

Abstract
A large database of copy number profiles from cancer genomes can facilitate the identification of recurrent chromosomal alterations that often contain key cancer-related genes. It can also be used to explore low-prevalence genomic events such as chromothripsis. In this study, we report an analysis of 8227 human cancer copy number profiles obtained from 107 array comparative genomic hybridization (CGH) studies. Our analysis reveals similarity of chromosomal arm-level alterations among developmentally related tumor types as well as a number of co-occurring pairs of arm-level alterations. Recurrent (“pan-lineage”) focal alterations identified across diverse tumor types show an enrichment of known cancer-related genes and genes with relevant functions in cancer-associated phenotypes (e.g., kinase and cell cycle). Tumor type-specific (“lineage-restricted”) alterations and their enriched functional categories were also identified. Furthermore, we developed an algorithm for detecting regions in which the copy number oscillates rapidly between fixed levels, indicative of chromothripsis. We observed these massive genomic rearrangements in 1%–2% of the samples with variable tumor type-specific incidence rates. Taken together, our comprehensive view of copy number alterations provides a framework for understanding the functional significance of various genomic alterations in cancer genomes.
Cancer genomes harbor various somatic forms of genetic alterations ranging from nucleotide-level changes (e.g., nucleotide substitutions and small insertions/deletions) (Greenman et al. 2007) to large chromosomal events (e.g., translocations and copy number alterations) (Albertson et al. 2003; Mitelman et al. 2007). As a comprehensive catalog of tumor-related chromosomal alterations can help identify genomic features with potential clinical benefits (Chin and Gray 2008; Meyerson et al. 2010), collecting and profiling tumor samples using genome-wide, high-throughput platforms have been major efforts during the last decade. The resulting accumulation of cancer genome studies has provided new mechanistic insights that aid in a more systematic understanding of human cancer and in identification of molecular targets for cancer therapy (Chin et al. 2011; Hanahan and Weinberg 2011). However, due to the intrinsic complexity and heterogeneity of human cancer genomes, there is still a large number of unresolved issues. For example, a majority of cancer-related genomic alterations are believed to be “passengers” that arise as a by-product during cancer genome evolution, without obvious advantage for the affected clones. The discrimination of such passenger alterations from “driver” alterations that contribute to tumor initiation and/or progression remains a difficult task.
In the past few years, array-based comparative genomic hybridization (array-CGH) has become a dominant tool for genome-wide detection of copy number changes in cancer (Pinkel et al. 1998; Snijders et al. 2001). It has been applied to a wide range of tumor types, with notable success in subtype classification and biomarker screening (Albertson and Pinkel 2003; Pinkel and Albertson 2005). Most array-CGH studies performed so far, however, focus on a specific tumor type with a limited number of samples. Some efforts have been made to construct a large-scale array-CGH database (Baudis and Cleary 2001; Beroukhim et al. 2010; Cao et al. 2010), with indications that a large compilation of cancer genomes may be advantageous in distinguishing driver alterations from passenger events. In particular, Beroukhim et al. (2010) reported analysis of ∼3000 cancer genome profiles that resulted in identification of numerous focal somatic alterations across tumor types, corroborating that a large database can help identify recurrent focal alterations that are likely to be oncogenic drivers. That analysis, however, was based on a single array-CGH platform from Affymetrix with 250K probes (Beroukhim et al. 2010). Considering the growing number of cancer data sets in public databases and continual improvement in platforms, it is imperative that the research community should take advantage of the statistical power and a wide range of tumor types that come with a larger compendium.
In this study, we present extensive computational analysis of 8227 copy number profiles gathered from 107 array-CGH human cancer studies. The data were collected from the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/), a public repository of microarray data sets (Barrett et al. 2009). Among the array-CGH studies available in GEO, we collected those profiled by high-resolution, oligonucleotide-based platforms (>100K probes) from two commercial vendors (Affymetrix and Agilent). After normalization and application of segmentation algorithms, the arm-level alteration frequencies across the samples were investigated for lineage-specific patterns in major tumor types and for concordant relationships between alterations. We also delineated the minimal common regions (MCRs) across the entire data set and in a given tumor type (“pan-lineage” and tumor type-specific MCRs, respectively). MCR (sometimes called MAR for “minimally altered region”) is defined as the minimal region of amplifications or deletions representing a common genomic alteration across the examined cancers (Santarius et al. 2010). MCRs were interpreted by their association with known cancer-related genes (Futreal et al. 2004) and Gene Ontology (GO) functional categories (Ashburner et al. 2000). Finally, we performed a survey of chromothripsis, a massive genomic rearrangement event that is thought to occur in a small fraction of tumors (Stephens et al. 2011). Overall, our study presents a global view of copy number alterations and demonstrates the utility of a large-scale copy number database in prioritizing candidate biomarkers and in exploring unique patterns of chromosomal imbalances such as chromothripsis.
Results
A compendium of copy number profiles from human cancer genomes
We searched the GEO database to obtain data from high-resolution, array-CGH platforms for human cancer samples (Fig. 1A). We focused on the five platforms comprising more than 100K oligonucleotide probes from two commercial vendors (Agilent and Affymetrix): Agilent 244K and Affymetrix 100K, 250K, 500K, and SNP6.0. The number of probes ranged from 115,417 (Affymetrix 100K) to 1.8 million (Affymetrix SNP6.0). From the 107 array-CGH studies based on these platforms, we collected a total of 8227 cancer genome copy number profiles after removing normal controls or duplicates. The sample numbers are shown for individual tumor types and array-CGH platforms in Figure 1B. The frequencies of individual tumor types in our data set were compared with the tumor incidence rates (Supplemental Fig. S1). A substantial level of correlation (r2 = 0.44) was observed, attributable to tumor types with higher incidence rates that are also well-represented in databases, such as breast and lung cancers. The detailed information on the data sets is available in Supplemental Tables S1 and S2. For subsequent analysis, we used the log2 ratio profiles as available in the GEO database for the Agilent 244K platforms (n = 1750) and derived log2 ratios (tumor/reference) using HapMap population data as a universal reference for the Affymetrix platform (n = 6477) (see Methods for segmentation steps and additional processing). The segmentation profiles of all tumor samples and other analysis files are available at http://compbio.med.harvard.edu/metaCGH/.
Figure 1.

Compilation of large-scale cancer genome copy number profiles. (A) A schematic of data collection is shown. Five high-resolution, array-CGH platforms used are listed with the corresponding GEO accession ID (GPL) and the number of associated samples. (B) Major tumor types (>100 samples for each type) are shown with their sample numbers with respect to the five platforms. (MDS) Myelodysplastic syndrome, (MM) multiple myeloma, (MPD) myeloproliferative disorder.
In our data set, we observed an average of 79.3 gains and 80.9 losses per sample, involving 9.7% and 11.6% of the reference genome, respectively. Among the five platforms, Affymetrix SNP6.0 (190.5 gains and 233.6 losses per sample) and Agilent 244K platform (108.5 gains and 107.4 losses per sample) showed a substantially higher number of alterations compared to Affymetrix 100K–500K platforms, with smaller alteration sizes on average (Supplemental Fig. S2). The small size and relative abundance of alterations in these platforms may be due to the higher sensitivity with increased resolution of the platforms (Affymetrix SNP6.0) or higher signal-to-noise ratio of longer oligonucleotides (60-bp oligonucleotides of the Agilent platform compared to Affymetrix 25-bp oligonucleotides). It may also be associated with hypersegmentation, which has been previously observed in regions with extreme copy numbers due to different attenuation curves of neighboring probes (Beroukhim et al. 2007; The Cancer Genome Atlas Research Network 2008).
