

Abstract
Patients with type 1 diabetes mellitus (T1DM) experience, on average, 2 to 3 hypoglycemic episodes per week. This study investigated the effect of hypoglycemia on cerebral glucose metabolism in patients with uncomplicated T1DM. For this purpose, hyperinsulinemic euglycemic and hypoglycemic glucose clamps were performed on separate days, using [1-13C]glucose infusion to increase plasma 13C enrichment. In vivo brain 13C magnetic resonance spectroscopy was used to measure the time course of 13C label incorporation into different metabolites and to calculate the tricarboxylic acid cycle flux (VTCA) by a one-compartment metabolic model. We found that cerebral glucose metabolism, as reflected by the VTCA, was not significantly different comparing euglycemic and hypoglycemic conditions in patients with T1DM. However, the VTCA was inversely related to the HbA1C and was, under hypoglycemic conditions, approximately 45% higher than that in a previously investigated group of healthy subjects. These data suggest that the brains of patients with T1DM are better able to endure moderate hypoglycemia than those of subjects without diabetes.
Introduction
Patients with type 1 diabetes mellitus (T1DM) experience on average 2 hypoglycemic episodes per week and 1 severe episode of hypoglycemia each year (1). Because the brain depends almost exclusively on glucose, recurrent hypoglycemia may be a threat for cognitive dysfunction and cerebral damage. Patients with T1DM are at increased risk for accelerated cognitive decline and possibly for dementia (2, 3). Interestingly, however, patients with T1DM enduring recurrent episodes of severe hypoglycemia do not appear at greater risk of developing cognitive function impairment than patients without such a history (4). This suggests that recurrent hypoglycemia can induce protective adaptations with respect to cerebral glucose metabolism or that hyperglycemia is at least as detrimental for the brain as hypoglycemia.
Recently, we showed that brain glucose metabolism in healthy subjects at glucose levels of approximately 3 mmol/l did not differ from that at normal glucose levels, as reflected by similar tricarboxylic acid (TCA) cycle rates (VTCA) (5). This remarkable maintenance of normal VTCA during symptomatic hypoglycemia indicates that the glucose threshold for effects on cerebral metabolism lies below 3 mmol/l, either because the brain can endure low glucose levels or because entrance of nonglucose energy substrates such as lactate compensates for the fall in glucose (6–9). Whether these findings can be extrapolated to patients with T1DM remains to be determined.
This study was therefore undertaken to investigate the effect of hypoglycemia on brain glucose metabolism in a representative group of patients with longstanding, uncomplicated, reasonably well-controlled T1DM. To do so, we used 13C magnetic resonance spectroscopy (MRS) of the human brain during euglycemic and hypoglycemic glucose clamps with infusion of [1-13C]glucose, as described previously (5, 10). With this approach, it is possible to directly assess the effects of hypoglycemia on cerebral glucose metabolism and therefore to compare VTCA under euglycemic and hypoglycemic conditions. In addition, we were able to compare the results acquired in the patient group to data previously obtained in subjects without diabetes, who had undergone a similar protocol (5).
Results
To assess the metabolic effects of hypoglycemia in patients with T1DM, a total of 8 hyperinsulinemic euglycemic and 8 hypoglycemic clamps were performed. Six patients completed both clamps (on separate days), 2 patients completed only the euglycemic clamp, and 2 other patients completed only the hypoglycemic clamp.
The clamps started with infusion of a 30 ml 100% [1-13C]glucose bolus to rapidly increase plasma 13C enrichment for steady-state conditions. As a consequence, plasma glucose and glucose 13C enrichment initially peaked (Figure 1). Thereafter, plasma glucose was maintained at 5.0 ± 0.2 mmol/l or 2.9 ± 0.2 mmol/l, as intended. Plasma glucose 13C enrichment stabilized at around 30% during both euglycemic and hypoglycemic clamps.
Figure 1. Plasma glucose concentration and 13C enrichment during clamp studies.

(A) Plasma glucose levels and (B) glucose 13C enrichment during euglycemic and hypoglycemic clamps. All data are expressed as mean ± SEM.
Hypoglycemia stimulated the release of the glucose counterregulatory hormones, adrenalin, noradrenalin, cortisol, and growth hormone (Figure 2), but not that of glucagon, as is characteristic for patients with longstanding T1DM. Although not formally addressed by questionnaires, all patients correctly identified the hypoglycemic clamp, indicating normal hypoglycemic awareness.
Figure 2. Counterregulatory hormone levels.

Levels of plasma glucagon, adrenalin, noradrenalin, growth hormone, and cortisol at baseline (BL) and at the end (End) of the euglycemic and hypoglycemic clamp. All data are expressed as mean ± SEM. *P < 0.05 compared with corresponding baseline value. †P < 0.05 compared with end value of euglycemic clamp.
