

Abstract
Tumor metastasis involves a series of biological steps during which the tumor cells acquire the ability to invade surrounding tissues and survive outside the original tumor site. During the early stages, the cancer cells undergo an epithelial-mesenchymal transition (EMT). Wnt/β-catenin signaling is known to drive EMT and metastasis. Here we report that Wnt/β-catenin signaling is hyperactivated in metastatic breast cancer cells that express microRNA 374a (miR-374a). In breast cancer cell lines, ectopic overexpression of miR-374a promoted EMT and metastasis both in vitro and in vivo. Furthermore, miR-374a directly targeted and suppressed multiple negative regulators of the Wnt/β-catenin signaling cascade, including WIF1, PTEN, and WNT5A. Notably, miR-374a was markedly upregulated in primary tumor samples from patients with distant metastases and was associated with poor metastasis-free survival. These results demonstrate that miR-374a maintains constitutively activated Wnt/β-catenin signaling and may represent a therapeutic target for early metastatic breast cancer.
Introduction
Distant metastasis and invasion of cancer are responsible for more than 90% of cancer-related deaths (1). Biologically, the process of tumor metastasis consists of multiple sequential steps, including invasion of cancer cells into surrounding tissue, intravasation, survival in the circulation, arrest at distant organ sites, extravasation, and growth of a macroscopic secondary tumor in distant organs (2). The initial stage of metastatic progression is essentially dependent on the prominent biological event referred to as epithelial-mesenchymal transition (EMT), which is characterized by specific morphogenetic changes, loss of cell-cell adhesion, and increased cell motility (3, 4).
Wnt/β-catenin signaling has been demonstrated to play an important role in the development and promotion of EMT and cancer metastasis (5–7). Using a mouse model, DiMeo et al. found that the downstream target genes of the Wnt/β-catenin pathway are significantly upregulated in early breast cancer metastatic cells in the lungs. Inhibition of β-catenin signaling by DKK1 or SFRP1 overexpression dramatically reduces the expression of the transcriptional repressors Slug and Twist, but upregulates the expression of breast epithelial markers (8). Meanwhile, numerous studies have revealed that β-catenin is upregulated in human cancers and correlates with poor clinical prognosis (9). Nuclear β-catenin accumulation in the invasive fronts of primary tumors further highlights the essential role of β-catenin in the process of metastasis (10).
From a molecular perspective, it is well recognized that Wnt/β-catenin signaling is subject to multiple levels of control. The canonical Wnt/β-catenin signaling pathway is initiated when Wnt ligands bind to its receptor(s), namely, Frizzled (Fzd) (11) and LDL receptor–related protein-5 or -6 (LRP5 or LRP6) (12). Consequently, cytoplasmic protein Dishevelled (Dvl) is phosphorylated and thus enabled to dissociate β-catenin from the “destruction complex” composed of Axin, adenomatous polyposis coli (APC), casein kinase 1α (CK1α), and glycogen synthase kinase 3β (GSK3β) (13). The stabilized β-catenin, which accumulates in the cytoplasm, is activated to translocate into the cell nucleus, where it forms a β-catenin–LEF/TCF transcriptional complex and induces transcription of downstream genes implicated in carcinogenesis. In the absence of Wnt ligand stimulation, however, β-catenin is sequestered in the “destruction complex,” leading to β-catenin degradation by the ubiquitin-proteasome mechanism and ultimate inactivation of β-catenin signaling (14–16). Another layer of regulation for the suppression of β-catenin signaling is through secretion of antagonists of the Wnt pathway, such as Wnt inhibitory factor-1 (WIF1), secreted Frizzled-related proteins (sFRPs), and Dickkopf1 (DKK1) (17). Moreover, the noncanonical pathway has also been linked to modulation of β-catenin signaling. For example, WNT5A, a member of the noncanonical Wnt family, suppresses the motility and invasiveness of breast cancer cells via CK1α activation, resulting in phosphorylation of β-catenin at Ser45 and subsequently increased formation of the β-catenin/E-cadherin complex at the cell membrane, without influencing the nuclear translocation and transcriptional activity of β-catenin (18). Additionally, cross-talk between β-catenin signaling and other pathways, such as the PI3K/Akt pathway, has been indicated in various cancers (19). The tumor suppressor gene phosphatase and tensin homolog (PTEN), a dual lipid and protein phosphatase that antagonizes PI3K/Akt signaling, is frequently silenced in numerous tumor types. It has also been demonstrated that inhibition of β-catenin activity through PI3K/Akt suppression can be a key mechanism by which PTEN suppresses tumor progression (20, 21). Although Wnt/β-catenin signaling is tightly controlled at various levels, as mentioned above, its constitutively activated form has been found in various human cancer types. Thus, understanding how these negative regulatory effects on the Wnt/β-catenin signaling pathway are concomitantly deregulated in cancers is biologically as well as clinically important for future development of antimetastatic strategies.
MicroRNAs (miRNAs), a class of endogenous noncoding small RNAs, are involved in the modulation of many biological processes by base-pairing, usually imperfectly, to the 3′ untranslated region (UTR) of a target mRNA, resulting in posttranscriptional inhibition, and sometimes mRNA cleavage (22). Accumulating evidence has extended the function of miRNAs to both physiological and pathological conditions, including cancer (23). Given that one specific miRNA is usually capable of targeting multiple mRNAs of different genes, it is of particular interest to identify miRNAs that might interfere with β-catenin signaling and thereby lead to cancer invasion and metastasis, particularly those miRNAs that can simultaneously interact with multiple regulators of the β-catenin pathway. We report that a human miRNA, miR-374a, activates the β-catenin pathway by suppressing the expression of WIF1, PTEN, and WNT5A, and consequently promotes EMT and metastasis of breast cancer.
Results
Identification of miR-374a as a metastasis-promoting miRNA in breast cancer.
To screen miRNAs that are deregulated in breast cancer metastases, we comparatively analyzed the miRNA profiles in parental MDA-MB-435 cells and the derivative cells recovered from lung metastases in a mouse model. As shown in Figure 1A, microarray analysis revealed that 12 miRNAs were elevated, and 11 were downregulated, in MDA-MB-435 cells and their lung metastatic derivatives. These miRNAs included upregulated miR-21, which has been reported to be elevated in breast cancer and to promote invasion and metastasis (24). Strikingly, miR-374a emerged as a highly upregulated miRNA in a series of tested human breast cancer cell lines as compared with that found in primary human normal breast epithelial cells (NBECs) and in the spontaneously immortal MCF10A cell line. Of particular note was the fact that its expression was most pronounced in cell lines known to be highly metastatic (MDA-MB-231, MDA-MB-435, and MDA-MB-468) compared with that in non- or low-metastatic breast cancer cell lines (MCF7, T47D, and BT474) (Figure 1B). By contrast, such a change was not observed in the expression of miR-330-3p or miR-489, suggesting the likelihood that miR-374a is associated with the development of breast cancer metastasis.
Figure 1. miR-374a is markedly overexpressed in metastatic breast cancer cell lines.

