Liddle’s syndrome. . J Clin Invest. 1999;103(5):667–673. doi: 10.1172/JCI5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harvey KF, Dinudom A, Cook DI, Kumar S. The Nedd4-like protein KIAA0439 is a potential regulator of the epithelial sodium channel. J Biol Chem. 2001;276(11):8597–8601. doi: 10.1074/jbc.C000906200. [DOI] [PubMed] [Google Scholar]
- 9.Debonneville C, et al. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J. 2001;20(24):7052–7059. doi: 10.1093/emboj/20.24.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hughey RP, et al. Maturation of the epithelial Na+ channel involves proteolytic processing of the α- and γ-subunits. J Biol Chem. 2003;278(39):37073–37082. doi: 10.1074/jbc.M307003200. [DOI] [PubMed] [Google Scholar]
- 11.Masilamani S, Kim G-H, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC α, β, and γ subunit proteins in rat kidney. J Clin Invest. 1999;104(7):R19–R23. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossier BC, Stutts MJ. Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu Rev Physiol. 2009;71:361–379. doi: 10.1146/annurev.physiol.010908.163108. [DOI] [PubMed] [Google Scholar]
- 13.Canessa CM, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. . Nature. 1994;367(6462):463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 14.Rubera I, et al. Collecting duct-specific gene inactivation of αENaC in the mouse kidney does not impair sodium and potassium balance. J Clin Invest. 2003;112(4):554–565. doi: 10.1172/JCI16956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ronzaud C, et al. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol. 2007;18(6):1679–1687. doi: 10.1681/ASN.2006090975. [DOI] [PubMed] [Google Scholar]
- 16.Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. 2005;85(2):423–493. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- 17.Wilson FH, et al. Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc Natl Acad Sci U S A. 2003;100(2):680–684. doi: 10.1073/pnas.242735399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang CL, Angell J, Mitchell R, Ellison DH. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest. 2003;111(7):1039–1045. doi: 10.1172/JCI17443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Louis-Dit-Picard H, et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet. 2012;44(4):456–460. doi: 10.1038/ng.2218. [DOI] [PubMed] [Google Scholar]
- 20.Boyden LM, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482(7383):98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faresse N, et al. Inducible kidney-specific Sgk1 knockout mice show a salt-losing phenotype. Am J Physiol Renal Physiol. 2012;302(8):F977–F985. doi: 10.1152/ajprenal.00535.2011. [DOI] [PubMed] [Google Scholar]
- 22.Wilson FH, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293(5532):1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 23.Mayan H, Vered I, Mouallem M, Tzadok-Witkon M, Pauzner R, Farfel Z. Pseudohypoaldosteronism type II: marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J Clin Endocrinol Metab. 2002;87(7):3248–3254. doi: 10.1210/jc.87.7.3248. [DOI] [PubMed] [Google Scholar]
- 24.Arroyo JP, et al. Nedd4-2 Modulates Renal Na+-Cl- Cotransporter via the Aldosterone-SGK1-Nedd4-2 Pathway. J Am Soc Nephrol. 2011;22(9):1707–1719. doi: 10.1681/ASN.2011020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi PP, et al. Salt-sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4-2. Am J Physiol Renal Physiol. 2008;295(2):F462–F470. doi: 10.1152/ajprenal.90300.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loffing-Cueni D, et al. Dietary sodium intake regulates the ubiquitin-protein ligase nedd4-2 in the renal collecting system. J Am Soc Nephrol. 2006;17(5):1264–1274. doi: 10.1681/ASN.2005060659. [DOI] [PubMed] [Google Scholar]
- 27.Traykova-Brauch M, et al. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med. 2008;14(9):979–984. doi: 10.1038/nm.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamynina E, Tauxe C, Staub O. Differential characteristics of two human Nedd4 proteins with respect to epithelial Na+ channel regulation. . Am J Physiol. 2001;281:F469–F477. doi: 10.1152/ajprenal.2001.281.3.F469. [DOI] [PubMed] [Google Scholar]
- 29.Ruffieux-Daidie D, Poirot O, Boulkroun S, Verrey F, Kellenberger S, Staub O. Deubiquitylation regulates activation and proteolytic cleavage of ENaC. J Am Soc Nephrol. 2008;19(11):2170–2180. doi: 10.1681/ASN.2007101130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pacheco-Alvarez D, et al. The Na+:Cl- cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem. 2006;Na(39):28755–28763. doi: 10.1074/jbc.M603773200. [DOI] [PubMed] [Google Scholar]
- 31.Boase NA, et al. Respiratory distress and perinatal lethality in Nedd4-2-deficient mice. Nat Commun. 2011;2:287. doi: 10.1038/ncomms1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loffing J, et al. Altered renal distal tubule structure and renal Na(+) and Ca(2+) handling in a mouse model for Gitelman’s syndrome. J Am Soc Nephrol. 2004;15(9):2276–2288. doi: 10.1097/01.ASN.0000138234.18569.63. [DOI] [PubMed] [Google Scholar]