13C MRS was performed to assess the tissue concentrations of 13C-labeled cerebral metabolites derived from glucose during the hyperinsulinemic clamps. Metabolic fluxes were calculated by fitting time courses of these metabolite concentrations with the one-compartment model, as presented elsewhere (5). This model was used to enable quantification and comparison of cerebral glucose metabolism under the two glycemic conditions. Input factors for the model included (a) plasma concentrations and plasma 13C isotopic enrichment of glucose and lactate and (b) the time courses of 13C incorporation into glutamate C4 (Glu4) and glutamate C3 (Glu3), derived from the 13C magnetic resonance (MR) spectra of the brain (Figure 3) that were measured sequentially, with a time resolution of 2.5 minutes.
Figure 3. 13C MR spectrum.

Representative MR spectrum acquired in 15 minutes after correction for baseline spectra to remove residual fat contamination (note that myo-inositol signal is also not visible because the concentration in the reference spectra is the same as in the dynamic spectra); this is an example of a spectrum fitted in jMRUI to derive concentrations of different labeled metabolites. ppm, parts per million.
Plasma lactate concentrations and lactate 13C enrichment increased over the first 20 minutes of the clamp. Thereafter, plasma lactate levels stabilized at around 1.42 ± 0.03 mmol/l. Plasma lactate 13C isotopic enrichment continued to increase during the euglycemic clamp and tended to stabilize during hypoglycemia (Figure 4).
Figure 4. Plasma lactate 13C enrichment during clamps.

Plasma lactate 13C enrichment during (A) euglycemic and (B) hypoglycemic clamps. Patients with T1DM are represented by black symbols; shaded areas represent values in healthy subjects, as reported in ref. 5. All data are expressed as mean ± SEM.
The time courses of isotope incorporation into glucose metabolites in the brain tissue showed greater increase of 13C Glu4 than of Glu3, consistent with Glu4 being labeled during the first turn of the TCA cycle and Glu3 being labeled during the second, irrespective of the glycemic condition. The concentrations of both Glu4 and Glu3 were lower during the hypoglycemic clamp than during the euglycemic clamp (Figure 5), as was expected from the slightly lower plasma glucose 13C enrichment values during hypoglycemia.
Figure 5. Brain glutamate 13C enrichment over time.

Brain Glu4 and Glu3 concentrations during (A) euglycemic and (B) hypoglycemic clamps, (C and D) together with the respective fits (solid lines) by the metabolic model. Patients with T1DM are represented by black symbols; shaded areas represent values in healthy subjects, as reported in ref. 5. All data were individually quantified and fitted and averaged afterward. All data are expressed as mean ± SEM.
The free flux parameters, VTCA, Vdil (representing the dilution of lactate), and Vgln (representing the efflux of glutamine), of the metabolic model were adjusted to best fit the time courses of Glu4 and Glu3 levels. Note that the model was applied to fit all individual data sets but that, for reasons of clarity, Figure 5, C and D, shows the averaged fits of the time courses of Glu4 and Glu3 on top of the averaged data.
In patients with T1DM, calculated values for VTCA were not different under euglycemic or hypoglycemic conditions (0.59 ± 0.19 μmol/g/min versus 0.62 ± 0.15 μmol/g/min, euglycemia versus hypoglycemia, respectively; P = 0.72; Figure 6). This did not materially change when only patients for whom there were complete data sets under both glycemic conditions were included (n = 6; 0.57 ± 0.21 μmol/g/min versus 0.58 ± 0.11 μmol/g/min, euglycemia versus hypoglycemia, respectively; P = 0.90). To explore the role of glycemic control as a surrogate measure of hypoglycemic burden, we investigated the relationship between the levels of VTCA and glycosylated hemoglobin (HbA1C) and found that lower HbA1C levels were associated with higher VTCA levels (r = 0.56, P < 0.01) (Figure 7).
Figure 6. VTCA.

VTCA during euglycemic and hypoglycemic clamps in patients with T1DM (this study) and healthy subjects (5). Individual symbols represent individual patients or healthy subjects; horizontal bars indicate the mean. *P = 0.014.
Figure 7. VTCA versus HbA1C.

Inverse correlation between the HbA1C and the VTCA (r = 0.56, P < 0.01). For paired measurements, the average VTCA was used. Individual symbols represent individual samples.
Compared with results obtained in healthy volunteers (published previously, ref. 5), the VTCA in patients with T1DM was significantly higher under hypoglycemic conditions (0.43 ± 0.08 μmol/g/min versus 0.62 ± 0.15 μmol/g/min; P = 0.014; Figure 6). There were no significant differences in Vdil or Vgln between healthy subjects and patients with T1DM or between glycemic states (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI62742DS1).
