

Abstract
We have identified variants present in high-coverage complete sequences of 36 diverse human Y chromosomes from Africa, Europe, South Asia, East Asia, and the Americas, representing eight major haplogroups. After restricting our analysis to 8.97 Mb of the unique male-specific Y sequence, we identified 6662 high-confidence variants, including single-nucleotide polymorphisms (SNPs), multi-nucleotide polymorphisms (MNPs), and indels. We constructed phylogenetic trees using these variants, or subsets of them, and recapitulated the known structure of the tree. Assuming a male mutation rate of 1 × 10−9 per base pair per year, the time depth of the tree (haplogroups A3-R) was ∼101,000–115,000 yr, and the lineages found outside Africa dated to 57,000–74,000 yr, both as expected. In addition, we dated a striking Paleolithic male lineage expansion to 41,000–52,000 yr ago and the node representing the major European Y lineage, R1b, to 4000–13,000 yr ago, supporting a Neolithic origin for these modern European Y chromosomes. In all, we provide a nearly 10-fold increase in the number of Y markers with phylogenetic information, and novel historical insights derived from placing them on a calibrated phylogenetic tree.
The human Y chromosome offers a unique perspective on human genetics because of its male-line inheritance and the very high resolution of its haplotype tree (Jobling and Tyler-Smith 2003). Insights it has provided include a recent African origin for paternal lineages (Hammer 1995; Cruciani et al. 2011), a single expansion of modern humans out of Africa (Underhill et al. 2000), evidence for Paleolithic movement back to Africa (Scozzari et al. 1999), and many examples of tracing events within historical times such as the Mongol (Zerjal et al. 2003) and Phoenician legacies (Zalloua et al. 2008) or the descendants of Thomas Jefferson (Foster et al. 1998). These conclusions have been reached after genotyping small numbers of known markers, or limited resequencing. Despite the valuable insights that have been obtained, there have been other areas of Y-chromosomal study where uncertainty or debate have persisted for decades, such as the understanding of the causal mutations underlying Y-linked spermatogenic failure (Tyler-Smith and Krausz 2009), the role of Y-chromosomal variation in phenotypes such as coronary artery disease (Charchar et al. 2012), or the origins of European male lineages. In the last case, opinions have included a Paleolithic origin for the major lineage (Semino et al. 2000), a Neolithic origin for the equivalent lineage (Balaresque et al. 2010), and the view that reliable age estimates for this lineage are currently impossible (Busby et al. 2011).
Studies of mitochondrial DNA, often regarded as a female-line counterpart of the Y, have benefited substantially from developments in data acquisition, moving from early restriction fragment length polymorphism (RFLP) typing (Cann et al. 1987) and control-region resequencing (Vigilant et al. 1991) to large-scale complete sequencing of chosen lineages or populations (Behar et al. 2008; Gunnarsdottir et al. 2011). It seemed likely that complete sequencing of Y chromosomes might lead to similar or even greater benefits. Although its 3000-fold greater length has made this more difficult, current technologies allow an enormous increase in sequence data collection. This is, however, accompanied by a cost: Short reads are generated, and these cannot be mapped accurately if they are derived from repetitive regions. Since the Y is richer in repeated sequences than any other chromosome, this is particularly problematic for sequencing the Y but can be overcome by concentrating on unique regions. This approach has allowed the sequencing of a pair of related Y chromosomes with sufficient accuracy to provide a measurement of the mutation rate (Xue et al. 2009), and lower coverage sequencing of 77 Y chromosomes to discover 2870 high-confidence Y-SNPs, 74% new (The 1000 Genomes Project Consortium 2010). Although low-coverage sequencing is an efficient way to discover variants, the interpretation of the resulting data is complicated by the incomplete ascertainment of variants in any single sample, and thus reliable information about some features, such as the time depth of the phylogeny, is difficult to extract. Sequencing technologies have now developed further and allow moderately sized samples to be sequenced at high coverage from either the complete genome or targeted regions (Drmanac et al. 2010; Hu et al. 2012), which should permit more complete ascertainment of variants and simplify interpretation.