Chromosomal arm-level alterations in human cancer genomes
We defined chromosomal arm-level alteration as a single alteration or an aggregate of alterations that encompass >50% of a chromosomal arm. The arm-level alteration frequency measured across the entire data set (n = 8227) (Fig. 2A) highlights the frequent gains at 1q, 5p, 7p, 8q, and 20q along with frequent losses at 4q, 6q, 8p, 13q, and 17p (for per-platform alteration frequencies, see Supplemental Fig. S3). The arm-level alteration frequencies were converted into chromosomal size-adjusted Z scores as described previously (Beroukhim et al. 2010) to show the extent of deviation from the background alteration rate (Supplemental Fig. S4). The distribution of alteration frequencies (negative correlation of r = −0.617 between arm-level gain and loss frequencies) (Fig. 2A) suggests that arm-level alteration-frequent chromosomes tend to be preferentially gained or lost, but rarely both (Beroukhim et al. 2010).
Figure 2.

Overview of chromosomal arm-level alteration frequency. (A) A scatter plot shows the arm-level alteration frequency measured across the entire data set (n = 8227). The top five most frequently gained or lost chromosomal arms are marked. Size-adjusted arm-level alteration frequencies are separately shown in Supplemental Figure S4. (B) Hierarchical clustering using the arm-level alteration frequency largely segregates 19 tumor types into three clusters of hematologic, epithelial, and neuroepithelial origins (from left to right). The heat map shows the frequency of chromosomal copy gains (above) and losses (below), ordered from 1p to 22q.
To investigate tumor-type specificity of arm-level gains or losses, we measured the arm-level alteration frequencies separately for the 19 tumor types that have more than 100 samples per tumor type. Hierarchical clustering segregates three main clusters of tumor types with similar developmental origins (Fig. 2B). In one cluster, five hematologic-origin tumors were observed together. A second cluster contained most of the epithelial tumors. A third cluster contained the four tumors arising from neuroepithelial origin (medulloblastoma, neuroblastoma, glioma, and melanoma) and two others. These clusters emphasize the strong relationship between embryogenesis and the post-embryonic development of human cancers at a molecular level. The similarity between sarcoma and renal cell carcinoma and their presence in the neuroepithelial cluster were unexpected. This pattern was previously reported in another large-scale study (Beroukhim et al. 2010) and was present even after the removal of the platform used in that study (Affymetrix 250K/StyI) (Supplemental Fig. S5), but its biological significance is not yet clear.
Next, we examined the extent of co-occurrence for arm-level alteration pairs. We distinguished concordant arm-level alteration pairs (gain-gain and loss-loss) (Fig. 3A) from discordant ones (gain-loss and loss-gain) (Fig. 3B). Among the potential concordant chromosomal arm pairs, we observed a cluster of pairs between short, gene-rich chromosomes, such as 16p, 17p/q, 19p/q, 20q, and 22q (gene density > 10 genes/Mb) (arrow indicated in Fig. 3A). We also observed two clusters of significant discordant pairs between short, gene-rich chromosomes listed above and gene-poor chromosomes such as 4q, 5p, 6q, 13q, and 18q (marked by two solid arrows in Fig. 3B).
Figure 3.

Concordant and discordant relationships between arm-level alterations. (A) The extent of concordance for chromosomal arm-level gain-gain and loss-loss is shown in the upper right and lower left triangles, respectively. The chromosomal arms are sorted by gene density (genes/Mb; e.g., 19q and 5p are the most gene-rich and gene-poor chromosomal arms, respectively). The heat map shows the multiple test-adjusted significance of concordance. The arrow marks a cluster of frequent gain-gain and loss-loss pairs between gene-rich chromosomal arms. (B) The extent of discordance between chromosomal arm-level gain-loss is shown. The two solid arrows indicate clusters of chromosomal arm pairs with frequent discordant changes between gene-rich and gene-poor chromosomal arms. The dotted arrow indicates a discordant pair between 7p gain and 10q loss.
The landscape of focal recurrent alterations across diverse tumor types
Genomic regions frequently altered across diverse tumor types are of primary interest, as they have elevated the likelihood of containing driver alterations. To identify significantly recurrent focal MCRs, we used the GISTIC algorithm to summarize multiple profiles and assign statistical significance (Beroukhim et al. 2007; Mermel et al. 2011). In this method, the average magnitude of copy number alteration (versus frequency alone) is used as a score, and a permutation-based test is used for estimating statistical significance. Across the entire data set, we identified a total of 94 amplification and 71 deletion MCRs (Supplemental Table S3). We termed these 165 recurrent alterations as pan-lineage MCRs.
Pan-lineage amplification and deletion MCRs comprise 0.69% and 0.50% of the reference genome, encompassing coding sequences of 421 and 156 known genes (out of 20,234 autosomal RefSeqs), respectively. Known cancer-related genes were significantly overrepresented in these MCRs (17 out of 264 autosomal cancer consensus genes; Fisher’s exact test P = 0.0015) (Futreal et al. 2004). Ten (MYCN, PDGFRB, EGFR, FGFR1, WHSC1L1, MYC, HOXC13, CDK4, NTRK3, and ERBB2) and seven cancer-related genes (CDKN2A, PTEN, ATM, ERC1, FOXO1, TP53, and TCF3) were associated with pan-lineage amplification and deletion MCRs, respectively. These 17 genes and their significance (GISTIC Q-values) are shown in Figure 4A.
Figure 4.

Recurrent chromosomal alterations across diverse tumor types. (A) The significance of recurrent amplification (left) and deletion (right) as measured by the GISTIC algorithm (GISTIC Q value; log-scaled) is plotted across the genome. Seventeen known targets (cancer consensus genes) are shown at the corresponding peaks. (B) The GO categories significantly enriched in pan-lineage amplification and deletion MCRs are shown. Similar functional categories (e.g., “kinase” or “kinase activity”) are grouped, and the representative function is shown with the number of related GO functions in parentheses. An asterisk indicates that the corresponding functional categories remained significant after removal of MCRs with known cancer genes.
GO analysis showed that 15 and 35 out of 1004 GO functional categories were significantly enriched in amplification and deletion MCRs (FDR < 0.01) (Fig. 4B; Supplemental Table S4), respectively. Among the 15 functions enriched in amplification MCRs, seven had functional annotations related to “protein tyrosine kinase.” “Cell cycle/apoptosis”-related functional categories comprised 10 out of 35 functions enriched in deletion MCRs. After excluding the peaks that contain cancer consensus genes, “microtubule,” “lyase,” and “transporter” (with amplification) and “cell junction/adhesion” and “metabolism” categories (with deletion) remained significant (Supplemental Table S4).
To identify MCRs that may have arisen due to increased local genomic fragility rather than selective advantage or functional significance, we compared the pan-lineage MCRs with 37 known fragile regions available in the literature (Bignell et al. 2010). Two (FRA6H/6p21 and FRA13E/13q22) and three fragile sites (FRA3B/3p14, FRA4F/4q22, and FRA10D/10q21) were associated with amplification and deletion MCRs, respectively. In addition, when we measured the overlap between pan-lineage MCRs and known germline copy number variations (CNVs) obtained from the Database of Genomic Variants (DGV) (http://projects.tcag.ca/variation/), eight MCRs showed substantially elevated CNV density (>100 CNVs/Mb; mean of all MCRs was 36.2 CNVs/Mb), indicative of potential germline origins for these recurrent alterations. The detailed information about the overlap with known fragile regions and CNVs is available in Supplemental Table S3.