- 33.Lalioti MD, et al. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet. 2006;38(10):1124–1132. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 34.Yang SS, et al. Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab. 2007;5(5):331–344. doi: 10.1016/j.cmet.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 35.Richardson C, et al. Activation of the thiazide-sensitive Na+-Cl- cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci. 2008;121(pt 5):675–684. doi: 10.1242/jcs.025312. [DOI] [PubMed] [Google Scholar]
- 36.Asher C, Wald H, Rossier BC, Garty H. Aldosterone-induced increase in the abundance of Na+ channel subunits. Am J Physiol. 1996;271(2 pt 1):C605–C611. doi: 10.1152/ajpcell.1996.271.2.C605. [DOI] [PubMed] [Google Scholar]
- 37.Rubera I, et al. Collecting duct-specific gene inactivation of alphaENaC in the mouse kidney does not impair sodium and potassium balance. J Clin Invest. 2003;112(4):554–565. doi: 10.1172/JCI16956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pradervand S, et al. Dysfunction of the epithelial sodium channel expressed in the kidney of a mouse model for Liddle syndrome. J Am Soc Nephrol. 2003;14(9):2219–2228. doi: 10.1097/01.ASN.0000080204.65527.E6. [DOI] [PubMed] [Google Scholar]
- 39.Staub O, Rotin D. Role of ubiquitylation in cellular membrane transport. Physiol Rev. 2006;86(2):669–707. doi: 10.1152/physrev.00020.2005. [DOI] [PubMed] [Google Scholar]
- 40.Brooks HL, et al. Profiling of renal tubule Na+ transporter abundances in NHE3 and NCC null mice using targeted proteomics. J Physiol. 2001;530(pt 3):359–366. doi: 10.1111/j.1469-7793.2001.0359k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hadchouel J, et al. Decreased ENaC expression compensates the increased NCC activity following inactivation of the kidney-specific isoform of WNK1 and prevents hypertension. Proc Natl Acad Sci U S A. 2010;107(42):18109–18114. doi: 10.1073/pnas.1006128107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Huysse JW, Amin MS, Yang B, Leenen FH. Salt-induced hypertension in a mouse model of liddle syndrome is mediated by epithelial sodium channels in the brain. Hypertension. 2012;60(3):691–696. doi: 10.1161/HYPERTENSIONAHA.112.193045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCormick JA, Nelson JH, Yang CL, Curry JN, Ellison DH. Overexpression of the sodium chloride cotransporter is not sufficient to cause familial hyperkalemic hypertension. Hypertension. 2011;58(5):888–894. doi: 10.1161/HYPERTENSIONAHA.110.167809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simon DB, et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet. 1996;12(1):24–30. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 45.Morris RG, Hoorn EJ, Knepper MA. Hypokalemia in a mouse model of Gitelman’s syndrome. Am J Physiol Renal Physiol. 2006;290(6):F1416–F1420. doi: 10.1152/ajprenal.00421.2005. [DOI] [PubMed] [Google Scholar]
- 46.Lin DH, et al. POSH stimulates the ubiquitination and the clathrin-independent endocytosis of ROMK1 channels. J Biol Chem. 2009;284(43):29614–29624. doi: 10.1074/jbc.M109.041582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khan SR. Is oxidative stress, a link between nephrolithiasis and obesity, hypertension, diabetes, chronic kidney disease, metabolic syndrome? Urol Res. 2012;40(2):95–112. doi: 10.1007/s00240-011-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cupisti A. Update on nephrolithiasis: beyond symptomatic urinary tract obstruction. J Nephrol. 2011;18:S25–S29. doi: 10.5301/JN.2011.7766. [DOI] [PubMed] [Google Scholar]
- 49.Reilly RF, Peixoto AJ, Desir GV. The evidence-based use of thiazide diuretics in hypertension and nephrolithiasis. Clin J Am Soc Nephrol. 2010;5(10):1893–1903. doi: 10.2215/CJN.04670510. [DOI] [PubMed] [Google Scholar]
- 50.Obligado SH, Goldfarb DS. The association of nephrolithiasis with hypertension and obesity: a review. Am J Hypertens. 2008;21(3):257–264. doi: 10.1038/ajh.2007.62. [DOI] [PubMed] [Google Scholar]
- 51.Hossain Khan MZ, et al. Phosphorylation of Na-Cl cotransporter by OSR1 and SPAK kinases regulates its ubiquitination. Biochem Biophys Res Commun. 2012;425(2):456–461. doi: 10.1016/j.bbrc.2012.07.124. [DOI] [PubMed] [Google Scholar]
- 52.Hummler E, et al. Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat Genet. 1996;12(3):325–328. doi: 10.1038/ng0396-325. [DOI] [PubMed] [Google Scholar]
- 53.Christensen BM, et al. Sodium and potassium balance depends on alphaENaC expression in connecting tubule. J Am Soc Nephrol. 2010;21(11):1942–1951. doi: 10.1681/ASN.2009101077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rafiqi FH, et al. Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol Med. 2010;2(2):63–75. doi: 10.1002/emmm.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wagner CA, et al. Mouse model of type II Bartter’s syndrome. II. Altered expression of renal sodium- and water-transporting proteins. Am J Physiol Renal Physiol. 2008;294:F1373–F1380. doi: 10.1152/ajprenal.00613.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.