

Abstract
The thin layer of cells that lines the interior of blood vessels, known as the endothelium, plays a complex role in vascular biology. The endothelium mediates blood vessel tone, hemostasis, neutrophil recruitment, hormone trafficking, and fluid filtration. Endothelial dysfunction, as defined by a lack of NO, has been linked to a variety of disease states, including atherosclerosis, diabetes mellitus, coronary artery disease, hypertension, and hypercholesterolemia. Indeed, restoration of endothelial function is one of the earliest recognizable benefits of statin therapy. In 1995, James Liao and colleagues published a study in the JCI demonstrating that NO is a vascular protective factor that limits endothelial activation and prevents leukocyte adhesion to the vessel wall.
Two converging concepts, endothelial dysfunction and endothelial activation, served as the basis for our 1995 JCI study (1). This study would not have been possible without the serendipitous contributions of our coinvestigators. At the time, we were interested in mechanisms underlying endothelial dysfunction, which is defined as the decreased synthesis, release, and/or activity of endothelium-derived NO, and how endothelial dysfunction could contribute to vascular disease. Gimbrone and colleagues were developing the concept of endothelial cell activation in response to nonlaminar flow as an early mediator of leukocyte trafficking in the vascular wall (2). Endothelial cell activation is defined by the endothelial expression of cell-surface adhesion molecules, such as VCAM-1, ICAM-1, and endothelial leukocyte adhesion molecule (ELAM, also known as E-selectin). Endothelial cell activation is typically induced by proinflammatory cytokines, such as TNF-α and IL-6, and facilitates the recruitment and attachment of circulating leukocytes to the vessel wall. In addition, Libby and colleagues were investigating signaling pathways that could lead to vascular inflammation and atherosclerosis (3). The close working relationship of our research laboratories provided us with the impetus to try to determine how endothelial dysfunction and endothelial cell activation were linked.
The idea that endothelium-derived NO could modulate leukocyte adhesion was first demonstrated by Kubes and colleagues, who showed that inhibitors of eNOS could increase neutrophil adherence to mesenteric venules (4). But the precise mechanism by which NO prevents leukocyte recruitment to the vascular wall was not known. To address this question, we first demonstrated that NO donors suppressed the expression of VCAM-1, ICAM-1, and E-selectin in response to a variety of proinflammatory cytokines. These results suggested a central mechanism for antiinflammatory effects of NO. Because the induction of these cellular adhesion molecules was mediated, in part, through the activation of the transcription factor, NF-κB (5), we postulated that NO prevented endothelial cell activation through inhibition of NF-κB. Using VCAM-1 expression as a marker of endothelial cell activation, we found that NO donors could inhibit cytokine-induced endothelial cell activation and monocyte adhesion under nonstatic conditions via inhibition of NF-κB (1). These findings indicated that exogenous NO could have therapeutic benefits in vascular inflammatory diseases, and subsequent studies have shown amelioration of atherosclerosis and other vascular diseases with NO donor therapy (6).
An important unresolved question at that time was whether endothelial dysfunction or endothelial cell activation was the primary initiator of atherogenesis. Endothelial cell activation by proinflammatory cytokines could clearly lead to endothelial dysfunction by inhibiting eNOS expression (7) and decreasing NO bioavailability through the induction of ROS (8). It was unclear how the endothelium was activated prior to the recruitment of inflammatory cells. A clue to this puzzle came from the seminal work of Li and colleagues, who showed that hypercholesterolemia brought on by an atherogenic diet could rapidly induce VCAM-1 expression in the rabbit endothelium (9). Additionally, hypercholesterolemia had previously been shown to produce endothelial dysfunction, in part, through inhibiting eNOS activity and/or NO bioavailability (10, 11).
Based upon these observations, we investigated whether endothelial dysfunction could lead to endothelial cell activation in the absence of cytokines. We found that suppression of basal eNOS activity by l-NG-arginine methylester (l-NAME) led to the rapid induction of VCAM-1 expression and increased monocyte adhesion (1), indicating that endogenous endothelium-derived NO could basally and tonically inhibit endothelial cell activation. These findings provided a mechanistic link by which mediators of endothelial dysfunction, such as hypercholesterolemia, smoking, or oxidative stress, could induce or enhance endothelial cell activation, leading to increased vasoconstriction, smooth muscle proliferation, platelet aggregation, leukocyte adhesion, LDL oxidation, and MMP activation (Figure 1). These results also suggested that areas of turbulent flow in the vessel wall, which would have less endothelium-derived NO, would be more prone to endothelial cell activation and atherosclerosis. Indeed, atherosclerotic lesions tend to develop in areas of vascular branching that are exposed to turbulent rather than laminar flow, and mice with targeted deletion of eNOS develop increased atherosclerosis and vascular inflammation (12).
Figure 1. Role of endothelial dysfunction and endothelial cell activation in vascular disease.