Here, we have analyzed a data set of 36 Y chromosome sequences in order to explore how effectively complete sequence data from the Y can be used to construct and calibrate a phylogeny, and the insights that may result from such an analysis.
Results
High-coverage sequences from 36 males were used, 35 released by Complete Genomics and an additional sequence from a haplogroup A individual (the most basal haplogroup branch) generated for this study. In all, there were nine males from Africa, 15 from Europe (including a three-generation family containing eight males), three from South Asia, two from East Asia, and seven from the Americas. Single-nucleotide polymorphism (SNP) genotyping data available from the HapMap3 Project (Altshuler et al. 2010) revealed that haplogroups A, D, E, G, I, N, Q, and R were represented, with many subdivisions of some of these, particularly E and R. Thus, despite the small number of individuals, there was good geographical representation of global populations and of the haplogroup tree. After QC and validation, we extracted 6662 high-confidence variants (i.e., sites that differ from the Y chromosome reference sequence), including both SNPs and indels, from 8.97 Mb of unique Y sequence (Supplemental 77s S1, S2). The variants were distributed evenly along the target regions of the chromosome (Fig. 1) and increase the number of high-quality variants with phylogenetic information by almost 10-fold.
Figure 1.

Regions of the Y chromosome analyzed and distributions of variants discovered. (A) Y chromosome. (B) Location of the centromere. (C) Scale (bp) used in sections D through L. (D) Y chromosome unique regions. (Yellow bar) Segment of the reference sequence derived from haplogroup G (14,328,588–15,370,586); (blue bar) Haplogroup R1b regions. (E) Landmark sequence-tagged sites (STSs) used to define unique regions. (F) Bases in the chimpanzee reference sequence that can be aligned with the human reference (gray), or gaps in the alignment (orange). (G) Published variants lying within the unique regions. (H) Published variants variable within the 36 individuals. (I) Region sequenced at high coverage in the haplogroup A individual. (J) Depth of coverage of the region in track I. (K) All variable sites discovered in this study.
We assigned ancestral states to 6271 of the variants and then constructed a rooted parsimony-based phylogenetic tree containing all 6662 variants (tree 1) (Supplemental Fig. 1) and additional trees containing subsets of these, consisting of SNPs only (tree 2) (Supplemental Fig. 2) or SNPs with ancestral state information, no recurrent mutations, and high coverage in all individuals (tree 3) (Fig. 2). The branch leading to the haplogroup A individual was shorter than any other, most likely reflecting the low-coverage sequencing of parts of this chromosome and consequent under-calling of variants; this effect was largely eliminated in tree 3, where only high-coverage regions were used (Fig. 2). The topology of all three trees was very similar, differing only by the lack of resolution of the haplogroup G and I branch order in the most highly filtered tree, because the relevant SNPs lay outside the region used. The structure recapitulated the known phylogeny of the Y chromosome (Karafet et al. 2008) and identified new splits within the E1b1a8a and I1* branches. In addition, the new markers allowed all unrelated chromosomes to be distinguished.
Figure 2.

Rooted maximum parsimony phylogenetic tree of 36 Y chromosomes. Sample codes and haplogroups are shown on the right, along with a number to distinguish different individuals who share the same haplogroup or pedigree. The length of the branch is proportional to the number of mutations, which is given along each branch.
Five thousand eight hundred sixty-five (88.0%) of the variants identified were SNPs, 56 (0.8%) MNPs, and 741 (11.1%) indels. Based on the phylogeny, we found that 172 (2.9%) SNPs, five (9%) multi-nucleotide polymorphisms (MNPs), and 85 (11.5%) indels showed evidence of recurrent mutation, either reverting to the ancestral state, recurring in more than one location on the tree, or showing more than two alleles. Indels, most of which lay in mononucleotide runs or short tandem repeats, were highly enriched for this behavior (P < 0.001, χ2 test). Among SNPs, 533 (9.1%) were present in CpG dinucleotides, and these were also enriched for recurrent mutations, but not significantly (18 of 533 in CpGs compared with 154 of 5332 outside; P = 0.30, Fisher exact test).