(A) miRNA array analysis showed differentially expressed miRNAs in parental MDA-MB-435 cells and their lung metastatic derivatives. Pseudo-color represents intensity scale of metastatic derivatives versus parental cells. (B) Real-time PCR analysis of the expression levels of miR-374a in human breast cancer cell lines compared with primary NBECs and spontaneously immortalized MCF10A cells. Error bars represent mean ± SD from 3 independent experiments. *P < 0.05.
MiR-374a promoted the development of prometastatic phenotype in vitro.
To understand the biological effect of miR-374a deregulation on the invasiveness of breast cancer cells, in vitro gain-of-function analyses were performed using an overexpression strategy through the retrovirally stable expression of miR-374a in human breast cancer cell line MCF-7, and 4T1, derived from spontaneous mammary tumor in a BALB/cfC3H mouse, which does not express endogenous miR-374a (25). As shown in Figure 2A, overexpression of miR-374a in MCF7 and 4T1 cells led to altered morphological characteristics of EMT, identified by a scattered distribution of cells in the culture and a spindle- or star-like morphology of the cells. Moreover, we found that epithelial markers, including E-cadherin, γ-catenin, and CK18, were drastically downregulated, but mesenchymal markers such as vimentin and N-cadherin were dramatically upregulated in miR-374a–transduced MCF7 and 4T1 cells (Figure 2B). These results suggest that miR-374a might promote a transition from epithelial to mesenchymal phenotype. Consistent with this postulation, Matrigel-coated (for invasion) or -uncoated (for migration) Transwell assay showed that miR-374a overexpression drastically increased the invasiveness and migration of the MCF7 and 4T1 breast cancer cell lines (Figure 2C). Furthermore, in 3D culture, miR-374a–transduced MCF7 and 4T1 cells grew into more structurally well-organized spheres with invasive projections emanating from the cells, when compared with the vector control cells (Figure 2D). In addition, wound healing assay showed that miR-374a overexpression enhanced the migratory speed of MCF7 and 4T1 cells compared with that of the vector control cells (Figure 2E).
Figure 2. Overexpression of miR-374a induces EMT of breast cancer cell lines in vitro.

(A) Effect of miR-374a overexpression or inhibition on cell morphology evaluated by phase-contrast microscopy. (B) Expression of epithelial cell markers (E-cadherin, γ-catenin, and CK18) and mesenchymal cell markers (vimentin and N-cadherin) in indicated cells were examined by WB analysis. GAPDH was used as a loading control. (C) Quantification of indicated invading or migrating cells in 5 random fields analyzed by Matrigel-coated or -noncoated Transwell assays, respectively. (D) Representative micrographs of indicated cells grown on Matrigel for 10 days in 3D spheroid invasion assay. (E) Representative micrographs of wound healing assay of the indicated cells. Wound closures were photographed at 0 and 24 hours after wounding. Experiments in A–E were repeated at least 3 times with similar results, and error bars in C represent mean ± SD. *P < 0.05. Original magnification, A and D, ×200; E, ×100. NC, negative control.
To further investigate the proinvasive role of miR-374a in breast cancer, we examined the effect of inhibiting miR-374a on the phenotype of a highly metastatic MDA-MB-435 breast cancer cell line that expresses miR-374a at an elevated level. As expected, inhibition of miR-374a markedly weakened EMT and the invasive capabilities of MDA-MB-435 cells (Figure 2, A–E). Collectively, our data suggest that miR-374a greatly contributes to the development of breast cancer metastasis and invasion.
miR-374a promoted distant metastasis.
Since the in vitro experiments revealed that miR-374a expression was associated with prometastatic traits, we next asked whether miR-374a could promote metastasis in vivo. The MCF7/miR-374a and the 4T1/miR-374a cells, as well as their corresponding vector control cells, were injected into the mammary gland fat pads of nude mice. Strikingly, mice bearing MCF7/miR-374a tumors displayed prominent lung metastasis, whereas no visible metastasis was found in mice transplanted with control MCF7 cells. We also found that 4T1/miR-374a xenotransplants generated an approximately 4-fold increase in the number of lung metastases than did vector control cells (Figure 3, A and B). Notably, ectopic miR-374a expression only slightly increased the weights of the MCF7 and 4T1 primary tumors (Figure 3C), and the enhanced metastatic capability of miR-374a was also demonstrated by the metastasis index (the number of metastases per primary tumor weight; Figure 3D) (26). To further validate whether miR-374a is required for the observed enhanced metastasis in vivo, antagomir-374a, an antisense-based specific inhibitor against miR-374a, was applied to inhibit endogenous expression of miR-374a in the experimental metastasis assay. As shown in Figure 3, A and B, the ability of MDA-MB-435 cells to cause lung metastasis was significantly impaired by intratumoral administration of antagomir-374a. Histological analyses and bioluminescence imaging further revealed that miR-374a could promote metastasis of breast cancer cell lines (Figure 3, E and F). Together, these data indicate that miR-374a plays a pivotal role in breast cancer metastasis in vivo.
Figure 3. miR-374a enhances metastasis of breast cancer cell lines in vivo.

(A) Representative bright-field imaging of the lungs. On days 60 (for MCF7), 15 (for 4T1), and 30 (for MDA-MB-435), mice receiving transplants of indicated cells were anesthetized and the lungs were collected. Arrows indicate surface metastatic nodules. (B) Number of visible surface metastatic lesions in mice (n = 6 per group) receiving an orthotopic injection of indicated cells. (C) Primary tumor weights in BALB/c mice that received transplants of indicated cells. (D) Metastasis index (number of metastases per primary tumor weight) in mice that were transplanted with indicated cells. (E) Lung metastases in the mice in which the indicated cells were implanted were confirmed by H&E staining. (F) Representative bioluminescence images of lung metastases in the mice that received an orthotopic injection of indicated cells. Error bars in B–D represent mean ± SD (n = 6 per group). *P < 0.05.
Wnt/β-catenin signaling mediated miR-374a–induced EMT and metastasis.
In light of the critical role of the Wnt/β-catenin pathway in tumorigenesis and metastasis, we then examined whether miR-374a activates Wnt/β-catenin signaling. Strikingly, subcellular fractionation and immunofluorescence staining assays showed that overexpression of miR-374a resulted in substantial nuclear accumulation of β-catenin in MCF7 and 4T1 breast cancer cell lines, suggesting that miR-374a might contribute to activation of Wnt/β-catenin signaling (Figure 4, A and B). As expected, miR-374a overexpression markedly increased the transactivating activity of β-catenin in MCF7 and 4T1 cells, as determined by β-catenin reporter assay (Figure 4C). Conversely, transfection of a miR-374a inhibitor reduced the nuclear translocation of β-catenin and subsequent TCF/LEF activities in MDA-MB-435 cells (Figure 4, A–C). These data suggest that miR-374a overexpression is able to enhance β-catenin nuclear translocation and, consequently, promote the transcriptional activity of TCF/LEF.
Figure 4. Wnt/β-catenin signaling mediates the effects of miR-374a.