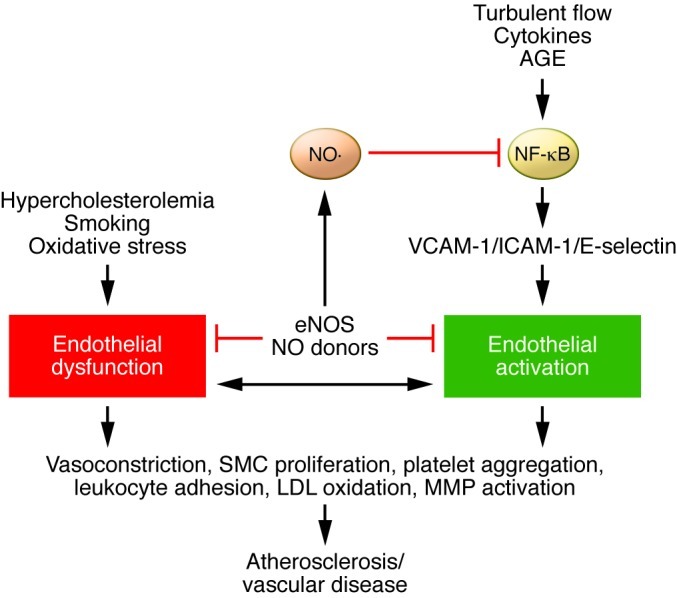
Cardiovascular risk factors, such as hypercholesterolemia, smoking, and oxidative stress, are important mediators of endothelial dysfunction, while proinflammatory cytokines, turbulent flow, and advanced glycation end-products (AGEs) are important mediators of endothelial cell activation via the activation of the transcription factor, NF-κB. NO from eNOS or NO donors reduces endothelial cell activation through inhibition of NF-κB. Loss of NO leads to increased endothelial cell activation. Likewise, endothelial cell activation can cause endothelial dysfunction. Both endothelial dysfunction and endothelial cell activation lead to atherosclerosis and vascular disease by increasing vasoconstriction, SMC proliferation, platelet aggregation, leukocyte adhesion, LDL oxidation, and MMP activation.
Since the publication of our study, the known vascular protective role of endothelium-derived NO has expanded beyond its inhibitory effects on endothelial-leukocyte interaction and leukocyte trafficking to include inhibitory effects on platelet reactivity, smooth muscle contraction and proliferation, and modulation of vascular matrix remodeling and angiogenesis. Conditions, such as laminar flow, or pharmacological agents, such as estrogens, HMG-CoA reductase inhibitors (statins), and angiotensin-converting enzyme inhibitors, which enhance endothelium-derived NO, have generally been found to be vascular protective and potentially antiatherogenic. In contrast, antiangiogenic therapies, which impair endothelium-derived NO through inhibition of VEGF signaling, occasionally cause endothelial dysfunction and hypertension in patients with cancer (13).
Acknowledgments
J.K. Liao is supported by grants from the NIH (HL052233, NS070001, DK086005).
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(2):540–541. doi:10.1172/JCI66843.
References
- 1.De Caterina R, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96(1):60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gimbrone MA, Jr, Nagel T, Topper JN. Biomechanical activation: An emerging paradigm in endothelial adhesion biology. J Clin Invest. 1997;99(8):1809–1813. doi: 10.1172/JCI119346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 4.Kubes P, Suzuki M, Granger DN. Nitric oxide: An endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A. 1991;88(11):4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baeuerle PA. Ikappab-nf-kappab structures: At the interface of inflammation control. Cell. 1998;95(6):729–731. doi: 10.1016/S0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]
- 6.Herman AG, Moncada S. Therapeutic potential of nitric oxide donors in the prevention and treatment of atherosclerosis. Eur Heart J. 2005;26(19):1945–1955. doi: 10.1093/eurheartj/ehi333. [DOI] [PubMed] [Google Scholar]
- 7.Yoshizumi M, Perrella MA, Burnett JC, Jr, Lee ME. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ Res. 1993;73(1):205–209. doi: 10.1161/01.RES.73.1.205. [DOI] [PubMed] [Google Scholar]
- 8.Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320(6061):454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Cybulsky MI, Gimbrone MA, Jr, Libby P. An atherogenic diet rapidly induces vcam-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler Thromb. 1993;13(2):197–204. doi: 10.1161/01.ATV.13.2.197. [DOI] [PubMed] [Google Scholar]
- 10.Liao JK, Shin WS, Lee WY, Clark SL. Oxidized low-density lipoprotein decreases the expression of endothelial nitric oxide synthase. J Biol Chem. 1995;270(1):319–324. doi: 10.1074/jbc.270.1.319. [DOI] [PubMed] [Google Scholar]
- 11.Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest. 1993;91(6):2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhlencordt PJ, et al. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein e/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104(4):448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 13.Mourad JJ, des Guetz G, Debbabi H, Levy BI. Blood pressure rise following angiogenesis inhibition by bevacizumab. A crucial role for microcirculation. Ann Oncol. 2008;19(5):927–934. doi: 10.1093/annonc/mdm550. [DOI] [PubMed] [Google Scholar]