Since our compendium is composed of genomic profiles from multiple array-CGH platforms, the reduction of potential platform biases may enhance the true biological signal. This is a challenging problem, especially because we do not have the same samples profiled on multiple platforms and the probe characteristics are variable across platforms. In our attempt to mitigate the impact of the platform-specific effects, we employed a linear mixed model on the profiles after they have been segmented (see Methods). The adjusted profiles show an overall improvement in the extent of overlap between the genomic peaks identified in each platform, compared to those from unadjusted profiles (Supplemental Fig. S6).
Tumor type-specific alterations and functional association map
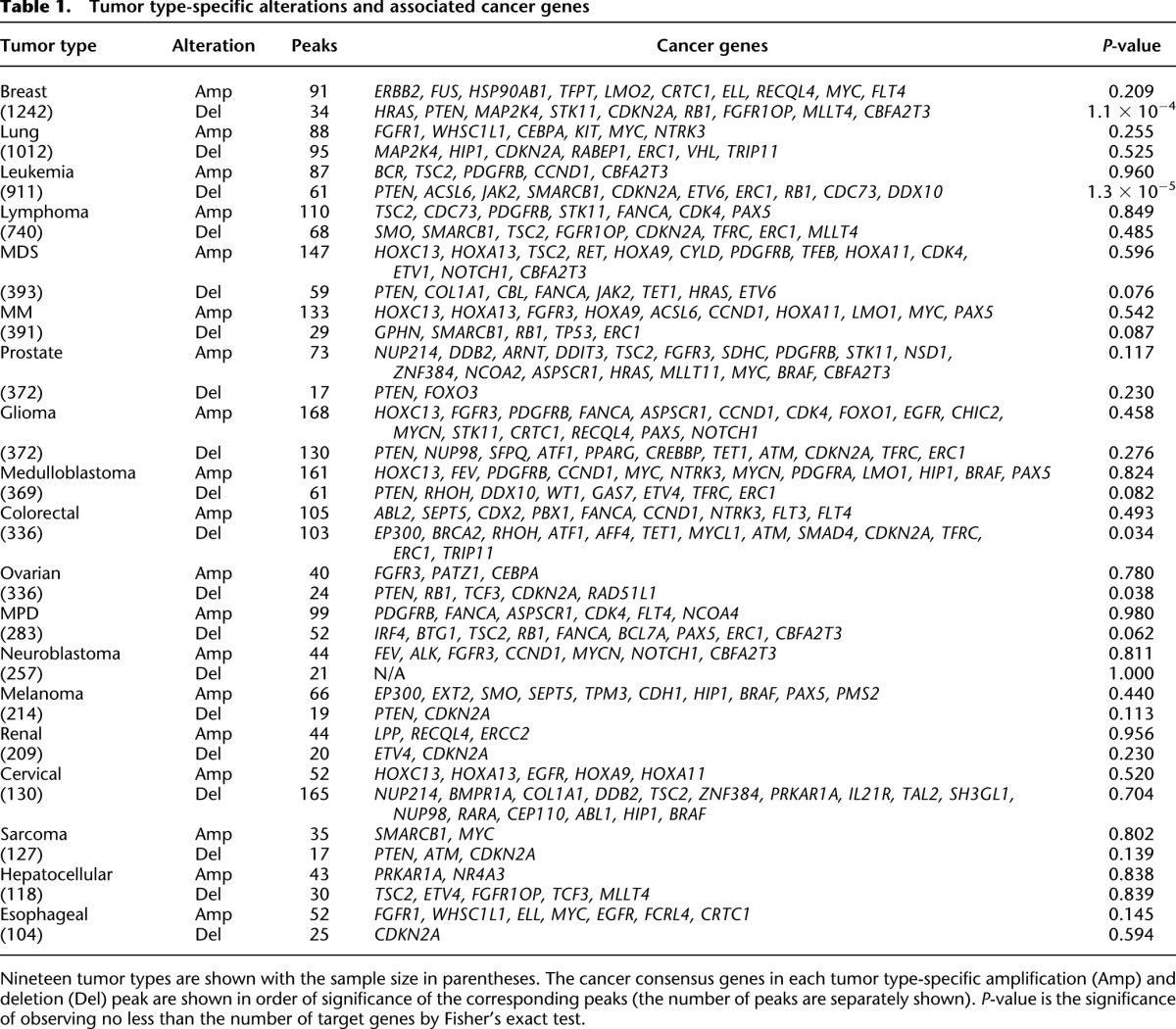
Our cancer genome database can be used to identify tumor type-specific alterations (“lineage-restricted” alterations) that may be important in a specific cellular context. We used the copy number profiles adjusted for platform effects to call tumor type-specific peaks in each of the 19 tumor types (those with >100 samples per tumor type). As we expected, the set of cancer genes observed in tumor type-specific alterations represents a mixture of cancer genes with broad tumorigenic potential such as CDKN2A, PTEN, ERC1, PDGFRB, MYC, and HOXC13 (for those in pan-lineage alterations) and CCND1, FGFR3, PAX5, and RB1 (not in pan-lineage alterations; all observed in five or more tumor types), as well as those with lineage-restricted functionality. Known cancer genes observed in tumor type-specific alterations with the significance of enrichment are listed in Table 1. Only four cancer type-specific alterations showed significant enrichment for known cancer genes (P < 0.05): deletion peaks of breast, leukemia, colorectal, and ovarian cancers. Although we observed some well-recognized pairs of cancer types and copy number altered genes such as breast cancer-ERBB2 (Slamon et al. 2001) and high-grade glioma-EGFR (The Cancer Genome Atlas Research Network 2008), it is likely that some genes whose oncogenic behavior is lineage-dependent are not on the list of known targets due to the incompleteness of this list. For example, although not listed as a cancer consensus gene, microphthalmia-associated transcription factor MITF (3p14.1), known as a master regulator of melanocyte development and a melanoma-specific oncogene (Garraway et al. 2005), was only included in the lineage-specific alterations of melanoma.
Table 1.
Tumor type-specific alterations and associated cancer genes
One of the 17 prostate cancer-specific deletions was observed within the intergenic region between ERG and TMPRSS2 loci. The average copy number profile of 372 prostate cancer genomes shows that the majority of genomic deletions involving the ERG locus in prostate cancer have ERG and TMPRSS2 loci at their 5′ and 3′ breakpoints, respectively (Supplemental Fig. S7). This deletion is known to give rise to the TMPRSS2-ERG gene fusion in prostate cancer (Kumar-Sinha et al. 2008), and the observed pattern is consistent with the “fusion breakpoint principle” of unbalanced translocation events (Wang et al. 2009). The co-occurrence of intragenic deletion breakpoints of ERG and TMPRSS2 loci was significant (n = 45 out of 372 prostate cancers; P = 9.1 × 10−25 by Fisher’s exact test).
To enhance biological interpretation of the tumor type-specific alterations and their associated genes, we measured the enrichment with respect to GO categories (Fig. 5). These maps show significant gene sets (based on tumor type-specific amplifications/deletions) and GO categories as nodes and significant overlaps between them as edges. Figure 5A lists the 31 gene sets showing significant enrichment with amplification peaks of nine tumor types. Genomic loci encoding kinase and signaling molecules are hotspots of amplification, especially in breast cancer, but are also common in other tumor types. Some enrichment can be explained by alterations on a few genomic loci affecting adjacent genes with similar functions (i.e., gene clusters). For example, gene clusters of chemokine ligands (CCL on 17q12) and fibroblast growth factors (FGF on 11q13) are responsible for the enrichment of “chemokine activity/immune” in colorectal cancers and “growth factor activity/signaling” in lung cancers, respectively (for details, see Supplemental Table S5). However, some enrichment such as “kinase activity” of breast cancer (20 genes on nine different chromosomal arms) and “MAP kinase activity” of renal cell carcinoma (MAPK9 on 5q35, MAPK11 and MAPK12 on 22q13, and MAPK15 on 8q24) are suggestive of functionally coordinated genomic alterations in the corresponding tumor types (Cooper et al. 2007). Tumor type-specific deletions of many tumor types (11 out of 19 tumor types examined) were linked to cell cycle and kinase-related gene sets, and they often share the enriched functions, especially for cell cycle- and adhesion-related functions (Fig. 5B). Some functional categories, such as genes encoding mitochondrial membrane components (“mitochondrial” in cervical cancer), DNA repair enzyme machineries (“transcription/DNA” in colorectal cancer) and metalloprotease (“enzyme/metabolism” in melanoma) highlight the relatively unique functionality driven by the genomic deletion in the corresponding tumor types.