Discussion
In this study, we examined the effect of hypoglycemia on brain glucose metabolism in patients with uncomplicated T1DM. The major observation of this study is that brain glucose metabolism, as reflected by the VTCA, was about similar under euglycemic and hypoglycemic conditions, analogous to previous results obtained in healthy subjects (5). However, when comparing patients with T1DM with controls, we found that the VTCA was approximately 45% higher in patients than in controls under hypoglycemic conditions. We also found that VTCA was inversely correlated with HbA1C in patients with T1DM. These findings suggest adaptations to endure hypoglycemia in the type 1 diabetic brain that are not present in the nondiabetic brain. Since deeper hypoglycemia will eventually cause brain glucose metabolism to deteriorate, these adaptations may shift such deterioration to occur at lower plasma glucose values in patients with T1DM than in subjects without diabetes.
A few studies have directly compared cerebral glucose metabolism in patients with T1DM with that in nondiabetic controls, yet in none of these studies was cerebral glucose metabolism directly assessed under hypoglycemic conditions. Henry et al. reported no differences in VTCA between patients with T1DM and hypoglycemia unawareness and healthy controls, as measured by 13C MRS. However, their study was conducted under hyperglycemic rather than under hypoglycemic conditions (11), clearly indicating that findings obtained during hyperglycemia cannot be extrapolated to include hypoglycemia. A PET study found no differences in brain glucose metabolism during hypoglycemia between patients with T1DM and controls (12), which is contrary to our findings. However, the measurement of glucose metabolism in this PET study was based on the rate of blood-to-brain glucose uptake, rather than on the quantification of glucose metabolites. The much milder level of hypoglycemia attained during the clamp (plasma glucose, 3.6 mmol/l) and the poor level of glycemic control of the patients enrolled (HbA1C 10.1%) could explain their findings and make a direct comparison with our study difficult.
Most patients with T1DM experience hypoglycemic events on a regular basis. Many patients and care providers are concerned that structural abnormalities in white and gray matter and the increased risk of dementia and cognitive dysfunction in T1DM may (in part) be due to recurrent (severe) hypoglycemia (1, 13–15). Although severe hypoglycemia may adversely affect brain anatomy and brain function, particularly in the very young (16) and potentially in the elderly, our findings provide some reassurance for patients that moderate hypoglycemia, such as tested here, has limited effects on cerebral energy metabolism. Whether such results extend to other aspects of brain structure or function cannot be derived from our data.
Since hypoglycemia threatens brain function, it seems plausible that recurrent hypoglycemia initiates an adaptive response to protect the brain against future insults. The inverse correlation between HbA1C and VTCA indeed suggests a role of prior hypoglycemic exposure to the higher VTCA in patients with T1DM with a similarly protective effect, although we cannot with certainty rule out an effect of diabetes per se. A variety of adaptive mechanisms have previously been suggested, including enhanced brain glucose uptake (17, 18), increased storage of glycogen in the brain (19, 20), or use of nonglucose compounds (6–9, 21, 22). However, most of these suggestions lack sufficient support from studies in humans. For instance, in contrast to rodent data (23, 24), studies in humans have generally failed to confirm that recurrent or prolonged hypoglycemia increases brain glucose uptake during subsequent hypoglycemia (25–27). Also, the potential contribution of glycogen supercompensation as a clinically meaningful adaptation to recurrent hypoglycemia in T1DM has recently been questioned (28).
Lactate can serve as an alternative fuel for the brain under conditions of glucopenia. Its metabolism shares pathways downstream from pyruvate into the TCA cycle with those of glucose. Net uptake of lactate by the brain may explain why 13C enrichment of plasma lactate continued to increase during euglycemia but not during hypoglycemia. Increased capacity to take up monocarboxylic acids, such as lactate, from the plasma into the brain during hypoglycemia has been observed in patients with T1DM with hypoglycemia unawareness, presumably as adaptation to recurrent hypoglycemia (29). Thus, enhanced lactate uptake may have contributed to the observed faster VTCA for patients with T1DM in our study, as we previously showed that brain glucose content and transport into the brain did not differ between patients and controls during hypoglycemia (30). Interestingly, VTCA was also increased in animals exposed to recurrent hypoglycemia, but only under euglycemic conditions, while it was reduced under hypoglycemic conditions and the consumption of nonglucose compounds appeared unaltered under either condition (31). The authors suggested that increased GABA levels during hypoglycemia might have inhibited neuronal metabolism (31). The discrepancy with our data may be due to differences in species but also to a different study design, since our patients were instructed to avoid hypoglycemia for at least 24 hours prior to the clamps and were exposed to less severe hypoglycemic conditions. Since patients with T1DM and hypoglycemia unawareness do not appear to have a greater VTCA under hyperglycemic conditions (11), it seems that the metabolic response we observed is specific for the particular experimental conditions.