(A) Altered nuclear translocation of β-catenin in response to deregulated miR-374a expression. Nuclear fractions of indicated cells were analyzed by WB analysis. EF-1α was used as a loading control. (B) Subcellular β-catenin localization in indicated cells was assessed by immunofluorescence staining. (C) Indicated cells transfected with TOPflash or FOPflash and Renilla pRL-TK plasmids were subjected to dual-luciferase assays 48 hours after transfection. Reporter activity detected was normalized by Renilla luciferase activity. (D) Depletion of TCF4 or LEF1 with specific siRNA in miR-374a–expressing cells confirmed by WB analysis. (E) Quantification of indicated invading cells in a Matrigel-coated Transwell assay. (F) Luciferase-reported TCF/LEF transcriptional activity in indicated cells. (G) Overexpression of TCF4 or LEF1 in miR-374a–silenced MDA-MB-435 cells confirmed by WB analysis. (H) Quantification of invading cells impacted by overexpression of TCF4 or LEF1 in Transwell assay. (I) Luciferase-reported TCF/LEF transcriptional activity in indicated cells. Experiments in A–I were repeated at least 3 times with similar results, and error bars in C, E, F, H, and I represent mean ± SD. *P < 0.05.
To further validate the role of β-catenin activation in miR-374a–induced cell invasion, we analyzed the impact of blocking Wnt/β-catenin signaling, via knocking down TCF4 or LEF1, on the invasive capability of miR-374a–transduced MCF7 and 4T1 cell lines (Figure 4D). As shown in Figure 4, E and F, inhibition of β-catenin signaling not only reduced TCF/LEF transcriptional activity, but also abrogated miR-374a–induced invasiveness. On the other hand, activation of β-catenin signaling by ectopically expressing TCF4 or LEF1 in miR-374a–inhibited MDA-MB-435 cells (Figure 4G) mimicked the effects of miR-374a, including increased cell invasion and TCF/LEF transcriptional activity (Figure 4, H and I). Taken together, our results indicate that Wnt/β-catenin signaling is a functional mediator for miR-374a–induced EMT and metastasis in the breast cancer cell lines.
miR-374a repressed multiple negative regulators of β-catenin signaling.
Next, using the DIANA-mirPath program (27), we investigated the mechanism by which miR-374a activates β-catenin signaling, and found that Wnt/β-catenin signaling was strikingly enriched by predicted targets of miR-374a. Among the predicted targets, WIF1, PTEN, and WNT5A were specifically noted as negative regulators of Wnt/β-catenin signaling (17, 18, 20). Moreover, two bioinformatics tools, TargetScan (28) and miRanda (29), were used to further confirm that these negative regulators were putatively potential targets of miR-374a (Figure 5A). Western blotting (WB) analysis consistently revealed that the expression levels of all 3 proteins were reduced in miR-374a–overexpressing cells, whereas miR-374a inhibition elevated the levels of these proteins (Figure 5C). Similar alterations were found at the mRNA level (Figure 5D). Furthermore, reporter assays showed that the activity of luciferase linked with the 3′ UTR of WIF1, PTEN, or WNT5A was repressed in a dose-dependent manner in miR-374a mimic–transfected MCF7 and 4T1 cells, compared with those in control cells (Figure 5E). Conversely, inhibition of miR-374a caused a significant increase in luciferase reporter activities under the control of the 3′ UTR of WIF1, PTEN, or WNT5A (Figure 5E). Of note, mutations brought into the seed sequence of miR-374a (Figure 5B) abolished its suppressive effects (Figure 5E). Collectively, these data suggest that miR-374a directly suppresses WIF1, PTEN, and WNT5A expression in breast cancer cell lines.
Figure 5. WIF1, PTEN, and WNT5A are direct targets of miR-374a.

(A) Schematic miR-374a putative target sites in 3′ UTRs of WIF1, PTEN, and WNT5. (B) Sequence of miR-374a-mut. (C) WB analysis of the protein levels of WIF1, PTEN, and WNT5A in response to deregulated miR-374a expression of indicated cells. (D) Real-time RT-PCR analysis of mRNA levels of WIF1, PTEN, and WNT5A in indicated cells. (E) Luciferase assay of pGL3-WIF1-3′-UTR, pGL3-WNT5A-3′-UTR, and pGL3-PTEN-3′-UTR reporters cotransfected with increasing amounts (10, 20, and 50 nM) of miR-374a mimic and mutant oligonucleotides in MCF7 and 4T1 cell lines, or with increasing amounts (20, 50, and 100 nM) of miR-374a inhibitor oligonucleotides. Experiments in C–E were repeated at least 3 times with similar results, and error bars in D and E represent mean ± SD. *P < 0.05.
Suppression of WIF1, PTEN, and WNT5A was functionally important for the biological effects of miR-374a.
To explore the functional significance of WIF1, PTEN, and WNT5A in the invasive capability of breast cancer cell lines and β-catenin activation induced by miR-374a, we studied the effects of their depletion using specific siRNAs. As shown in Figure 6, A and B, individually silencing WIF1, PTEN, or WNT5A in MCF7 and 4T1 cells led to increased cell invasiveness, as evidenced by Matrigel-coated Transwell assay. Moreover, siRNA silencing of WIF1 or PTEN expression enhanced the TCF/LEF transcriptional activity (Figure 6C). In line with the previous studies demonstrating that WNT5A suppressed the mobility of breast cancer cell lines by increasing β-catenin/E-cadherin complex formation, but not by influencing the nuclear translocation and transcriptional activity of β-catenin (18, 30), we did not find a detectable effect of WNT5A depletion on the luciferase activity driven by β-catenin signaling (data not shown). Importantly, when the WIF1, PTEN, or WNT5A open reading frame, which lacks the corresponding 3′ UTR, was ectopically overexpressed in miR-374a–transduced MCF7 and 4T1 cells (Figure 6D), miR-374a–induced cell motility and invasion were, at least partly, antagonized (Figure 6E). Moreover, reintroduction of WIF1 or PTEN, but not WNT5A, abrogated TCF/LEF transcriptional activity (Figure 6F). In addition, depletion of WIF1, PTEN, or WNT5A dramatically suppressed the invasiveness of miR-374a–inhibited MDA-MB-435 cells (Figure 6, G and H). Silencing WIF1 or PTEN also increased TCF/LEF transcriptional activity in MDA-MB-435 cells whose miR-374a was suppressed (Figure 6I). These results demonstrated that WIF1, PTEN, and WNT5A were functionally important for miR-374a–induced cell motility and invasiveness in breast cancer cell lines.
Figure 6. WIF1, PTEN, and WNT5A are functionally involved in miR-374a–induced invasion and TCF/LEF transcriptional activity of breast cancer cell lines.

(A) WB analysis confirming the transfection of WIF1-, PTEN-, or WNT5A-targeting siRNAs in MCF7 and 4T1 cells. (B) Quantification of invading MCF7 and 4T1 cells transfected with indicated siRNAs. (C) Luciferase assay of TCF/LEF transcriptional activity in MCF7 and 4T1 cells transfected with indicated siRNAs. (D) Ectopic expression of WIF1, PTEN, or WNT5A in indicated cells was confirmed by WB analysis using GAPDH as a loading control. (E) Quantification of invading cells impacted by overexpression of WIF1, PTEN, or WNT5A in Transwell assay. (F) Luciferase assay of TCF/LEF transcriptional activity in indicated cells. (G) WB analysis confirmed the transfection of specific siRNAs in miR-374a–silenced cells. (H) Quantification of indicated invading cells with specific siRNA transfection. (I) Luciferase assay of TCF/LEF transcriptional activity in indicated cells transfected with specific siRNA. Error bars in B, C, E, F, H, and I represent mean ± SD from 3 independent experiments. *P < 0.05.