Figure 5.

Functional association map of tumor type-specific alterations. (A) The genes belonging to tumor type-specific amplifications are shown as red nodes in a circular layout. (CRC) Colorectal cancer, (HCC) hepatocellular carcinoma, and (RCC) renal cell carcinoma. Significantly enriched GO categories are shown as nodes with different color schemes according to their functional annotations below. The size of each node is proportional to the number of genes in the gene set. (B) The association map of tumor type-specific deletions and their enriched GO categories is shown. GO categories associated with more than one tumor type and those with single connections are shown in and out of the cancer node circle, respectively. The full list of functional annotations and of individual GO categories and the genes responsible for the enrichment are available in Supplemental Table S5.
Large-scale survey of genomic hallmark for chromothripsis
Chromothripsis refers to a genomic instability-generating phenomenon in which tens to hundreds of chromosomal rearrangements occur in a “one-off” cellular event (Stephens et al. 2011). This has been observed so far in cancer cell lines and sarcomas (Stephens et al. 2011), multiple myeloma (Magrangeas et al. 2011), and colorectal cancers (Kloosterman et al. 2011). The proposed mechanism for chromothripsis is a massive fragmentation of one or a few chromosomes, followed by rejoining of the fragments (Stephens et al. 2011). During this rearrangement, the fragments can be lost or retained, which will appear as a series of copy number losses and gains, respectively, along the chromosome. To identify copy number changes associated with chromothripsis, we developed a statistical method that measures the extent of structured oscillations in segmented copy ratios and the deviation from the expected distribution of the segment sizes per chromosome (see Methods and Supplemental Fig. S8). Using this method, we performed a large-scale survey of chromothripsis in our compendium and observed that 124 samples (1.5% out of 8227 samples) may harbor genomic evidence of chromothripsis. We find that the tumors of epithelial origins have a wide range of frequencies, with prostate and lung cancers as the most and least frequent (5.6% and 1.1%, respectively) (Fig. 6A). The hematologic malignancies showed lower frequencies compared to those of epithelial origin, with multiple myeloma (1.9%) and leukemia (0.3%) as the most and least frequent in this group. Among the 19 tumor types examined (>100 supporting samples per tumor type), renal cell and hepatocellular carcinomas showed no evidence of chromothripsis. Initial reports estimated the prevalence of chromothripsis to be 2%–3% based on ∼700 cancer cell lines (Stephens et al. 2011) and 1.7% based on ∼700 primary multiple myeloma cases (Magrangeas et al. 2011). The incidence of chromothripsis estimated in our data set is largely consistent with these previous estimates. Our calculations show the prevalence to be 2.1% in cell lines (16 out of 748 cell line data) and 2.0% in multiple myeloma (eight out of 391 multiple myeloma cases).
Figure 6.

The prevalence of chromothripsis and examples of local chromothripsis involving known cancer genes. (A) The prevalence of chromothripsis measured across different tumor types is shown with 95% confidence intervals. The number of samples showing genomic evidence of chromothripsis is shown in parentheses with the total sample number associated with the tumor type. A dashed line indicates the average frequency across the entire data set. (B) Three neuroblastoma cases with evidence of chromothripsis on chromosome 2 are shown. Arrows indicate the MYCN locus, and insets show a more detailed pattern of copy number changes around the locus. (C) Three examples of local chromothripsis involving known cancer genes of EGFR, PTEN, and CCND1 loci are shown, with the log2 ratios at the cancer gene loci in parentheses.
We observed a number of examples in which localized chromothripsis events involved known cancer-related genes. For example, all of the three candidate neuroblastoma cases showing chromothripsis in chromosome 2 have amplifications in MYCN (Fig. 6B). Although the first case (GSM313805) showed a chromosome-wide chromothripsis that may be independent of the MYCN amplification, the other two cases (GSM333824 and GSM314024) have MYCN amplification that is embedded within the localized chromothripsis events in chromosome 2. In addition, we observed signatures of localized chromothripsis involving EGFR (chr7; prostate cancer), PTEN (chr10; prostate cancer), and CCND1 (chr11; esophageal cancer) (Fig. 6C).
Discussion
This study presents a meta-analysis of a compendium of copy number profiles for more than 8000 human cancer genomes. Two types of recurrent alterations, chromosomal arm-level and focal MCRs, were analyzed separately across the entire data set and within individual tumor types. Our results show that hierarchical clustering of arm-level alteration frequencies can largely segregate the tumor types according to their developmental lineages (e.g., hematologic, epithelial, and neuroepithelial clusters) (Fig. 2B). Although clustering of multiple tumor types based on developmental lineages was demonstrated using a large-scale transcriptome data set (Ramaswamy et al. 2001), our results are suggestive of a substantial embryological influence on the pattern of large chromosomal alterations that arises during development.
In a pairwise correlation analysis between arm-level alterations, we observed significant concordant pairs between small, gene-rich chromosomes. The higher contact probability between these chromosomes as measured in a long-range interaction map (Lieberman-Aiden et al. 2009) raises the intriguing possibility that these chromosomes share physical domains in the nucleus and that this physical proximity may be responsible for the observed copy number changes. Some of the discordant pairs (gain-loss) appear to be examples of functional synergism between known gene targets, e.g., gain of 7p and loss of 10q associated with potential EGFR gain and PTEN loss (marked by a dotted arrow in Fig. 3B; von Deimling et al. 1992). However, we also observed clusters of discordant pairs between gene-rich and gene-poor chromosomes. Although speculative, one hypothesis is that the gain of a driver alteration in a gene-rich chromosome is followed by dosage-compensating losses of gene-poor chromosomes, or vice versa. Restoration of copy number states by the original allele may be preferred (e.g., copy number-neutral loss of heterozygosity) (Makishima and Maciejewski 2011), but gene-poor chromosomes may be substituted in order to minimize the perturbation of essential genes. Biological significance of these concordant/discordant pairs of chromosomal changes needs to be investigated further, as previously explored for glioblastoma and hematologic malignancies (Bredel et al. 2009; Klijn et al. 2010).
MCRs identified from the entire data set (pan-lineage MCRs) showed a significant enrichment for known cancer-related genes as well as with genomic loci encoding kinases (amplification) and cell-cycle/apoptosis-related molecules (deletion). This suggests that a selective advantage during clonal evolution of tumor cells can be largely derived from common genomic alterations. We also observed that “microtubule” and “transporter” (in amplification MCRs) along with “cell junction” and “metabolism” (in deletion MCRs) are overrepresented GO categories in pan-lineage MCRs. The enrichment of these molecular functions remained significant after removal of recurrent alterations containing known targets (i.e., cancer consensus genes). Microtubules constituting the mitotic spindle in dividing cells have been major targets of chemotherapeutic agents such as taxane (Dumontet and Jordan 2010). The role of genomic dosage imbalances in microtubule-encoding genes is less well-established in tumorigenesis; however, point mutations have been shown to influence the drug susceptibility to microtubule-binding agents (Giannakakou et al. 1997; Kavallaris et al. 2001). Further investigation is needed to evaluate the relationship between copy number and the functional status of these genes, as well as their potential utility as biomarkers in monitoring the efficacy of microbutule binding agents. “Cell junction/adhesion” was another enriched function associated with universal deletion. These genomic alterations can lead to dysfunction in cell-to-cell integrity, which has been associated with tumorigenesis, especially in the context of invasion and metastasis (Martin and Jiang 2009; Escudero-Esparza et al. 2011).