A potential benefit of increased brain metabolism under hypoglycemia may be increased ability to endure hypoglycemia. Studies in rats have indicated that exposure to recurrent (moderate) hypoglycemia protects against the development of brain damage or cognitive decline from subsequent severe hypoglycemia (32). Indirect evidence for this notion is provided by a nested case-control study showing that patients with T1DM with at least one severe episode of hypoglycemia during follow-up tended to be at lower risk of cardiovascular events, including strokes, than patients without such events (33). A recent report found that hypoglycemia caused less regional brain deactivation during working memory tasks in patients with T1DM and recurrent hypoglycemia than in healthy subjects (34). The authors interpreted this as a form of cerebral inefficiency, in that the patients needed to engage more brain regions to preserve cognitive function during hypoglycemia. However, it could also be interpreted as greater ability to resist hypoglycemia, which would be consistent with the findings of this study. Indeed, there was an inverse correlation between HbA1C and brain activation, very similar to the relationship we observed between HbA1C and VTCA.
The strength of our study is that we were able to measure brain glucose metabolism with 13C MRS under hypoglycemic conditions in vivo in a relevant population at high risk of recurrent hypoglycemia. There are also limitations; our measurements were done in a voxel in the occipital cortex, a general limitation of brain 13C MRS, so that we cannot vouch for other brain regions. Also, the metabolic modeling requires a number of assumptions on cerebral metabolite concentrations and fluxes. These assumptions were identical to those used before (5), and therefore the same limitations apply. It was not possible to assess cognitive function during the experiments, as movement artifacts would have disturbed the highly vulnerable MR measurements. Besides, it should be acknowledged that very complex tasks would have been required to detect cognitive changes during moderate hypoglycemia. No attempt was made to assess overall cognitive function in the patients or in the control subjects investigated earlier.
In conclusion, hypoglycemia does not affect brain glucose metabolism in patients with longstanding, uncomplicated T1DM, indicating that alternative sources of energy, such as lactate, can be used by the brain when glucose delivery falls. We also found that the VTCA during hypoglycemia was higher in patients with T1DM than in healthy controls, which we postulate to be the result of cerebral adaptations to chronic recurrent hypoglycemic episodes.
Methods
Subjects
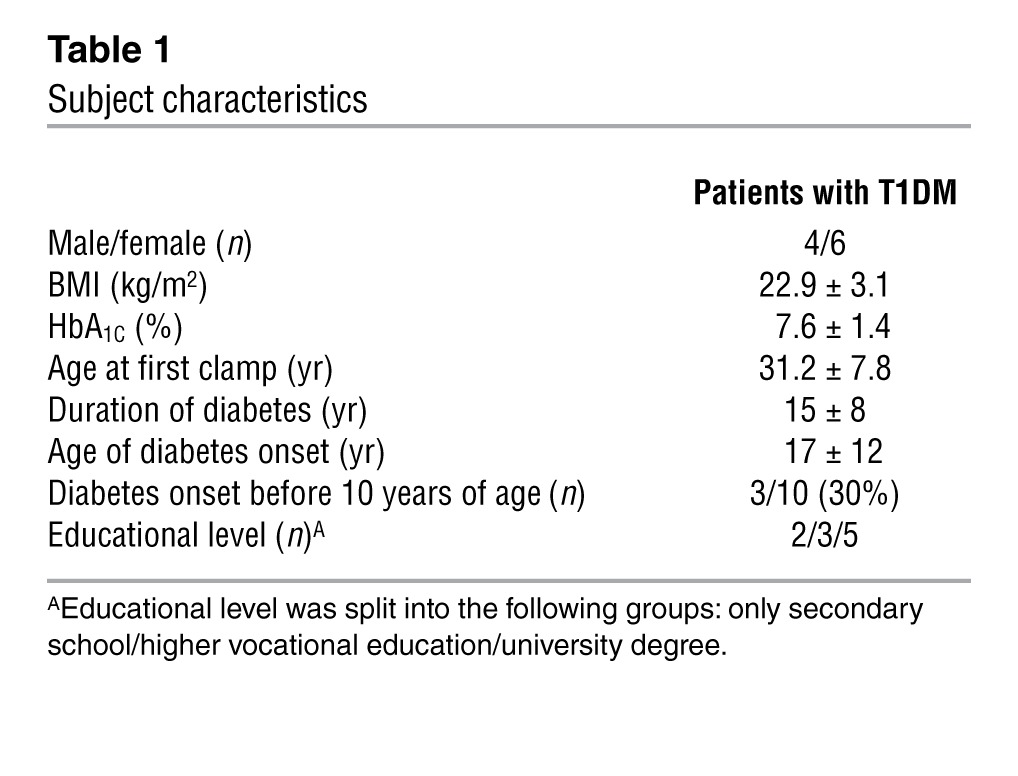
Ten patients with T1DM were included in this study (see Table 1 for patient characteristics). All were free from microvascular complications, except for background retinopathy, and they used no medications other than insulin, thyroxin supplementation (provided that both the dose and serum thyroxin levels were stable), or oral contraceptives. All subjects had at least finished secondary school, and all but one were in professional jobs. Hypoglycemia unawareness was excluded by a Dutch translation of the Cox questionnaire (35, 36). Patients who participated in both the euglycemic and the hypoglycemic clamp had these experiments scheduled in random order at least 2 weeks apart. Female subjects were tested at 4- or 8-week intervals to ensure that experiments took place during corresponding periods of the menstrual cycle.