It is in general difficult to identify functionally important variants from DNA sequence data alone, but variants that lead to loss of function or altered amino acid sequence in protein-coding genes can readily be identified and often influence function. No variants in our list were predicted to lead to loss of function, but there were six missense SNPs (Supplemental Table S3), two of which had been reported in a previous study examining 16 Y genes in 105 men (Rozen et al. 2009). As in the earlier study, none lay within haplogroup I and so do not provide any functional insights into the coronary artery disease (Charchar et al. 2012) and HIV progression (Sezgin et al. 2009) associated with this haplogroup. Five of the six SNPs were predicted not to damage the protein, but one, in the gene USP9Y in the R1b family of European origin, was predicted to be highly damaging to the protein.
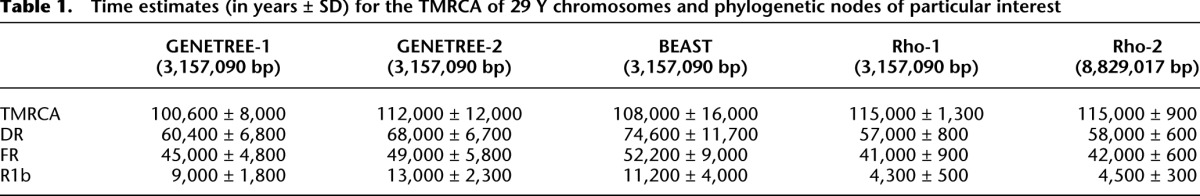
In addition to carrying large numbers of new markers informative about tree topology, the trees based on sequence data have branch lengths that are informative about the times when lineages diverged. We made use of this information by estimating times for the entire tree, and divergences of particular interest corresponding to the out-of-Africa movement, a topologically striking but poorly understood Paleolithic expansion, and to the expansion of R1b in Europe (Table 1). We made five estimates for each of the nodes of interest. Three of these used coalescent modeling either with a simple model of a constant-sized subdivided population (GENETREE-1) or with more complex models, including variation in population size and migration rate (GENETREE-2 and BEAST; for details, see Methods) (Bahlo and Griffiths 2000; Drummond et al. 2012). The other two estimates were based on the phylogeny (Rho-1 and Rho-2) (Forster et al. 1996; Saillard et al. 2000). These times were all broadly consistent and are relevant to debates about human expansions and migrations and so are discussed further below.
Table 1.
Time estimates (in years ± SD) for the TMRCA of 29 Y chromosomes and phylogenetic nodes of particular interest
Discussion
Many aspects of the approach adopted in this study, including complete sequencing of Y chromosomes, using data from public sequence resources, generating additional data from lineages of particular interest, and filtering of the Y-chromosomal regions and variants, are likely to become standard in future analyses of this kind. The current study has provided insights into several relevant methodological topics and also into the way that a calibrated Y-chromosomal phylogeny can provide insights into recent human evolution. We consider issues arising in these areas in this Discussion.
Next-generation sequence data are error-prone, with errors contributed by both base calling and mapping. The former can largely be overcome by high coverage and so are minimized in this study; in order to exclude the latter, we stringently excluded repeated regions, which are most prone to mapping errors. This resulted in a high-quality data set but at the cost of not detecting and using all variants. Future studies should investigate the possibility of extracting reliable variant calls from additional regions of the chromosome, and this will be facilitated by the longer reads expected as sequencing technologies improve. An additional source of biological error is the mutations that occur somatically in the donor or during cell culture, which are relevant here since all sequences were derived from lymphoblastoid cell lines. We can estimate this number from the sequences of the three-generation family. The grandfather and father carry 13 and 11 specific variants, respectively, two of which are absent from the grandfather but present in the father and transmitted to all his sons and are thus likely to represent in vivo de novo mutations, while the remaining 22 are likely to be somatic (Supplemental Fig. 1). This observation of two germline mutations in two transmissions of 8.97 Mb is consistent with the expectation of ∼0.6 mutations in two transmissions (0.3 variants observed per meiosis in 10.5 Mb) (Xue et al. 2009). In addition, the sons carry from zero to 17 individual-specific variants each. If we assume that all the nontransmitted variants are somatic, we can estimate an upper limit to the number of somatic variants at eight per individual (three SNPs per individual). These somatic variants are thus highly enriched for indels compared with total variants (42 of 67 compared with 699 of 6595; P < 0.001, Fisher exact test). Three somatic SNPs per individual would have a negligible effect on the analyses presented.