Our analysis includes the copy number profiles from cancer cell lines that comprise ∼10% of the total (n = 748). In spite of the technical advantages of cell lines (e.g., free from normal cell contamination), cell lines may harbor passenger alterations acquired during in vitro culture. Consistent with this, we observed a higher number of alterations in cancer cell lines compared to primary samples (Supplemental Fig. S9). Although the overall fraction of cell lines is not substantial for most tumor types, some with a higher proportion of cell lines (e.g., 76% in melanoma) may require caution when interpreting the segmentation results.
Identification of driver alterations can be confounded by several genomic and technical factors. For example, we observed that five pan-lineage MCRs overlapped with known fragile sites (Bignell et al. 2010). These recurrent alterations may have arisen due to increased local mutational rates rather than as driver alterations with selective advantages. It is also important to filter germline alterations. Although we removed known CNVs from the HapMap population (Redon et al. 2006; McCarroll et al. 2008) in the peak-calling step of GISTIC, patient-specific germline alterations, especially those shared by many samples, may be mistakenly called as MCRs. To compare the performance of our strategy with that using matched normal controls, we collected Affymetrix SNP6.0 genotype data for two cancer types from The Cancer Genome Atlas (TCGA) for which paired blood DNA was also profiled: 377 glioblastoma multiforme (GBM) and 514 serous ovarian cancer pairs (OV) (The Cancer Genome Atlas Research Network 2008, 2011). Using the HapMap reference produced more segments (246 and 279 per sample for GBM and OV, respectively) compared to using matched normal controls (141 and 207 for GBM and OV, respectively). The comparison of genomic peaks showed only moderate concordance (67%/56% and 73%/60% of the amplification/deletion peaks have overlaps for GBM and OV data, respectively) (Supplemental Fig. S10). This indicates that, not surprisingly, not having the matched control may lead to spurious peaks that may correspond to germline CNVs and that filtering based on the HapMap population is incomplete. Although the ideal strategy is to use the normal reference DNA from the same individuals as controls, such samples may not be available and, if available, doubles the cost of experiments. A practical solution is to use a public database of germline alterations, as we have done in our analysis.
In the case of tumor type-specific alterations, we have recovered some of the well-established lineage-specific genes such as MITF in melanoma (Garraway et al. 2005). Current understanding of the tumor type-specific or context-dependent oncogenic roles of known cancer genes is limited. Nevertheless, we expect that the tumor type-specific alterations and their associated functional categories observed in our study can serve as a resource in prioritizing candidates for potential biomarkers. For example, our analysis revealed that colorectal cancers show frequent deletions of multiple DNA repair enzyme-associated loci such as REV1 (2q11), SUMO1 (2q33), RAD50 (5q31), GTF2H1 (11p15), RAD52 (12p13), BRCA2 (13q13), ATXN3 (14q32), RAD51C (17q22), and XRCC6 (22q13) (Supplemental Table S5). DNA repair enzymes have been suggested as potential selective targets in cancer treatments, and the activity of these enzymes has been frequently associated with drug resistance to conventional platinum-based chemotherapy (Kelley and Fishel 2008). Although the relationship between the genomic imbalances and the functional status of these genes has not been well-described, our results suggest that the colorectal cancers have frequent deletions involving these multiple loci with functional consequences.
A well-known translocation event, TMPRSS2-ERG gene fusion in prostate cancer (known to occur in about half of the primary cases) (Kumar-Sinha et al. 2008), was recognized in our data set in that TMPRSS2 and ERG were associated with prostate cancer-specific deletions in a highly concordant manner (P = 9.1 × 10−25) (Supplemental Fig. S7). It has been proposed that the chromosomal translocation events, especially those accompanying genomic imbalances, have unique copy number signatures (Wang et al. 2009), and some of them can be identified from copy number profiles (Ritz et al. 2011). These results suggest that a large-scale copy number database may serve as a potential source in search of chromosomal copy number changes associated with translocation events.
Our large database enabled a survey of the low-prevalence genomic event called chromothripsis, and our estimates of the incidence rates are largely consistent with the current literature (Magrangeas et al. 2011; Stephens et al. 2011), despite the variable power for detection among platforms (Supplemental Fig. S11). It was reported (Stephens et al. 2011) that the frequency of chromothripsis could be exceptionally high in some specific tumor types (e.g., ∼33% for osteosarcoma). However, in our data set, the evidence of chromothripsis was present in only one of 127 sarcoma genomes (differentiated liposarcoma; GSM486220) and in none of the seven osteosarcoma cases. Therefore, additional samples are needed to obtain confident estimates for some tumor types.
We observed examples of local chromothripsis involving known cancer genes such as MYCN, EGFR, PTEN, and CCND1, suggesting that chromothripsis falls within the general spectrum of DNA alterations affecting cancer-relevant genes. The more accurate method for examining chromothripsis is using paired-end information from whole-genome sequencing to reconstruct the event, but the cost for high-coverage sequencing is still high. For now, our statistical method using the large number of accumulated array-based copy number profiles is valuable in estimating the prevalence and unique nature of chromothripsis.
A unique pattern of copy number changes has been previously reported (Hicks et al. 2006; Russnes et al. 2010), but it should be noted that these observations do not necessarily relate to chromothripsis. For example, Hicks et al. reported a phenomenon called “firestorm” to describe chromosomes with multiple closely spaced amplicons (Hicks et al. 2006), estimating that they occur in ∼25% of the cases. In our data, we find that the fast oscillation between copy number states is not restricted to amplicons, with the confident cases involving only deletions (Fig. 6C). The frequency of the chromothripsis events we detect based on this is much rarer (1%–5%). Russnes et al. introduced an index called CAAI (complex arm aberration index) for copy number profiles (Russnes et al. 2010) to measure the local complexity of CNV regions. However, this does not measure an oscillating pattern indicative of chromothripsis and their examples of chromosomes with high CAAI had little evidence of an alternating pattern in the copy number.
While we have assembled a large compendium of array-CGH profiles, not all subtypes are present for each tumor type, and data for some tumors are gathered from only one or two studies. Thus, the interpretation of some tumor type-specific results should be tempered until verified using additional data sets. Importantly, although researchers are encouraged to make their data and metadata available in public databases once published, the currently available data sets represent only a fraction of the profiles generated by the community. An increase in the data deposition rate into public databases would facilitate meta-analyses such as this one as well as reanalyses for confirming specific hypotheses, and this would be critical for making efficient use of limited resources for cancer research. Another caveat related to our study is that this analysis only focused on copy number profiles, whereas known oncogenes or tumor suppressor genes can be activated or inhibited by several alternative mechanisms (Chin et al. 2011). Thus, integrative, multidimensional analysis using different genomic data types will be critical. The potential of such analysis was recently demonstrated, for example, in identifying novel cancer genes (Akavia et al. 2010) and cancer classification (Kim et al. 2011). The use of additional data sources (e.g., mRNA and microRNA expression, somatic mutation, and promoter methylation), especially from large-scale cancer genome projects such as the TCGA consortium, will enable these integrative, multidimensional analyses (The Cancer Genome Atlas Research Network 2008, 2011).