Table 1.
Subject characteristics
Hyperinsulinemic glucose clamps
The experimental procedure has been described in detail before (5, 10). All patients came to the MR research facility at 8 AM, after an overnight fast and after having abstained from alcohol and caffeine-containing substances for 24 hours. Patients were instructed to reduce the basal insulin dose by 25% the evening before the clamp to avoid hypoglycemic incidents, to check their blood glucose level at approximately 2 AM, to omit the insulin dose on the morning of the clamp, and (if applicable) to disconnect the insulin pump at 7 AM. If hypoglycemia occurred in the 24 hours before the clamp, experiments were rescheduled. After arrival at the MR facility, the brachial artery of the nondominant arm was cannulated under local anesthesia for frequent blood sampling. An intravenous catheter was inserted in the antecubital vein of the contralateral arm for administration of 13C-glucose and insulin. Before patients were placed in the scanner, low-dose insulin was infused to normalize plasma glucose levels (5–8 mmol/l). MR reference measurements were acquired for 30 minutes without administration of exogenous 13C-labeled glucose, followed by a hyperinsulinemic (60 mU/min/m2) euglycemic (5.0 mmol/l) or hypoglycemic (3.0 mmol/l) glucose clamp. A 30 ml bolus of 100%-enriched [1-13C]glucose (20% w/w) (Campro Scientific) was given 5 minutes after the start of insulin administration to rapidly increase plasma glucose 13C enrichment, followed by variable infusions of 40%- or 50%-enriched [1-13C]glucose (20% w/w) during the euglycemic and hypoglycemic experiments, respectively. Blood was sampled every 5 minutes for immediate determination of plasma glucose (Beckman Glucose Analyzer II, Coulter, or Biosen C-line, EKF Diagnostics) and lactate (Biosen C-line, EKF Diagnostics) and for later measurement of plasma 13C isotopic enrichment of glucose and lactate by high-resolution proton nuclear MR (1H NMR) at 500 MHz (37, 38). At baseline, at t = 30 minutes after the start of the clamp and at the end of the clamp, additional blood was sampled for measurement of plasma insulin and counterregulatory hormones, as described previously (10). The clamps were terminated after 100 minutes.
MR protocol
MR experiments were conducted with the same protocol applied to the healthy volunteers (5) on a 3T MR system (Magnetom Trio, Siemens) equipped with a home-built 1H volume coil, with a circularly polarized 13C surface coil inserted into it (39). Transversal T2-weighted images were acquired to locate the voxel position for the spectroscopy experiment. Shimming was performed with an automatic gradient-echo–based shim sequence. 13C MR spectra were acquired with the ISIS-DEPT sequence (40), which combines 1H-ISIS localization with 1H-13C polarization transfer. A 45° alpha pulse was used to observe 13C-MR signals from CH, CH2, and CH3 groups simultaneously. Broadband proton decoupling (WALTZ-16; ref. 41) was applied to simplify the MR spectra. MR spectra were acquired from a voxel of approximately 125 ml in the occipital brain tissue. Each spectrum consisted of 72 scans with a TR of 2 seconds, resulting in a total duration of 2.5 minutes. Acquisition of 13C MR spectra started 20 minutes before [1-13C]glucose infusion to obtain 8 reference spectra and continued throughout the entire clamp. Visual stimulation was avoided by dimming the lights in the magnet room during the experiments.
Data analysis
13C MRS data processing and quantification.
To remove contamination of natural abundance 13C MR signals, averaged reference spectra were subtracted from the dynamic spectra acquired during 13C-labeled glucose infusion. After application of a moving average of 15 minutes, the peaks of interest in the spectra were fitted with the advanced MR (AMARES) algorithm (42) in the Java-based MR user interface (jMRUI) software package (ref. 43 and see Figure 3 for representative spectrum). To quantify the concentrations of 13C-labeled metabolites (i.e., Glu4 and Glu3), the natural abundance myo-inositol (mI) concentration was used, assuming that mI peaks equaled 1.1% of 6 μmol/g (44). The ratio between the mI concentration and the peak integral was derived from the sum of all spectra before baseline correction. Results from phantom measurements were used to correct for the effect of the pulse profiles on the signal intensities.
Metabolic modeling.