Clinical association of miR-374a with metastasis in human breast cancer.
To further understand the clinical relevance of the above findings in human breast cancer, the expression of miR-374a was examined in 166 human breast cancer tissue specimens (Table 1; mean follow-up time was 51 months). Patients were first divided into 2 groups: those who developed distant metastasis and those who were metastasis-free within the follow-up period, respectively. As shown in Figure 7A, the expression level of miR-374a was significantly elevated in the primary tumors of 33 patients with distant metastasis, as compared with that in primary tumors removed from 133 patients without detectable distant metastasis. Samples from a total of 166 breast cancer patients were collected in our current study and divided into 2 groups: a high miR-374a expression group (48 patients, including 33 patients with distant metastasis and 15 patients without metastasis), and a low miR-374a expression group (118 patients without metastasis), Histological analysis showed that high miR-374a expression level was significantly associated with short metastasis-free survival (defined as the time from diagnosis to the presence of first evidence of distant metastasis) (Figure 7B). The results of this analysis suggest a strong correlation between miR-374a and distant metastasis, as well as the consequent death of breast cancer patients.
Table 1.
Clinicopathologic characteristics of breast cancer patients
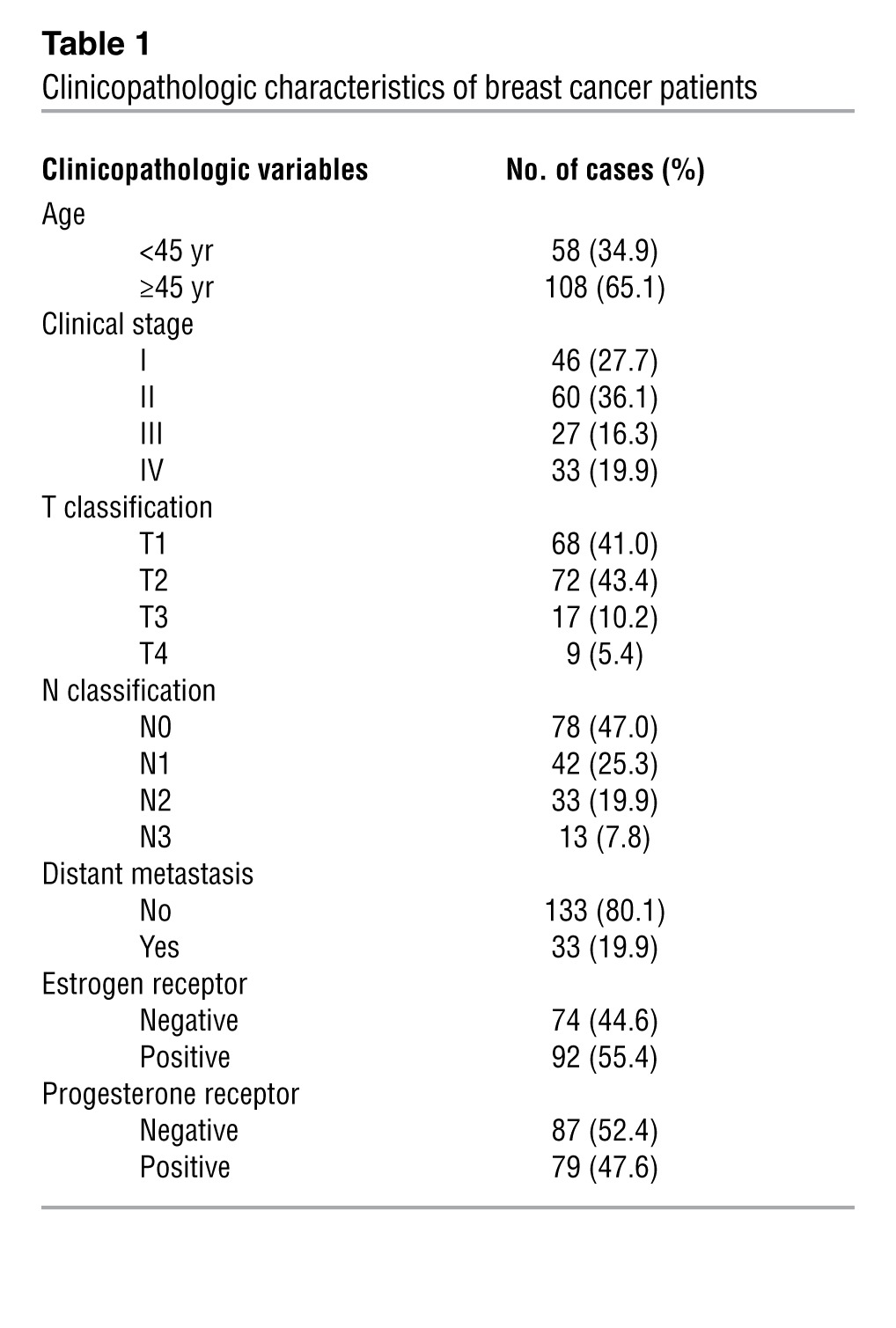
Figure 7. Clinical association of miR-374a with metastasis of human breast cancer.

(A) Real-time RT-PCR analysis of the expression levels of miR-374a in tumors with metastatic relapse (n = 33) and in tumors without metastasis (n = 133). (B) Kaplan-Meier metastasis-free survival curve comparing 2 groups of high (greater than the mean; n = 48) and low (less than the mean; n = 116) miR-374a expression level in the primary tumors of 166 breast cancer patients; P value is based on a log-rank test. (C) Expression of miR-374a is associated with WIF1, PTEN, WNT5A, E-cadherin, and N-cadherin expression levels and β-catenin localization in clinical breast cancer specimens. Two representative cases are shown. Original magnification, ×400. (D) Percentage of specimens showing low or high miR-374a expression in relation to the expression levels of WIF1, PTEN, WNT5A, E-cadherin, N-cadherin, and cytoplasmic/nuclear or membrane of β-catenin. *P < 0.05.