Methods
Compendium of human cancer genome copy number profiles
We collected human cancer array-CGH data sets from a public microarray database (GEO; http://www.ncbi.nlm.nih.gov/geo/) (Barrett et al. 2009). We limited our analysis to studies using high- resolution, array-CGH platforms containing more than 100K oligonucleotide probes from two commercial vendors (Agilent and Affymetrix). For the Agilent platform, we used the GEO platform ID of GPL4091 and GPL9128 (both 244K probes). For the Affymetrix platform, we used GPL2004/2005 (100K probes), GPL3718/3720 (500K probes), and GPL6801 (Affymetrix SNP6.0; 1.8 million probes). For the 100K Affymetrix platform, we only used the paired data (i.e., a sample is profiled by both GPL2004/50K-HindIII and GPL2005/50K-XbaI to achieve 100K resolution). For the 500K platforms (GPL3718/250K-NspI and GPL3720/250K-StyI), we included samples genotyped either by GPL3718 or GPL3720 and designated them as “Affymetrix 250K.” For five platforms (Agilent 244K and Affymetrix 100K, 250K, 500K, and SNP6.0), we collected a total of 107 GEO studies. The GEO accession of 107 studies and related information is available in Supplemental Table S1. In this study, we only collected the genomic profiles corresponding to tumor genomes, removing control profiles. We also searched for duplicate samples (most of which are dye-swap cases) or samples that were included in multiple GEO data sets. Although we have done our best, these are sometimes difficult to identify, and it is still possible that our data set contains biological or technical duplicates across different data sets. Detailed information for the final 8227 tumor samples is available in Supplemental Table S2.
Data processing and segmentation
For Agilent platforms, we downloaded the probe-level log2 ratio profiles of individual samples from the GEO database. For the Affymetrix platform, we used the CRMA algorithm (Bengtsson et al. 2009) for probe summarization and normalization to obtain probe-level intensity values from individual CEL files. We also processed the normal HapMap population CEL files (available in http://www.affymetrix.com). The average intensity values of the HapMap population were calculated for individual probes and used as a reference to calculate tumor/reference log2 ratios. We ensured that the genomic coordinates used in this study are hg18/build 36 using UCSC Genome Browser liftover tools (Fujita et al. 2011). Segmentation was performed using the Circular Binary Segmentation (CBS) algorithm available as an R package (Olshen et al. 2004).
The segment values were median-centered by extracting the median of autosomal segment values per sample. For each study, we calculated MAD (median absolute deviation) for the 50th percentile of autosomal segments whose absolute log2 ratios were close to zero and rescaled the segment values of the samples in the corresponding study. The resulting segment values were further rescaled so that the standard deviation of autosomal segment values of the entire data set was equal to one. We verified that our preprocessing step of median-centering does not impact the analysis results by comparing the results obtained with and without the matched controls samples in the GBM and OV TCGA data sets (data not shown). However, for cancer types with a substantial fraction of the genome altered, it is conceivable that median-centering can cause a slight bias in segmentation calls. The processed segmentation profiles and associated analysis files are available at http://compbio.med.harvard.edu/metaCGH/.
Analysis of chromosomal arm-level alterations
We defined alterations as segments with a predefined threshold for the rescaled segment values (>+0.2 and <−0.2 for copy number gain and loss, respectively). In this study, we defined chromosomal arm-level alterations as a single alteration or an aggregate of alterations that exceeds half of the size of the corresponding chromosomal arm. To take into account the background arm-level alteration frequency, we converted the arm-level alteration frequency into size-adjusted Z scores as described previously (Beroukhim et al. 2010). The expected frequency of gain and loss was determined by linear regression, and the Z score was calculated using the normal approximation to the binomial distribution. For hierarchical clustering, tumor type-specific arm-level alteration frequencies were calculated as arm-level gain minus arm-level loss frequencies for 19 tumor types supported by more than 100 samples per tumor type. Hierarchical clustering was performed using 1 – Pearson correlation as the distance with average linkage. In concordance analysis, four possible co-occurrence scenarios between arm-level alterations were separated into concordant (gain/gain and loss/loss) and discordant pairs (gain/loss and loss/gain). The significance of gain/gain co-occurrence between two chromosomal arms of A and B was calculated using Fisher’s exact test:
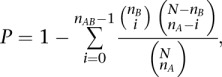 |
where nAB, nA, nB, and N represent the number of samples showing gain for both chromosomal arms of A and B (nAB), chromosomal arm A (nA) and B (nB), and the total number of samples (N), respectively. To account for multiple test adjustment, we permuted the calls of arm-level alterations in each sample across the entire data set. For each permuted data set, we calculated the significance of co-occurrence for all possible chromosomal arm pairs to find the minimal P-value. This permutation was repeated 10,000 times separately for gain-gain, loss-loss, and gain-loss pairs. The empirical P-value for each arm-level alteration pair was computed as the proportion of permutations whose minimum P-value is smaller than the observed P-value of the corresponding alteration pair.
Pan-lineage alterations and functional analysis
To identify MCR, we used the peak-calling algorithm of GISTIC (Beroukhim et al. 2007). The algorithm calculates the probe-level sum of log2 ratios above or below a given threshold (amplification and deletion, separately) across the samples. Then, the algorithm determines significantly altered regions as sets of consecutive probes with a predefined significance threshold (Q < 0.25). From significantly altered regions, it identifies the highest scoring peak corresponding to the MCR. Because the peak or MCR can be displaced from true targets due to neighboring passenger events or noise (Beroukhim et al. 2007), we allowed for 100 kb of confidence intervals at both flanking regions of the identified peak. To suppress the known germline alterations of the HapMap population, we filtered probes corresponding to 2333 autosomal HapMap CNVs obtained from two previous studies using Affymetrix 500K Early Access and SNP6.0 platforms (Redon et al. 2006; McCarroll et al. 2008). For known cancer-related genes, we used 264 autosomal cancer consensus genes available in the literature (Futreal et al. 2004). The significance of enrichment of 165 pan-lineage MCRs with known cancer-related genes was calculated by Fisher’s exact test. We also selected 421 and 156 genes associated with pan-lineage amplification and deletion MCRs, respectively, and measured the enrichment with GO functional categories (http://www.broadinstitute.org/gsea/msigdb/index.jsp; c5 GO categories) (Subramanian et al. 2005). The significance of enrichment was calculated by Fisher’s exact test and corrected for multiple hypothesis testing using the Benjamini-Hochberg false discovery method. Thirty-seven common fragile regions were obtained elsewhere (Bignell et al. 2010). We also downloaded a list of hg18-compatible CNVs from a public database (variation.hg18.v10.nov.2010; http://projects.tcag.ca/variation/). A total of 57,706 CNVs were collected from 17 studies (>1000 CNVs per study), and regional CNV density was calculated for each pan-lineage MCR.