A standard one-compartment metabolic model (11, 45, 46) was used to determine rates of metabolic fluxes representing the VTCA, the loss of label through exchange with unlabeled glutamine (Vefflux), and the exchange of intracellular lactate with plasma lactate (Vdil). The model was adapted to include inflow from labeled plasma lactate. A series of differential equations describe the fluxes of 13C labels from and to different metabolite pools. Plasma glucose and lactate concentrations and 13C isotopic enrichments were used as input for the metabolic model. The time courses of label incorporation into glutamate at the C4 (Glu4) and C3 (Glu3) positions were fitted with a nonlinear least-squares minimization algorithm in MATLAB 2008 (MathWorks). This results in values for the flux parameters. This analysis was performed on all data sets of individual measurements. Note that since the current model used the exact lactate concentration input curves, rather than approximations, the flux values in the healthy volunteers differed slightly, but not significantly, from those reported previously (5).
Statistics.
All data are expressed as mean ± SD, unless mentioned otherwise. Differences in means were tested by 2-tailed Student’s t tests, and the relationship between HbA1C and VTCA was derived with linear regression analysis. Graphpad Prism 4 (Graphpad) was used for statistical analysis, and a P value of less than 0.05 was considered statistically significant.
Study approval
All patients gave written informed consent before participating in this study, which was approved by the institutional review board of the Radboud University Nijmegen Medical Centre.
Supplementary Material
Acknowledgments
We are indebted to Karin Saini, Cindy Frentz, and Bas Schouwenberg for assistance during the hyperinsulinemic clamps and Angelina Goudswaard, Frederique Vermeulen, and Udo Engelke for help with high-resolution NMR of plasma samples. This work was financially supported by the Dutch Diabetes Research Foundation (grant 2004.00.012), the framework of the Center for Translational Molecular Medicine ( http://www.ctmm.nl/pro1/general/home.asp; project PREDICCt grant 01C-104), the Netherlands Heart Foundation, and the Dutch Kidney Foundation.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(2):623–629. doi:10.1172/JCI62742.
References
- 1.Strachan MWJ. Hypoglycemia In Clinical Diabetes. 1999. Frequency, causes and risk factors for hypoglycaemia in type 1 diabetes. In: Frier BM, Fisher M, eds. pp. 49–82. 2nd ed. New York, New York, USA: John Wiley and Sons; [Google Scholar]
- 2.Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5(1):64–74. doi: 10.1016/S1474-4422(05)70284-2. [DOI] [PubMed] [Google Scholar]
- 3.Sims-Robinson C, Kim B, Rosko A, Feldman EL. How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol. 2010;6(10):551–559. doi: 10.1038/nrneurol.2010.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frier BM. Cognitive functioning in type 1 diabetes: the Diabetes Control and Complications Trial (DCCT) revisited. Diabetologia. 2011;54(2):233–236. doi: 10.1007/s00125-010-1983-6. [DOI] [PubMed] [Google Scholar]
- 5.Van de Ven KC, et al. Effect of acute hypoglycemia on human cerebral glucose metabolism measured by 13C magnetic resonance spectroscopy. Diabetes. 2011;60(5):1467–1473. doi: 10.2337/db10-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boumezbeur F, et al. The contribution of blood lactate to brain energy metabolism in humans measured by dynamic 13C nuclear magnetic resonance spectroscopy. J Neurosci. 2010;30(42):13983–13991. doi: 10.1523/JNEUROSCI.2040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maran A, Cranston I, Lomas J, Macdonald I, Amiel SA. Protection by lactate of cerebral function during hypoglycaemia. Lancet. 1994;343(8888):16–20. doi: 10.1016/S0140-6736(94)90876-1. [DOI] [PubMed] [Google Scholar]
- 8.Smith D, Pernet A, Hallett WA, Bingham E, Marsden PK, Amiel SA. Lactate: a preferred fuel for human brain metabolism in vivo. J Cereb Blood Flow Metab. 2003;23(6):658–664. doi: 10.1097/01.WCB.0000063991.19746.11. [DOI] [PubMed] [Google Scholar]