We next asked whether there was an association between the expression levels of miR-374a and its targets, as well as the subsequent β-catenin activation in the clinical specimens. As shown in Figure 7, C and D, 48.3% (57 cases), 49.2% (58 cases), and 64.4% (76 cases) of samples with low miR-374a expression (118 cases), respectively, exhibited high levels of WIF1, PTEN, and WNT5A, whereas 81.3% (39 cases), 77.1% (37 cases), and 75% (36 cases) of samples with high miR-374a expression (48 cases) showed low expression of WIF1, PTEN, and WNT5A, respectively (P < 0.05). Moreover, breast cancer samples with high miR-374a expression showed higher β-catenin activation levels (32 of 48 samples; 66.7%) than those with low miR-374a expression (41 of 118 samples; 34.7%) (Figure 7 C and D; P < 0.05). Importantly, expression of the epithelial marker E-cadherin was strong in 95 of 118 (80.5%) specimens expressing low levels of miR-374a, and in contrast, high E-cadherin expression was only observed in 21 of 48 (43.7%) samples with high miR-374a. Concurrently, an inverse expression pattern of the mesenchymal marker N-cadherin was found in the same patient cohort, suggesting that miR-374a expression significantly correlated (P < 0.05) with altered presentation of EMT-specific markers. Taken together, our data indicate that miR-374a overexpression in breast cancer lesions was associated with downregulation of WIF1, PTEN, and WNT5A, activation of β-catenin, and induction of EMT, which further contributed to the metastasis of breast cancer.
Discussion
Tumor invasion and metastasis are complex, multistep processes underlain by genetic and/or epigenetic changes within probably a fraction of malignant cells in the tumor (31, 32). The regulatory networks that control such molecular alterations, however, have not been defined. The upstream regulators of the metastatic process appear to be subject to therapeutic targeting, and miRNAs are attractive candidates for such regulation due to the capacity of one miRNA to coordinately suppress multiple target genes and thereby modulate multiple steps of metastatic progression in various cancer types (33). In this context, some miRNAs have been found to be metastasis inhibitors. For example, Valastyan et al. have demonstrated that miR-31 inhibits tumor invasion and metastasis by repressing multiple metastasis-promoting genes (34). In our current study, miRNA microarray profiling revealed that miR-374a represents a miRNA species most strikingly upregulated in lung metastatic derivatives of MDA-MB-435 breast cancer cell lines. Moreover, miR-374a evidently induces nonmetastatic MCF7 cells to invade and metastasize, and it causes further, dramatic enhancement of the invasive and metastatic capabilities of the 4T1 breast cancer cell, which has been widely known as a metastatic cell line. Furthermore, examination of clinical samples of breast cancer showed that miR-374a levels correlate with metastatic recurrence in breast cancer patients. Thus, our current study provides new insights into this area of research by identifying miR-374a as a clinically relevant promoter of tumor metastasis.
Molecular mechanisms underlying the distant metastasis of breast cancer have been intensively studied. Cellular signaling pathways, including the Wnt/β-catenin, ERK/MAPK, and PI3K/Akt pathways, have been found to be aberrantly activated and play vital roles in the development and progression of breast cancer (35–37). Among these pathways, the prometastatic functions of Wnt/β-catenin signaling in breast cancer have been well documented (35, 38). Notably, unlike in other cancer types, the mutations in β-catenin are rare, but the expression and/or nuclear localization of β-catenin is often abnormal in breast cancer, indicating a constitutive activation of Wnt/β-catenin signaling (9, 39, 40). Thus, understanding whether and how a specific factor in breast cancer cells can simultaneously interact with multiple regulators of β-catenin signaling may provide new insights into the molecular mechanisms underlying cancer invasion and metastasis, and facilitate the identification of novel antimetastatic targets. Our current data show that downregulation of WIF1, PTEN, and WNT5A mediates the effects of miR-374a on β-catenin activation. The 3 identified mediating molecules have all been suggested to play important roles in the modulation of β-catenin signaling. First, WIF1 has proven to act as a secreted Wnt inhibitor that binds directly to Wnt, and thus constrains the binding of Wnt ligands to the Frizzled receptor (41). Functioning as a tumor suppressor, the expression of WIF1 has been found to be downregulated in various human cancers, including breast cancer (42). Furthermore, Rubin et al. have reported that the functional loss of WIF1 could trigger Wnt/β-catenin signaling and thereby contributes to tumor invasion and metastasis (43). Second, as a well-known tumor suppressor, PTEN is silenced in various types of cancers (44). Moreover, it has been well characterized that the PTEN/Akt axis is involved in tumor metastasis via β-catenin signaling (45). Third, WNT5A, a nontransforming protein, is a ligand for noncanonical WNT signaling. While the role of WNT5A in tumorigenesis is still controversial, its low expression has been associated with poor clinical outcome in breast cancer patients (46). A study by Medrek et al. has elegantly demonstrated that WNT5A inhibits the motility and invasion of breast cancer cell lines via activation of CK1α, leading to β-catenin phosphorylation at Ser45, and a subsequent increased formation of the β-catenin/E-cadherin complex on the cell membrane (18). Our finding that upregulated miR-374a may confer simultaneous suppression of multiple inhibitors of β-catenin signaling strongly supports the notion that the miRNA represents a potent activator of the pathway and subsequent tumor metastasis. While it remains to be clarified whether other mediating signals may also contribute to the prometastatic effect of miR-374a, taken together, our data provide a biological basis for the potential use of miR-374a as an antimetastatic target in breast cancer.
In addition to its role in cell cycle regulation, apoptosis, and angiogenesis, evidence from recent animal and clinical studies indicates that PTEN also contributes to suppression of tumor metastasis (47, 48). Reduction of PTEN is likely to facilitate metastasis by enhancing proliferation and inhibiting apoptosis at the metastatic site (49). Of note, it has been recognized that loss of PTEN is involved in the activation of β-catenin signaling in breast cancer and is also associated with development of breast tumor invasion, indicating a possibility that the prometastatic effect of PTEN loss might be mediated, at least in part, via β-catenin activation (20). Consistent with these previous reports, we also found that both activation of β-catenin and promotion of cell invasion can result from knockdown of PTEN, further supporting the notion that the β-catenin–mediated proinvasive function of PTEN loss, aside from its proliferation-promoting and apoptosis-inhibiting effects, might also contribute to miR-374a–induced EMT and metastasis in breast cancer. In this context, an observation made in this study — namely, that silencing WIF1 or WNT5A cannot fully recapitulate the miR-374a–induced invasive phenotype in breast cancer cell lines — suggests that suppression of PTEN is required for the prometastatic property of miR-374a upregulation.
Previously, extensive research has demonstrated that antagomirs might represent a class of potentially efficient, specific, and long-lasting silencers of endogenous miRNAs in vivo (50, 51). Our current study shows that intratumoral administration of an antagomir against miR-374a could effectively suppress miR-374a expression and, consequently, pulmonary metastasis of experimental breast cancer, suggesting that a direct miRNA-targeting antagonistic strategy might be of promising therapeutic efficacy. These results also underscore the significance of identifying such a prometastatic miRNA as a potential target for cancer therapy. Furthermore, due to the importance of β-catenin signaling in many physiological and pathological processes, as well as the multitarget capabilities of miR-374a as an activator of this pathway, in vivo antagomir-based strategies might also be useful for studying the basic and translational significance of β-catenin signaling.
In summary, our studies have found that elevated expression of miR-374a promotes EMT and metastasis in breast cancer by simultaneously suppressing multiple inhibitors of β-catenin signaling, including WIF1, PTEN, and WNT5A. These findings uncover a novel molecular mechanism that maintains the constitutive activation of the Wnt/β-catenin pathway and may prove clinically useful for developing a new prognostic biomarker and therapeutic target for breast cancer invasion and metastasis.
Methods
Cell culture.