This study identified 6662 Y variants and placed them on a phylogenetic tree. Seventy-three percent of the variants appear to be novel, in that they are not in dbSNP 135, which already includes the 1000 Genomes Pilot data (The 1000 Genomes Project Consortium 2010), and 97% have not been used in published phylogenetic studies. They thus represent a valuable resource for future studies. Variants on a branch represented by a single chromosome, such as A, D, G, N, and Q, will include everything from ancient haplogroup-defining variants to those private to the individual sequenced, and additional work is needed to refine their phylogenetic positions. When multiple individuals within a haplogroup were sequenced, the shared variants already provide useful markers. For example, the geographical origin (south or west Asia) and time depth of haplogroup R1a are disputed, and few useful markers within this haplogroup have previously been available (Underhill et al. 2010); typing the 173 shared variants identified here in additional geographically diverse R1a samples would be of great interest.
Although this study was not aimed at investigating Y gene function, it was striking that a variant predicted to be highly damaging to protein structure was discovered in the USP9Y gene. USP9Y loss of function has been associated with variable phenotypes ranging from azoospermia to oligoasthenoteratozoospermia and normal sperm production (Tyler-Smith and Krausz 2009). The transmission of this variant through three generations demonstrates that it is compatible with male fertility, providing further evidence for the phenotypic diversity linked to variation in this gene.
A novel aspect of using extensive sequence data is that it is possible to investigate the time depth of the entire Y-chromosomal phylogeny represented by the samples or of any subset of lineages. Times are determined in mutational steps, and in order to convert these into a more useful unit such as years, a calibration metric has to be applied. We used a calibration based on direct measurement of the Y-chromosomal SNP mutation rate in both years and generations from a deep-rooting family (Xue et al. 2009) since this requires the minimum number of assumptions and has already been adopted in the literature (Cruciani et al. 2011). The measurement does, however, have wide confidence intervals since only a small number of mutations were observed, and these confidence intervals were not included in our consideration of times, which used the point estimate. Two lines of reasoning suggest that we may have more confidence in the point estimate than simple consideration of the number of mutations might suggest. First, it is consistent with other direct measurements of the human mutation rate, allowing for the expected higher mutation rate on the Y because of its permanent location in the mutation-prone male germ line (e.g., Roach et al. 2010). Second, it is consistent with the rate inferred from human–chimpanzee comparisons of the same sections of the Y chromosome: 1.3 × 10−9 mutations per nucleotide per year for a 6.5 million yr Y-chromosomal divergence time (Xue et al. 2009; Scally et al. 2012). Nevertheless, additional measurements of mutation rate are urgently needed to improve calibration.