- 9.Herzog RI, Sherwin RS, Rothman DL. Insulin-induced hypoglycemia and its effect on the brain: unraveling metabolism by in vivo nuclear magnetic resonance. Diabetes. 2011;60(7):1856–1858. doi: 10.2337/db11-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van de Ven KC, van der Graaf M, Tack CJ, Klomp DW, Heerschap A, de Galan BE. Optimized [1-(13)C]glucose infusion protocol for 13C magnetic resonance spectroscopy at 3T of human brain glucose metabolism under euglycemic and hypoglycemic conditions. J Neurosci Methods. 2010;186(1):68–71. doi: 10.1016/j.jneumeth.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henry PG, Criego AB, Kumar A, Seaquist ER. Measurement of cerebral oxidative glucose consumption in patients with type 1 diabetes mellitus and hypoglycemia unawareness using (13)C nuclear magnetic resonance spectroscopy. Metabolism. 2010;59(1):100–106. doi: 10.1016/j.metabol.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fanelli CG, et al. Blood-to-brain glucose transport and cerebral glucose metabolism are not reduced in poorly controlled type 1 diabetes. Diabetes. 1998;47(9):1444–1450. doi: 10.2337/diabetes.47.9.1444. [DOI] [PubMed] [Google Scholar]
- 13.Seaquist ER. The final frontier: how does diabetes affect the brain? Diabetes. 2010;59(1):4–5. doi: 10.2337/db09-1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Musen G, et al. Effects of type 1 diabetes on gray matter density as measured by voxel-based morphometry. Diabetes. 2006;55(2):326–333. doi: 10.2337/diabetes.55.02.06.db05-0520. [DOI] [PubMed] [Google Scholar]
- 15.Brands AM, et al. Cognitive performance, psychological well-being, and brain magnetic resonance imaging in older patients with type 1 diabetes. Diabetes. 2006;55(6):1800–1806. doi: 10.2337/db05-1226. [DOI] [PubMed] [Google Scholar]
- 16.Hershey T, Perantie DC, Warren SL, Zimmerman EC, Sadler M, White NH. Frequency and timing of severe hypoglycemia affects spatial memory in children with type 1 diabetes. Diabetes Care. 2005;28(10):2372–2377. doi: 10.2337/diacare.28.10.2372. [DOI] [PubMed] [Google Scholar]
- 17.Heikkila O, Makimattila S, Timonen M, Groop PH, Heikkinen S, Lundbom N. Cerebellar glucose during fasting and acute hyperglycemia in nondiabetic men and in men with type 1 diabetes. Cerebellum. 2010;9(3):336–344. doi: 10.1007/s12311-010-0166-9. [DOI] [PubMed] [Google Scholar]
- 18.Kreis R, Ross BD. Cerebral metabolic disturbances in patients with subacute and chronic diabetes mellitus: detection with proton MR spectroscopy. Radiology. 1992;184(1):123–130. doi: 10.1148/radiology.184.1.1319074. [DOI] [PubMed] [Google Scholar]
- 19.Oz G, et al. Human brain glycogen metabolism during and after hypoglycemia. Diabetes. 2009;58(9):1978–1985. doi: 10.2337/db09-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herzog RI, Chan O, Yu S, Dziura J, McNay EC, Sherwin RS. Effect of acute and recurrent hypoglycemia on changes in brain glycogen concentration. Endocrinology. 2008;149(4):1499–1504. doi: 10.1210/en.2007-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B. In vivo evidence for lactate as a neuronal energy source. J Neurosci. 2011;31(20):7477–7485. doi: 10.1523/JNEUROSCI.0415-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veneman T, Mitrakou A, Mokan M, Cryer P, Gerich J. Effect of hyperketonemia and hyperlacticacidemia on symptoms, cognitive dysfunction, and counterregulatory hormone responses during hypoglycemia in normal humans. Diabetes. 1994;43(11):1311–1317. doi: 10.2337/diabetes.43.11.1311. [DOI] [PubMed] [Google Scholar]
- 23.McCall AL, Fixman LB, Fleming N, Tornheim K, Chick W, Ruderman NB. Chronic hypoglycemia increases brain glucose transport. Am J Physiol. 1986;251(4 pt 1):E442–E447. doi: 10.1152/ajpendo.1986.251.4.E442. [DOI] [PubMed] [Google Scholar]
- 24.Simpson IA, et al. Blood-brain barrier glucose transporter: effects of hypo- and hyperglycemia revisited. J Neurochem. 1999;72(1):238–247. doi: 10.1046/j.1471-4159.1999.0720238.x. [DOI] [PubMed] [Google Scholar]
- 25.Criego AB, Tkac I, Kumar A, Thomas W, Gruetter R, Seaquist ER. Brain glucose concentrations in healthy humans subjected to recurrent hypoglycemia. J Neurosci Res. 2005;82(4):525–530. doi: 10.1002/jnr.20654. [DOI] [PubMed] [Google Scholar]
- 26.Segel SA, et al. Blood-to-brain glucose transport, cerebral glucose metabolism, and cerebral blood flow are not increased after hypoglycemia. Diabetes. 2001;50(8):1911–1917. doi: 10.2337/diabetes.50.8.1911. [DOI] [PubMed] [Google Scholar]
- 27.Bingham EM, et al. Differential changes in brain glucose metabolism during hypoglycaemia accompany loss of hypoglycaemia awareness in men with type 1 diabetes mellitus. An [11C]-3-O-methyl-D-glucose PET study. Diabetologia. 2005;48(10):2080–2089. doi: 10.1007/s00125-005-1900-6. [DOI] [PubMed] [Google Scholar]