Breast cancer cell lines MCF7, T47D, BT474, MDA-MB-231, MDA-MB-435, MDA-MB-468, and 4T1 were purchased from ATCC and maintained in DMEM supplemented with 10% FBS. Spontaneously immortalized MCF10A epithelial cells were maintained in keratinocyte serum-free medium. NBECs were established as described previously (52).
Human breast cancer specimens.
This study was conducted with a total of 166 paraffin-embedded human breast cancer specimens that were histopathologically diagnosed at the First Affiliated Hospital of Sun Yat-sen University and Sun Yat-sen University Cancer Center from 2000 to 2005. Clinical information about the samples is presented in Table 1.
Plasmids and generation of stably engineered cell lines.
The miR-374a expression plasmid was generated by cloning the genomic pre–miR-374a gene, with a 300-bp sequence on each flanking side, into retroviral transfer plasmid pMSCV-puro (Clontech Laboratories Inc.) to generate plasmid pMSCV-miR-374a. pMSCV-miR-374a was cotransfected with the pIK packaging plasmid in 293FT cells using the standard calcium phosphate transfection method, as previously described (53). Thirty-six hours after the cotransfection, supernatants were collected and incubated with cells to be infected for 24 hours in the presence of polybrene (2.5 μg/ml). After infection, puromycin (1.5 μg/ml) was used to select stably transduced cells over a 10-day period. The ORFs of LEF1, TCF4, WIF1, PTEN, and WNT5A generated by PCR amplification were cloned into mammalian expression vector pcDNA 3.1 (Invitrogen; Life Technologies). The 3′ UTRs of WIF1, PTEN, and WNT5A, respectively, were amplified and cloned downstream to the luciferase gene in a modified pGL3 control vector. The reporter plasmids containing wild-type (CCTTTGATC; TOPflash) or mutated (CCTTTGGCC; FOPflash) TCF/LEF DNA binding sites were purchased from Upstate Biotechnology.
RNA extraction, reverse transcription, and real-time RT-PCR.
Total RNA was extracted from cultured cells using the mirVana miRNA Isolation Kit (Ambion; Life Technologies), and from formalin-fixed and paraffin-embedded normal and malignant human breast tissue specimens using the RecoverAll Total Nucleic Acid Isolation Kit (Ambion; Life Technologies). cDNA was synthesized from total RNA with specific stem-loop primers and the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems; Life Technologies). Expression of miRNAs was analyzed by real-time PCR using the TaqMan MicroRNA Assay kit (Applied Biosystems; Life Technologies). Detection of mRNA was performed as described previously (54). The sequences of the primers were as follows: c-Myc forward, 5′-TCAAGAGGCGAACACACAAC-3′; c-Myc reverse, 5′-GGCCTTTTCATTGTTTTCCA-3′; TCF4 forward, 5′-TCTCCATAGTTCCTGGACGG-3′; TCF4 reverse, 5′-CCAACTTCTTTGGCAAGTGG-3′; LEF1 forward, 5′-CACTGTAAGTGATGAGGGGG-3′; LEF1 reverse, 5′-TGGATCTCTTTCTCCACCCA-3′; MMP7 forward, 5′-GCATCTCCTTGAGTTTGGCT-3′; MMP7 reverse, 5′-GAGCTACAGTGGGAACAGGC-3′; VEGF forward, 5′-AGCTGCGCTGATAGACATCC-3′; VEGF reverse, 5′-CTACCTCCACCATGCCAAGT-3′; E-cadherin forward, 5′-GACCGGTGCAATCTTCAAA-3′; E-cadherin reverse, 5′-TTGACGCCGAGAGCTACAC-3′; BMP4 forward, 5′-GCATTCGGTTACCAGGAATC-3′; BMP4 reverse, 5′-TGAGCCTTTCCAGCAAGTTT-3′; FoxN1 forward, 5′-CTCCACCTTCTCGAAGCACT-3′; FoxN1 reverse, 5′-CGGAGCACTTTCCTTACTTCA-3′; and GAPDH forward, 5′-GACTCATGACCACAGTCCATGC-3′; GAPDH reverse, 5′-AGAGGCAGGGATGATGTTCTG-3′.
WB.
WB was performed according to a standard method, as described previously (54). The following primary antibodies were used: anti–E-cadherin, anti–α-catenin, anti–β-catenin, anti–γ-catenin, anti-vimentin, and anti–N-cadherin (Pharmingen; BD Biosciences); anti-WIF1, anti-TCF4, and anti-PTEN (Epitomics Inc.); anti-EF1α (Upstate Biotechnology); and anti-WNT5A (Abcam). Nuclear extracts were prepared using the Nuclear Extraction Kit (Active Motif), according to the manufacturer’s instructions.
Wound healing, cell invasion, and migration assays.
Indicated cells were plated to confluence in 6-well plates. Streaks were created in the monolayer with a pipette tip. Progression of migration was observed and photographed at 24 hours after wounding. Cell invasion and migration assays were analyzed using the Transwell chambers assay (Costar; Corning Inc.), with or without coated Matrigel (BD Biosciences). The lower chamber of the Transwell device was filled with 500 μl DMEM supplemented with 10% FBS. After 24 hours of incubation, cells invading into the bottom side of the inserts were fixed, stained, photographed, and quantified by counting them in 5 random high-power fields.
Luciferase reporter assay.
Cells were seeded in triplicate in 24-well plates and allowed to settle for 24 hours. Indicated plasmids plus 1 ng pRL-TK Renilla plasmid were transfected into the cells using Lipofectamine 2000 Reagent (Life Technologies). Forty-eight hours after transfection, Dual-Luciferase Reporter Assay (Promega) was performed according to the manufacturer’s instructions, as previously described (55).
3D spheroid invasion assay.
Indicated cells (5 × 103) were trypsinized and seeded on 2% Matrigel coated in 24-well plates, and medium was refreshed every other day. Cells forming a 3D spherical structure (spheres) were photographed at 2-day intervals for 10 days.
Immunohistochemistry.
Immunohistochemistry assays were performed and quantified as previously described (56). The degree of immunostaining of indicated proteins was evaluated and scored by 2 independent observers, as previously described, scoring both the proportion of positive staining tumor cells and the staining intensities. Scores representing the proportion of positively stained tumor cells were graded as: 0 (no positive tumor cells); 1 (<10%); 2 (10%–50%); and 3 (>50%). The intensity of staining was determined as: 0 (no staining); 1 (weak staining = light yellow); 2 (moderate staining = yellow brown); and 3 (strong staining = brown). The staining index (SI) was calculated as the product of staining intensity × percentage of positive tumor cells, resulting in scores of 0, 1, 2, 3, 4, 6, and 9. Cutoff values for high and low expression of the protein of interest were chosen based on a measurement of heterogeneity using the log-rank test with respect to overall survival. The optimal cutoff was identified as an SI score of greater than or equal to 4, which was considered to be high expression, and less than or equal to 3, which was considered to be low expression.