We based our time estimates solely on SNPs, because indel mutation rates are poorly known and likely to be complex. We used either the complete set of SNPs (with the rho estimator) or a reduced set where coverage was high in all samples, ancestral state was known, and recurrent mutations were absent, and related individuals were excluded, required by GENETREE, and also used with BEAST and rho. As a result, we obtained five estimates for each time, which were similar (Table 1). Point estimates of the time to the most recent common ancestor (TMRCA) for the complete set of chromosomes examined were 101,000–115,000 yr ago. This is consistent with the published estimate of 105,000 yr ago for haplogroup A3 (Cruciani et al. 2011). We identified three additional nodes in the tree as being of particular interest. The first of these was DR, corresponding to the expansion of Y chromosomes following the out-of-Africa migration (Underhill et al. 2000; Jobling and Tyler-Smith 2003). The time of 57,000–74,000 yr ago is consistent with abundant nongenetic evidence, for example, of the first colonization of Australia around 50,000 yr ago (Roberts et al. 1990). The agreement of these two times with previous work strengthens our confidence in the remaining estimates. The second internal node examined was the multifurcation of haplogroups I, G, and NR (i.e., K), the representation in this reduced set of individuals of a larger multifurcation also involving several F sublineages, H and J (Karafet et al. 2008). The minimal resolution of the lineages, even in a phylogeny based on sequencing of 8.97 Mb, implies a rapid expansion, which we date here at 41,000–52,000 yr ago. This could correspond to an expansion into the interior of Europe and Asia following adaptation to these novel environments soon after the initial rapid coastal migration. The third internal node was that of R1b, a well-documented expansion in Europe, but with a much-debated time depth. Here, we estimate a time of 4,300–13,000 yr ago, the most uncertain of the dates. Despite the range of estimates, all these dates favor a Neolithic (Balaresque et al. 2010), more than a Paleolithic (Semino et al. 2000) or Mesolithic, expansion of this lineage. The three haplogroup I individuals, representing the next most frequent haplogroup in Europe, show signs of an expansion at approximately the same time (Fig. 2), although the number of individuals is too low to present any clear conclusion about whether or not this lineage was influenced by the same demographic events as R1b. Nevertheless, the rapid expansion of R1b (and possibly I1) in Europe contrasts with the less starlike expansion of E1b1a in Africa, which has been associated with the spread of farming, ironworking, and the Bantu languages in Africa over the last 5000 yr (Berniell-Lee et al. 2009). Both R1b and E1b1a samples are from a mixture of indigenous donors (from Europe and Africa, respectively) and admixed American donors, so sampling strategy does not provide an obvious explanation for the difference. Instead, the different phylogenetic structure, with far more resolution of the individual E1a1a branches, may reflect expansion starting from a larger and more diverse population and thus retaining more ancestral diversity.
In conclusion, our study identifies the methodological steps necessary to obtain reliable biological insights from current next-generation sequence data and the novel information that is available. It reveals, for example, how rapid some expansions of the Y phylogeny were, so that even extensive sequence data do not resolve all multifurcations. During the expansion of the six unrelated R1b lineages examined, only one mutation has arisen, despite a mutation rate of 0.3 per generation per 10 Mb. The study also poses challenges for the field, among which are (1) integrating sequence data generated by different technologies with different error modes on different samples; (2) the question of whether an inferred ancestral reference sequence would be more useful than the current hybrid of modern sequences; (3) the need to develop a compact and useful nomenclature system that can accommodate extensive sequence information, for example, based on major haplogroups and significant subclusters that provide a memorable but not complete indication of the location in the phylogeny; and (4) the difficulty of understanding the archaeological events associated with the male expansions detected, as well as the standards of evidence that should be required in order to accept links.
Methods
Definition of Y-specific unique regions
We identified unique regions within the male-specific part of the Y chromosome reference sequence (GRCh37 http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/) where we expected read mapping and variant detection to escape complications introduced by repeated sequences. This was achieved by excluding the pseudoautosomal, heterochromatic, X-transposed, and ampliconic segments (Skaletsky et al. 2003), leaving nine separate regions (Fig. 1; Supplemental Table S1). Together, these spanned 8.97 Mb. All analyses in this study were restricted to these regions or to subsets of them.
Y-chromosomal sequence data sources
High-coverage Y-chromosomal sequence data from 35 males were downloaded from the Complete Genomics database (ftp://ftp2.completegenomics.com/vcf_files/Build37_2.0.0/). Coverage of the Y unique regions ranged from 19× to 35× (Supplemental Table S4). These males did not include any individual belonging to the most basal haplogroup, haplogroup A. We therefore generated additional sequence data from a haplogroup A individual, NA21313. This chromosome was derived for the markers M32, M190, M220, M144, M202, M305, and M219 and ancestral for P97, placing it in haplogroup A3b2*. Sequencing included both low-coverage (mean, 5×) whole-genome data and high-coverage data from long-PCR products between chrY:13,798,579–19,720,738 (GRCh37).
NA21313 Low-coverage sequencing
Low-coverage reads were mapped and variants called in combination with 1000 Genomes samples using samtools and bcftools (Li et al. 2009) and then filtered (StrandBias 1 × 10−5; EndDistBias 1 × 10−7; MaxDP 10000; MinDP 2; Qual 3; SnpCluster 5,10; MinAltBases 2; MinMQ 10; SnpGap 3).