- 28.Öz G, Tesfaye N, Kumar A, Deelchand DK, Eberly LE, Seaquist ER. Brain glycogen content and metabolism in subjects with type 1 diabetes and hypoglycemia unawareness. J Cereb Blood Flow Metab. 2012;32(2):256–263. doi: 10.1038/jcbfm.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mason GF, Petersen KF, Lebon V, Rothman DL, Shulman GI. Increased brain monocarboxylic acid transport and utilization in type 1 diabetes. Diabetes. 2006;55(4):929–934. doi: 10.2337/diabetes.55.04.06.db05-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van de Ven KC, van der Graaf M, Tack CJ, Heerschap A, de Galan BE. Steady-state brain glucose concentrations during hypoglycemia in healthy humans and patients with type 1 diabetes. Diabetes. 2012;61(8):1974–1977. doi: 10.2337/db11-1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang L, et al. Recurrent antecedent hypoglycemia alters neuronal oxidative metabolism in vivo. Diabetes. 2009;58(6):1266–1274. doi: 10.2337/db08-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puente EC, et al. Recurrent moderate hypoglycemia ameliorates brain damage and cognitive dysfunction induced by severe hypoglycemia. Diabetes. 2010;59(4):1055–1062. doi: 10.2337/db09-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gruden G, Barutta F, Cavallo Perin P, Bruno G. Severe hypoglycemia and cardiovascular disease incidence in type 1 Diabetes: The EURODIAB Prospective Complications Study. Diabetes Care. 2012;35(12):e89. doi: 10.2337/dc12-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolo NR, et al. Brain activation during working memory is altered in patients with type 1 diabetes during hypoglycemia. Diabetes. 2011;60(12):3256–3264. doi: 10.2337/db11-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke WL, Cox DJ, Gonder-Frederick LA, Julian D, Schlundt D, Polonsky W. Reduced awareness of hypoglycemia in adults with IDDM. A prospective study of hypoglycemic frequency and associated symptoms. Diabetes Care. 1995;18(4):517–522. doi: 10.2337/diacare.18.4.517. [DOI] [PubMed] [Google Scholar]
- 36.De Galan BE, De Mol P, Wennekes L, Schouwenberg BJ, Smits P. Preserved sensitivity to beta2-adrenergic receptor agonists in patients with type 1 diabetes mellitus and hypoglycemia unawareness. J Clin Endocrinol Metab. 2006;91(8):2878–2881. doi: 10.1210/jc.2006-0528. [DOI] [PubMed] [Google Scholar]
- 37.Serlie MJ, et al. Glycogen synthesis in human gastrocnemius muscle is not representative of whole-body muscle glycogen synthesis. Diabetes. 2005;54(5):1277–1282. doi: 10.2337/diabetes.54.5.1277. [DOI] [PubMed] [Google Scholar]
- 38.Van Den Bergh AJ, Tack CJ, Van Den Boogert HJ, Vervoort G, Smits P, Heerschap A. Assessment of human muscle glycogen synthesis and total glucose content by in vivo 13C MRS. Eur J Clin Invest. 2000;30(2):122–128. doi: 10.1046/j.1365-2362.2000.00603.x. [DOI] [PubMed] [Google Scholar]
- 39.Klomp DW, Renema WK, van der Graaf M, de Galan BE, Kentgens AP, Heerschap A. Sensitivity-enhanced 13C MR spectroscopy of the human brain at 3 Tesla. Magn Reson Med. 2006;55(2):271–278. doi: 10.1002/mrm.20745. [DOI] [PubMed] [Google Scholar]
- 40.Klomp DW, Kentgens AP, Heerschap A. Polarization transfer for sensitivity-enhanced MRS using a single radio frequency transmit channel. NMR Biomed. 2008;21(5):444–452. doi: 10.1002/nbm.1208. [DOI] [PubMed] [Google Scholar]
- 41.Shaka AJ, Keeler J, Frenkiel T, Freeman R. An improved sequence for broadband decoupling: WALTZ-16. J Magn Reson. 1983;52:335–338. [Google Scholar]
- 42.Vanhamme L, van den Boogaart A, Van Huffel S. Improved method for accurate and efficient quantification of MRS data with use of prior knowledge. J Magn Reson. 1997;129(1):35–43. doi: 10.1006/jmre.1997.1244. [DOI] [PubMed] [Google Scholar]
- 43.Naressi A, et al. Java-based graphical user interface for the MRUI quantitation package. MAGMA. 2001;12(2–3):141–152. doi: 10.1007/BF02668096. [DOI] [PubMed] [Google Scholar]
- 44.Ross B, Lin A, Harris K, Bhattacharya P, Schweinsburg B. Clinical experience with 13C MRS in vivo. NMR Biomed. 2003;16(6–7):358–369. doi: 10.1002/nbm.852. [DOI] [PubMed] [Google Scholar]
- 45.Henry PG, et al. In vivo 13C NMR spectroscopy and metabolic modeling in the brain: a practical perspective. Magn Reson Imaging. 2006;24(4):527–539. doi: 10.1016/j.mri.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 46.Henry PG, Lebon V, Vaufrey F, Brouillet E, Hantraye P, Bloch G. Decreased TCA cycle rate in the rat brain after acute 3-NP treatment measured by in vivo 1H-[13C] NMR spectroscopy. J Neurochem. 2002;82(4):857–866. doi: 10.1046/j.1471-4159.2002.01006.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.