Transient transfection.
miR-374a inhibitors and their corresponding control oligonucleotides were transfected into MDA-MB-435 cells cultured in 6-well plates using Lipofectamine 2000 (Life Technologies), according to the manufacturer’s instructions. siRNA transfection was performed following the same procedure. The sense strand sequences of siRNAs, which were designed to target both human and mouse cells, except where specifically noted, were as follows: siTCF4, 5′-AAGUCCGAGAAAGGAAUCUGA-3′; hsiLEF1, 5′-UCAGAUGUCAACUCCAAACAA-3′; msiLEF1, 5′-AUCUUCGCCGAGAUCAGUCAU-3′; siWIF1-1, 5′-CUCAUAGGAUUUGAAGAAG-3′; siWIF1-2, 5′-GGAUAAAGGCAUCAUGGCA-3′; siWNT5a-1, 5′-UUGGUGGUCGCUAGGUAUGAA-3′; siWNT5a-2, 5′-GGUCGCUAGGUAUGAAUAA-3′; siPTEN-1, 5′-GAGCGUGCAGAUAAUGACA-3′; and siPTEN-2, 5′-GGCGCUAUGUGUAUUAUUA-3′.
Animal studies.
In vivo selection and establishment of new cell lines applied in a microarray assay were performed as described previously (57). Briefly, 3 × 106 MDA-MB-435 cells were injected into mammary fat pads of BALB/c nude mice. After 30 days, the animals were sacrificed, their pulmonary metastatic nodules were isolated and minced, and cell culture was established using the obtained explants. For spontaneous metastasis assays, MCF7/pMSCV-miR-374a; MCF7/vector (1 × 107); 4T1/pMSCV-miR-374a; and 4T1/vector (1 × 106) cells were injected into the mammary fat pads of age-matched female NOD/SCID mice. All mice injected with MCF7/pMSCV-miR-374a and MCF7/vector cells were supplemented with estrogen pellets (Innovative Research of America). For in vivo antagomir administration, 3 × 106 MDA-MB-435 cells were injected into mammary fat pads of BALB/c nude mice. One hundred microliters of miR-374a antagomir (diluted in PBS at 2 mg/ml) or control antagomir was administrated intratumorally 3 times per week for 2 weeks, starting when the average volume of grown tumors reached approximately 50 mm3. Sixty days (for MCF7), 15 days (for 4T1), or 30 days (for MDA-MB-435) after tumor implantation, the mice were killed, the mammary tumors were removed and weighed, and the lungs were collected to count surface metastases under a dissecting microscope. Lungs were fixed in formalin and embedded in paraffin using the routine method. H&E staining was performed on sections from paraffin-embedded samples for histological evidence of the tumor phenotype.
For bioluminescence imaging assay, cells to be tested were injected subcutaneously into the dorsal region near the thigh of the BALB/c nude mice. Bioluminescence imaging was performed using the Xenogen IVIS Spectrum Imaging System (Caliper Life Sciences). Fifteen minutes prior to imaging, mice were injected i.p. with 150 mg/kg luciferin. Following anesthesia, images were taken and analyzed using Spectrum Living Image 4.0 Software (Caliper Life Sciences).
miRNA arrays.
Microarray analysis and preparation of RNA samples were performed commercially by the Shanghai Biochip Corp. according to standard Agilent protocols. Bioinformatics analysis and visualization of microarray data were performed with the MeV v4.4 program ( http://www.tm4.org/mev/) (58).
Accession numbers.
Microarray data described herein have been deposited in the Gene Expression Omnibus database (GEO GSE39358).
Bioinformatics analysis.
The following on-line software programs were used for bioinformatics analysis: DIANA-mirPath ( http://diana.cslab.ece.ntua.gr/pathways/); TargetScan 4.1 ( http://targetscan.org/vert_40/); and miRanda ( http://www.microrna.org/microrna/getGeneForm.do).
Statistics.
All statistical analyses were carried out using SPSS 13.0 statistical software. The Kaplan-Meier method was used to establish survival curves, and the survival differences were compared using the log-rank test. Continuous data were compared using Student’s 2-tailed t test. In all cases, P < 0.05 was considered statistically significant.
Study approval.
For the use of materials for research purposes, prior patient consent was obtained, and study approval was granted by the Sun Yat-sen University and Cancer Center Institution Board. All animal studies were approved by the Sun Yat-sen University Institutional Animal Care and Use Committee.
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology of China (973-2011CB11305); The Natural Science Foundation of China (81071647, 81071762, 30900415, 81272339, 81272417, and 81001190); Guangdong Recruitment Program of Creative Research Groups (2009010058 and 2010B030600003); and National Science and Technique Major Project (201005022-2, 2012ZX10004213, and 311030).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(2):566–579. doi:10.1172/JCI65871.
References
- 1.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 2.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12(8):895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 3.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 4. Sleeman JP, Thiery JP. SnapShot: the epithelial-mesenchymal transition. Cell. 2011. 145 1 162 .e1. 10.1016/j.cell.2011.03.029 [DOI] [PubMed] [Google Scholar]
- 5.Fu Y, et al. β-catenin as a potential key target for tumor suppression. Int J Cancer. 2011;129(7):1541–1551. doi: 10.1002/ijc.26102. [DOI] [PubMed] [Google Scholar]
- 6.Valenta T, Hausmann G, Basler K. The many faces and functions of β-catenin. EMBO J. 2012;31(12):2714–2736. doi: 10.1038/emboj.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anson M, et al. Oncogenic β-catenin triggers an inflammatory response that determines the aggressiveness of hepatocellular carcinoma in mice. J Clin Invest. 2012;122(2):586–599. doi: 10.1172/JCI43937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiMeo TA, et al. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009;69(13):5364–5373. doi: 10.1158/0008-5472.CAN-08-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin SY, et al. Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci U S A. 2000;97(8):4262–4266. doi: 10.1073/pnas.060025397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brabletz T, et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A. 2001;98(18):10356–10361. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhanot P, et al. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996;382(6588):225–230. doi: 10.1038/382225a0. [DOI] [PubMed] [Google Scholar]
- 12.Tamai K, et al. LDL-receptor-related proteins in Wnt signal transduction. Nature. 2000;407(6803):530–535. doi: 10.1038/35035117. [DOI] [PubMed] [Google Scholar]
- 13.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 14.Kishida S, et al. Axin, a negative regulator of the wnt signaling pathway, directly interacts with adenomatous polyposis coli and regulates the stabilization of beta-catenin. J Biol Chem. 1998;273(18):10823–10826. doi: 10.1074/jbc.273.18.10823. [DOI] [PubMed] [Google Scholar]
- 15.Sakanaka C, Weiss JB, Williams LT. Bridging of beta-catenin and glycogen synthase kinase-3beta by axin and inhibition of beta-catenin-mediated transcription. Proc Natl Acad Sci U S A. 1998;95(6):3020–3023. doi: 10.1073/pnas.95.6.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Behrens J, et al. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280(5363):596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- 17.Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003;116(pt 13):2627–2634. doi: 10.1242/jcs.00623. [DOI] [PubMed] [Google Scholar]