Removal of platform-specific biases
We employed a linear mixed effect model to remove the potential platform biases. A natural idea is to model the intensity value of each probe in the platforms to adjust for the local bias. However, as probe sets are different across platforms, a probe in one platform may not exist in another platform. We will thus have many missing values for these platform-specific probes. A solution to this “missing-value problem” is to use intensity values of the nearby probes, but this will require determining the size of the neighborhood of the probe, and this choice may have an important influence on the model fitting and bias removal. In this paper, we choose to perform the bias removal based on the segmentation data. Suppose that 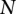 samples were profiled on
samples were profiled on 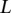 platforms, and each sample has been processed with a segmentation algorithm. For clarity, we only consider segmentations for one chromosome in the discussion. The segmentation of one sample will correspond to one set of breakpoints. Collect the breakpoints of all samples and denote the breakpoints as
platforms, and each sample has been processed with a segmentation algorithm. For clarity, we only consider segmentations for one chromosome in the discussion. The segmentation of one sample will correspond to one set of breakpoints. Collect the breakpoints of all samples and denote the breakpoints as 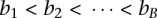 . Given the
. Given the 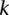 th interval
th interval 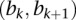 (
( ) and the
) and the 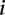 th sample profiled on the
th sample profiled on the 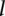 th platform (call this sample
th platform (call this sample 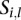 ;
; 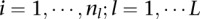 ;
;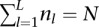 ), there will be a unique segment from the segmentation of the sample
), there will be a unique segment from the segmentation of the sample 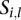 overlapping with the interval
overlapping with the interval 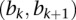 . Denote the “segmean” value (i.e., the mean of probe intensity values in the corresponding segment) of this segment as
. Denote the “segmean” value (i.e., the mean of probe intensity values in the corresponding segment) of this segment as 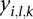 . We fit a linear mixed effect model with
. We fit a linear mixed effect model with 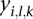 as the response variable and the platform as a random effect. Since the tumor type may be an important factor influencing the values of
as the response variable and the platform as a random effect. Since the tumor type may be an important factor influencing the values of 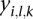 , we also include the tumor type as a predictor (fixed effect) in the model. Assume that there are
, we also include the tumor type as a predictor (fixed effect) in the model. Assume that there are 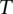 types of tumors. Given a sample
types of tumors. Given a sample 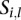 , let
, let 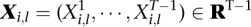 be the vector such that
be the vector such that 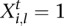 if the sample
if the sample 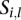 belongs to the
belongs to the  th tumor type (
th tumor type ( ) and 0 for all the other
) and 0 for all the other  . Then, we have the following linear mixed effect model:
. Then, we have the following linear mixed effect model:
where  corresponds to the random platform effect,
corresponds to the random platform effect, 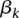 is the fixed tumor type effect, and
is the fixed tumor type effect, and 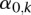 is the intercept coefficient in the linear model. For each
is the intercept coefficient in the linear model. For each 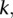 we then can use the restricted maximum likelihood estimation (RMLE) to estimate the parameters in the above model. In particular, we can get an estimate
we then can use the restricted maximum likelihood estimation (RMLE) to estimate the parameters in the above model. In particular, we can get an estimate 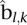 of
of 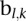 . From this estimate, we can get the platform effect-corrected value
. From this estimate, we can get the platform effect-corrected value 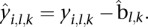 Given a segment
Given a segment 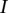 in the segmentation of the sample
in the segmentation of the sample 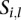 , suppose that it overlaps with
, suppose that it overlaps with 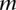 > intervals like
> intervals like 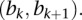 Assume that these intervals are the
Assume that these intervals are the  th intervals and that
th intervals and that  is the corresponding overlapping length with the segment
is the corresponding overlapping length with the segment 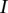 . We use
. We use 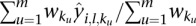 as the platform effect-corrected intensity value for the segment
as the platform effect-corrected intensity value for the segment 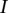 of the sample
of the sample 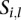 .
.
Lineage- or tumor type-specific alterations and functional association map
GISTIC-based peak calling was performed for individual tumor type-specific subsets (19 tumor types supported by more than 100 samples). The extent of enrichment with cancer consensus genes and GO functional categories was calculated using Fisher’s exact test. For the functional association map, we collected the GO categories that showed substantial enrichment (adjusted Fisher’s exact test P < 0.3) with any of the tumor type-specific alterations. In the network, nodes are sets of genes belonging to tumor type-specific alterations or selected GO categories. The edges are significant gene overlaps between them. An association map was separately generated for tumor type-specific amplifications deletions. We used Cytoscape software for network visualization (Shannon et al. 2003).
Chromothripsis
To identify the genomic signatures representing chromothripsis, we focused on chromosomes with at least 10 alterations. Furthermore, the breakpoints in chromothripsis should occur randomly (Stephens et al. 2011). Thus, the sizes of neighboring segments in a chrompthripsis region should be roughly the same or at least at the same order of magnitude. If the sizes of neighboring segments are very different from each other, we would expect that those are not from chromothripsis. Therefore, we designed a score to measure the “extremeness” of the neighboring segments in a copy number profile. Suppose that a chromosome has  segments and their corresponding sizes are
segments and their corresponding sizes are 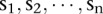 . If the sizes of the segments
. If the sizes of the segments 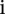 and
and 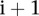 are very different, we would have that
are very different, we would have that 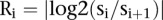 is large. The median
is large. The median 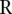 of the
of the 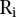 ’s would be a good measure of the extremeness of the neighboring segments. To account for the number of segments in a copy number profile, we normalized the score
’s would be a good measure of the extremeness of the neighboring segments. To account for the number of segments in a copy number profile, we normalized the score 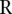 by its expected value
by its expected value 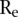 under the hypothesis that the breakpoints are randomly distributed in the chromosome, i.e., we use
under the hypothesis that the breakpoints are randomly distributed in the chromosome, i.e., we use 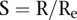 as the score. We used
as the score. We used 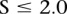 to filter out the profiles that are unlikely to be involved with chromothripsis. If one chromosome of a sample involves chromothripsis, we would expect to see an alternating pattern of the copy ratio (log2 copy ratio) estimates. If we view each segment in the copy number profile as a point and plot the log2 copy ratio estimates, the alternating copy number states will appear as many peaks and valleys that correspond to copy number gains and losses, respectively. The peak and valley count can then be used as a measurement of the alternating pattern. Given a copy number profile of a chromosome, if chromothripsis occurs, its peak and valley count would be significantly higher than expected. We measure the significance with a permutation test. In detail, we randomly permute the order of the segments and count the peaks and valleys in the permuted profile. The permutation is performed 100,000 times, and a P-value is assigned as the proportion of the permutations whose peak and valley counts are greater than or equal to the observed peak and valley count. Because the peak and valley counts as well as the significant calls of chromothripsis will be dependent on the segmentation, we also segmented the copy number profiles using the GLAD algorithm (Hupe et al. 2004). Confident calls of chromothripsis were identified as chromosomes showing significant (FDR < 0.1) fluctuation in both of the CBS and GLAD segmentation profiles. We observed a total of 209 chromothripsis events in 124 tumor samples. The chromosomal profiles of 209 significant events are available at http://compbio.med.harvard.edu/metaCGH/.
to filter out the profiles that are unlikely to be involved with chromothripsis. If one chromosome of a sample involves chromothripsis, we would expect to see an alternating pattern of the copy ratio (log2 copy ratio) estimates. If we view each segment in the copy number profile as a point and plot the log2 copy ratio estimates, the alternating copy number states will appear as many peaks and valleys that correspond to copy number gains and losses, respectively. The peak and valley count can then be used as a measurement of the alternating pattern. Given a copy number profile of a chromosome, if chromothripsis occurs, its peak and valley count would be significantly higher than expected. We measure the significance with a permutation test. In detail, we randomly permute the order of the segments and count the peaks and valleys in the permuted profile. The permutation is performed 100,000 times, and a P-value is assigned as the proportion of the permutations whose peak and valley counts are greater than or equal to the observed peak and valley count. Because the peak and valley counts as well as the significant calls of chromothripsis will be dependent on the segmentation, we also segmented the copy number profiles using the GLAD algorithm (Hupe et al. 2004). Confident calls of chromothripsis were identified as chromosomes showing significant (FDR < 0.1) fluctuation in both of the CBS and GLAD segmentation profiles. We observed a total of 209 chromothripsis events in 124 tumor samples. The chromosomal profiles of 209 significant events are available at http://compbio.med.harvard.edu/metaCGH/.
Acknowledgments
This work was supported by grants from the National Institutes of Health to P.J.P. (R01GM082798, U24CA144025) and by grants from the National Institutes of Health to M.D.J. (NIH Director’s New Innovator Award [DP2OD002319]; NINDS NS062219-01).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.140301.112.