NA21313 High-coverage sequencing
High-coverage sequence information was generated by amplifying 5- to 6-kb overlapping fragments by long PCR. Approximately equimolar amounts of PCR fragments were pooled and used for library preparation and paired-end sequencing (54 bp) on an Illumina GAII Genome Analyzer to obtain 475× median coverage (European Nucleotide Archive [ENA] sample accession no.: ERS006694; www.ebi.ac.uk/ena/). MAQ (Li et al. 2008) was used for mapping and SNP calling in the high -coverage data. Read depth coverage (>1/2 mean depth) and mapping (consensus > 30, mapping > 63) filters reduced the raw variant calls from a total of 5684 to 2233. Subsequent filters included removal of heterozygous calls and sites that were within 4 bp of each other to create a high-confidence filtered list of 615 SNPs, none of which were discordant with the established Y phylogeny.
Ancestral states
We extracted the ancestral allele for each position that was variable in humans (assumed to be the allele present in chimpanzee) using the Ensembl-Compara pipeline (Vilella et al. 2009), release 66, and obtained calls for 6271 of the total number of 6662 variable sites (Fig. 1; Supplemental Table S2).
Data QC and filtering
We extracted the variants from the Y-specific unique regions present in the 35 male individuals from the database of Complete Genomics, including both SNPs and indels. The number of differences from the reference sequence ranged from 210–1257. Examination of the distribution of variants along the chromosome revealed an excess in one sample, GS19649, in the region 28,670,244–28,735,914 compared with the other individuals; this was associated with low coverage of this region, so we hypothesized that the excess arose from a combination of deletion of the region in this individual and mismapping of reads originating from other parts of the chromosome. We excluded from further consideration all variable sites from all individuals that fell into this region.
We identified 631 variable sites mapping within the 8.97-Mb region that had been described in the literature and placed on the 2008 Y Chromosome Consortium phylogenetic tree (Karafet et al. 2008) and its subsequent partial updates (Cruciani et al. 2008, 2010, 2011; Chiaroni et al. 2009; Sims et al. 2009; Trombetta et al. 2010, 2011; Debnath et al. 2011; Jota et al. 2011; Mendez et al. 2011). These were used for assigning a standard haplogroup to each sample and in validation.
From all 36 individuals, we identified a total of 6662 variable sites. Missing calls ranged from 31–165 per individual; call rates were on average 98.9%. We imputed the missing sites based on the tree structure and the assumption of parsimony; the only errors introduced by this procedure will be missed reversions or recurrent mutations, both very rare.
Validation
SNP validation
We performed in silico validation of the SNP calls using three approaches. First, calls from 101 SNPs based on the intersection of the Affymetrix Human SNP array 6.0 and the Illumina Human 1M beadchip were available for 11 individuals from the HapMap3 genotyping of the same samples (Altshuler et al. 2010). We observed 100% concordance at these 1111 positions (Supplemental Table S5). Second, high-coverage Illumina GAII sequence data were available for two individuals, NA19239 and NA12891, the trio fathers in the 1000 Genomes Pilot Project (The 1000 Genomes Project Consortium 2010). There was 85.1% (553 of 650) and 96.5% (694 of719) concordance between the two data sets. In the light of the other validation results, we ascribe the relatively low concordance in this test to the early sequencing technologies used in this part of the 1000 Genomes Pilot project, much of it single-ended and particularly susceptible to mapping problems. Third, using the literature SNP (see Data QC and Filtering above) with its derived allele furthest from the root in each individual to assign a haplogroup, we could predict the allelic states expected for the remaining 630 literature sites. Here, after correcting some typographical errors in site positions (Supplemental Table S6), there was 99.8% concordance between the prediction and observation (22,680 genotypes); discrepancies are listed at the top of Supplemental Table S6, and several may even represent mistakes in the locations of published markers on the phylogeny or recurrent mutations not previously recorded rather than genotyping errors in the present data set. Validation rates in the two reliable tests are thus very high.