- 18.Medrek C, Landberg G, Andersson T, Leandersson K. Wnt-5a-CKI{alpha} signaling promotes {beta}-catenin/E-cadherin complex formation and intercellular adhesion in human breast epithelial cells. J Biol Chem. 2009;284(16):10968–10979. doi: 10.1074/jbc.M804923200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perry JM, et al. Cooperation between both Wnt/{beta}-catenin and PTEN/PI3K/Akt signaling promotes primitive hematopoietic stem cell self-renewal and expansion. Genes Dev. 2011;25(18):1928–1942. doi: 10.1101/gad.17421911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korkaya H, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009;7(6):e1000121. doi: 10.1371/journal.pbio.1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mulholland DJ, Dedhar S, Wu H, Nelson CC. PTEN and GSK3beta: key regulators of progression to androgen-independent prostate cancer. Oncogene. 2006;25(3):329–337. doi: 10.1038/sj.onc.1209020. [DOI] [PubMed] [Google Scholar]
- 22.Djuranovic S, Nahvi A, Green R. A parsimonious model for gene regulation by miRNAs. Science. 2011;331(6017):550–553. doi: 10.1126/science.1191138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esquela-Kerscher A, Slack FJ. Oncomirs — microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 24.Zhu S, Wu H, Wu F, Nie D, Sheng S, Mo YY. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008;18(3):350–359. doi: 10.1038/cr.2008.24. [DOI] [PubMed] [Google Scholar]
- 25.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52(6):1399–1405. [PubMed] [Google Scholar]
- 26.Ma L, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12(3):247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papadopoulos GL, Alexiou P, Maragkakis M, Reczko M, Hatzigeorgiou AG. DIANA-mirPath: integrating human and mouse microRNAs in pathways. Bioinformatics. 2009;25(15):1991–1993. doi: 10.1093/bioinformatics/btp299. [DOI] [PubMed] [Google Scholar]
- 28.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–798. doi: 10.1016/S0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 29.Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5(1):R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dejmek J, Safholm A, Kamp Nielsen C, Andersson T, Leandersson K. Wnt-5a/Ca2+–induced NFAT activity is counteracted by Wnt-5a/Yes-Cdc42-casein kinase 1alpha signaling in human mammary epithelial cells. Mol Cell Biol. 2006;26(16):6024–6036. doi: 10.1128/MCB.02354-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369(9574):1742–1757. doi: 10.1016/S0140-6736(07)60781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 33.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 34.Valastyan S, et al. A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell. 2009;137(6):1032–1046. doi: 10.1016/j.cell.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Chen Y, Shi HY, Stock SR, Stern PH, Zhang M. Regulation of breast cancer-induced bone lesions by β-catenin protein signaling. J Biol Chem. 2011;286(49):42575–42584. doi: 10.1074/jbc.M111.294595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rathore K, Choudhary S, Odoi A, Wang HC. Green tea catechin intervention of reactive oxygen species-mediated ERK pathway activation and chronically induced breast cell carcinogenesis. Carcinogenesis. 2012;33(1):174–183. doi: 10.1093/carcin/bgr244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riggio M, et al. PI3K/AKT pathway regulates phosphorylation of steroid receptors, hormone independence and tumor differentiation in breast cancer. Carcinogenesis. 2012;33(3):509–518. doi: 10.1093/carcin/bgr303. [DOI] [PubMed] [Google Scholar]
- 38.Yan D, Avtanski D, Saxena NK, Sharma D. Leptin-induced epithelial-mesenchymal transition in breast cancer cells requires β-catenin activation via Akt/GSK3- and MTA1/Wnt1 protein-dependent pathways. J Biol Chem. 2012;287(11):8598–8612. doi: 10.1074/jbc.M111.322800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abraham SC, et al. Fibromatosis of the breast and mutations involving the APC/beta-catenin pathway. Hum Pathol. 2002;33(1):39–46. doi: 10.1053/hupa.2002.30196. [DOI] [PubMed] [Google Scholar]
- 40.Jonsson M, Borg A, Nilbert M, Andersson T. Involvement of adenomatous polyposis coli (APC)/beta-catenin signalling in human breast cancer. Eur J Cancer. 2000;36(2):242–248. doi: 10.1016/S0959-8049(99)00276-2. [DOI] [PubMed] [Google Scholar]
- 41.Hsieh JC, et al. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature. 1999;398(6726):431–436. doi: 10.1038/18899. [DOI] [PubMed] [Google Scholar]
- 42.Wissmann C, et al. WIF1, a component of the Wnt pathway, is down-regulated in prostate, breast, lung, and bladder cancer. J Pathol. 2003;201(2):204–212. doi: 10.1002/path.1449. [DOI] [PubMed] [Google Scholar]
- 43.Rubin EM, Guo Y, Tu K, Xie J, Zi X, Hoang BH. Wnt inhibitory factor 1 decreases tumorigenesis and metastasis in osteosarcoma. Mol Cancer Ther. 2010;9(3):731–741. doi: 10.1158/1535-7163.MCT-09-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 45.Wang H, Quah SY, Dong JM, Manser E, Tang JP, Zeng Q. PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 2007;67(7):2922–2926. doi: 10.1158/0008-5472.CAN-06-3598. [DOI] [PubMed] [Google Scholar]
- 46.Jonsson M, Dejmek J, Bendahl PO, Andersson T. Loss of Wnt-5a protein is associated with early relapse in invasive ductal breast carcinomas. Cancer Res. 2002;62(2):409–416. [PubMed] [Google Scholar]
- 47.Wikman H, et al. Relevance of PTEN loss in brain metastasis formation of breast cancer patients. Breast Cancer Res. 2012;14(2):R49. doi: 10.1186/bcr3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mulholland DJ, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012;72(7):1878–1889. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res. 2001;264(1):29–41. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- 50.Krutzfeldt J, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438(7068):685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 51.Ma L, et al. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010;28(4):341–347. doi: 10.1038/nbt.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J, et al. Astrocyte elevated gene-1 is a novel prognostic marker for breast cancer progression and overall patient survival. Clin Cancer Res. 2008;14(11):3319–3326. doi: 10.1158/1078-0432.CCR-07-4054. [DOI] [PubMed] [Google Scholar]
- 53.Hahn WC, et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22(7):2111–2123. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guan H, et al. Sphingosine kinase 1 regulates the Akt/FOXO3a/Bim pathway and contributes to apoptosis resistance in glioma cells. PLoS One. 2011;6(5):e19946. doi: 10.1371/journal.pone.0019946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li J, et al. Oncoprotein Bmi-1 renders apoptotic resistance to glioma cells through activation of the IKK-nuclear factor-kappaB pathway. Am J Pathol. 2010;176(2):699–709. doi: 10.2353/ajpath.2010.090502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang L, et al. MicroRNA-30e* promotes human glioma cell invasiveness in an orthotopic xenotransplantation model by disrupting the NF-kappaB/IkappaBalpha negative feedback loop. J Clin Invest. 2012;122(1):33–47. doi: 10.1172/JCI58849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Y, et al. Stepwise metastatic human hepatocellular carcinoma cell model system with multiple metastatic potentials established through consecutive in vivo selection and studies on metastatic characteristics. J Cancer Res Clin Oncol. 2004;130(8):460–468. doi: 10.1007/s00432-004-0564-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saeed AI, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34(2):374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]