References
- Akavia UD, Litvin O, Kim J, Sanchez-Garcia F, Kotliar D, Causton HC, Pochanard P, Mozes E, Garraway LA, Pe’er D 2010. An integrated approach to uncover drivers of cancer. Cell 143: 1005–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertson DG, Pinkel D 2003. Genomic microarrays in human genetic disease and cancer. Hum Mol Genet 12: R145–R152 [DOI] [PubMed] [Google Scholar]
- Albertson DG, Collins C, McCormick F, Gray JW 2003. Chromosome aberrations in solid tumors. Nat Genet 34: 369–376 [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. 2000. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, et al. 2009. NCBI GEO: Archive for high-throughput functional genomic data. Nucleic Acids Res 37: D885–D890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudis M, Cleary ML 2001. Progenetix.net: An online repository for molecular cytogenetic aberration data. Bioinformatics 17: 1228–1229 [DOI] [PubMed] [Google Scholar]
- Bengtsson H, Wirapati P, Speed TP 2009. A single-array preprocessing method for estimating full-resolution raw copy numbers from all Affymetrix genotyping arrays including GenomeWideSNP 5 & 6. Bioinformatics 25: 2149–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, Vivanco I, Lee JC, Huang JH, Alexander S, et al. 2007. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc Natl Acad Sci 104: 20007–20012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. 2010. The landscape of somatic copy-number alteration across human cancers. Nature 463: 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, et al. 2010. Signatures of mutation and selection in the cancer genome. Nature 463: 893–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredel M, Scholtens DM, Harsh GR, Bredel C, Chandler JP, Renfrow JJ, Yadav AK, Vogel H, Scheck AC, Tibshirani R, et al. 2009. A network model of a cooperative genetic landscape in brain tumors. JAMA 302: 261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network 2008. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network 2011. Integrated genomic analyses of ovarian carcinoma. Nature 474: 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Zhou M, Wang X, Meyer CA, Zhang Y, Chen Z, Li C, Liu XS 2010. CaSNP: A database for interrogating copy number alterations of cancer genome from SNP array data. Nucleic Acids Res 39: D968–D974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Gray JW 2008. Translating insights from the cancer genome into clinical practice. Nature 452: 553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Hahn WC, Getz G, Meyerson M 2011. Making sense of cancer genomic data. Genes Dev 25: 534–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Nickerson DA, Eichler EE 2007. Mutational and selective effects on copy-number variants in the human genome. Nat Genet 39: S22–S29 [DOI] [PubMed] [Google Scholar]
- Dumontet C, Jordan MA 2010. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat Rev Drug Discov 9: 790–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudero-Esparza A, Jiang WG, Martin TA 2011. The Claudin family and its role in cancer and metastasis. Front Biosci 16: 1069–1083 [DOI] [PubMed] [Google Scholar]
- Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, et al. 2011. The UCSC Genome Browser database: Update 2011. Nucleic Acids Res 39: D876–D882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR 2004. A census of human cancer genes. Nat Rev Cancer 4: 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, et al. 2005. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436: 117–122 [DOI] [PubMed] [Google Scholar]
- Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JT, Fojo T, Poruchynsky MS 1997. Paclitaxel-resistant human ovarian cancer cells have mutant β-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem 272: 17118–17125 [DOI] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. 2007. Patterns of somatic mutation in human cancer genomes. Nature 446: 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA 2011. Hallmarks of cancer: The next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Hicks J, Krasnitz A, Lakshmi B, Navin NE, Riggs M, Leibu E, Esposito D, Alexander J, Troge J, Grubor V, et al. 2006. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res 16: 1465–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hupe P, Stransky N, Thiery JP, Radvanyi F, Barillot E 2004. Analysis of array CGH data: From signal ratio to gain and loss of DNA regions. Bioinformatics 20: 3413–3422 [DOI] [PubMed] [Google Scholar]
- Kavallaris M, Tait AS, Walsh BJ, He L, Horwitz SB, Norris MD, Haber M 2001. Multiple microtubule alterations are associated with Vinca alkaloid resistance in human leukemia cells. Cancer Res 61: 5803–5809 [PubMed] [Google Scholar]
- Kelley MR, Fishel ML 2008. DNA repair proteins as molecular targets for cancer therapeutics. Anticancer Agents Med Chem 8: 417–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TM, Huang W, Park R, Park PJ, Johnson MD 2011. A developmental taxonomy of glioblastoma defined and maintained by microRNAs. Cancer Res 71: 3387–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klijn C, Bot J, Adams DJ, Reinders M, Wessels L, Jonkers J 2010. Identification of networks of co-occurring, tumor-related DNA copy number changes using a genome-wide scoring approach. PLoS Comput Biol 6: e1000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Hoogstraat M, Paling O, Tavakoli-Yaraki M, Renkens I, Vermaat JS, van Roosmalen MJ, van Lieshout S, Nijman IJ, Roessingh W, et al. 2011. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol 12: R103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar-Sinha C, Tomlins SA, Chinnaiyan AM 2008. Recurrent gene fusions in prostate cancer. Nat Rev Cancer 8: 497–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrangeas F, Avet-Loiseau H, Munshi NC, Minvielle S 2011. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood 118: 675–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima H, Maciejewski JP 2011. Pathogenesis and consequences of uniparental disomy in cancer. Clin Cancer Res 17: 3913–3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TA, Jiang WG 2009. Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta 1788: 872–891 [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, et al. 2008. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet 40: 1166–1174 [DOI] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G 2011. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12: R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson M, Gabriel S, Getz G 2010. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet 11: 685–696 [DOI] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F 2007. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 7: 233–245 [DOI] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M 2004. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5: 557–572 [DOI] [PubMed] [Google Scholar]
- Pinkel D, Albertson DG 2005. Array comparative genomic hybridization and its applications in cancer. Nat Genet (Suppl) 37: S11–S17 [DOI] [PubMed] [Google Scholar]
- Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, et al. 1998. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 20: 207–211 [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Tamayo P, Rifkin R, Mukherjee S, Yeang CH, Angelo M, Ladd C, Reich M, Latulippe E, Mesirov JP, et al. 2001. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci 98: 15149–15154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. 2006. Global variation in copy number in the human genome. Nature 444: 444–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz A, Paris PL, Ittmann MM, Collins C, Raphael BJ 2011. Detection of recurrent rearrangement breakpoints from copy number data. BMC Bioinformatics 12: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russnes HG, Vollan HK, Lingjaerde OC, Krasnitz A, Lundin P, Naume B, Sørlie T, Borgen E, Rye IH, Langerød A, et al. 2010. Genomic architecture characterizes tumor progression paths and fate in breast cancer patients. Sci Transl Med 2: 38ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS 2010. A census of amplified and overexpressed human cancer genes. Nat Rev Cancer 10: 59–64 [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T 2003. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, et al. 2001. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344: 783–792 [DOI] [PubMed] [Google Scholar]
- Snijders AM, Nowak N, Segraves R, Blackwood S, Brown N, Conroy J, Hamilton G, Hindle AK, Huey B, Kimura K, et al. 2001. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat Genet 29: 263–264 [DOI] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. 2011. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144: 27–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Deimling A, Louis DN, von Ammon K, Petersen I, Hoell T, Chung RY, Martuza RL, Schoenfeld DA, Yasargil MG, Wiestler OD, et al. 1992. Association of epidermal growth factor receptor gene amplification with loss of chromosome 10 in human glioblastoma multiforme. J Neurosurg 77: 295–301 [DOI] [PubMed] [Google Scholar]
- Wang XS, Prensner JR, Chen G, Cao Q, Han B, Dhanasekaran SM, Ponnala R, Cao X, Varambally S, Thomas DG, et al. 2009. An integrative approach to reveal driver gene fusions from paired-end sequencing data in cancer. Nat Biotechnol 27: 1005–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]