Indel validation
We used two approaches to assess the quality of the indel calls. First, using a logic similar to the third SNP validation method above, we identified 40 reported indel sites within the unique regions of the Y chromosome (Karafet et al. 2008). Genotype calls were made at 28 of these in our data set, and we observed a 99.7% concordance between the observed calls and those expected from the phylogeny (974 of 977). The false-negative call rate was therefore low. Second, most indel mutations occur in simple sequences such as mononucleotide runs or short tandem repeats. We therefore examined the sequences flanking the calls and observed a preponderance of these motifs: 471 poly A or poly T, 17 poly C or poly G, 87 short tandem repeats, and 166 other (Supplemental Table S7). This shows that, although these variants have not been experimentally tested, at least 575 of 741 of the indels (78%) are highly plausible candidates. In all, we can have reasonable confidence in this indel callset.
Constructing the Y-chromosomal haplogroup tree
FASTA formatted sequence files were used to generate haplogroup trees. Sequence alignments were built using CLUSTALW2 (http://www.clustal.org/), and a maximum parsimony (MP) phylogenetic tree was created using the PHYLIP software (http://evolution.gs.washington.edu/phylip.html). We generated three trees. First, we used all the 6662 sites in 36 individuals to construct a haplogroup tree, which was rooted using the chimpanzee Y sequence. The resulting tree (tree 1) is shown in Supplemental Figure 1. Second, we excluded all indels and MNPs to generate a second haplogroup tree based only on SNPs (tree 2, Supplemental Fig. 2). Third, we removed all sites that were recurrent in this set of 36 males or lacked ancestral information, and restricted the region considered to the 3.2 Mb with high coverage in the haplogroup A individual, NA21313 (tree 3, Fig. 2). Stringent sites of this kind are required by GENETREE (Bahlo and Griffiths 2000), one of the approaches used to estimate times on the tree.
Estimating the TMRCA and ages of nodes of the haplogroup tree
To estimate the TMRCA of the complete tree and the ages of nodes of particular interest, DR, FR, and R1b, we applied two broad approaches that provided five individual estimates for each timepoint. The first was to use coalescent modeling implemented either in GENETREE (Bahlo and Griffiths 2000) or BEAST (Drummond et al. 2012) using the sites in tree 3. To avoid the influence of related individuals, we removed seven out of the eight individuals who were part of the same three-generation pedigree. This resulted in 2004 SNPs from 29 individuals.
GENETREE
We were unable to successfully run GENETREE using all 2004 sites. We therefore divided the data into nonoverlapping sets of 90–99 SNPs according to their position on the chromosome, resulting in 21 sets and thus 21 GENETREE runs. GENETREE version 8.3 was run in two ways. The first used a demographic model of a constant-sized but subdivided population with fixed migration rates. Run details are as follows: An initial theta value was calculated using the formula θ = Ne × μ × length of region spanning 90–99 SNPs (Ne = 2,000; μ = 3 × 10−8 mutations per nucleotide per generation); 1 million coalescent simulations and 101 surface points were used to find the optimal value. The migration rate between Africa and Europe was set at 3.2 × 10−5 per generation and between Africa and Asia at 0.8 × 10−5 per generation (Schaffner et al. 2005) to generate the migration file. From this calculation, a best estimate of theta was obtained and used to calculate the TMRCA and other times (Table 1, GENETREE-1; Supplemental Figs. S3.1–3.21). The second used a demographic model of exponential growth and a subdivided population, with growth and migration rates derived from the data. Run details are as follows: An initial theta value was calculated as before and used to generate the migration file; initial exponential growth rates for Europe (0.20%), Asia (0.41%), and Africa (0.74%) were used (Shi et al. 2010). One million coalescent simulations and 101 surface points were used to find the optimal value for theta, the migration rate, and the growth rate. From these, a best estimate of theta, migration rate, and exponential growth rate were obtained and used to calculate the TMRCA and other times (Table 1, GENETREE-2).
BEAST
We also used BEAST version 1.7.2 (http://beast.bio.ed.ac.uk) to estimate the TMRCA and divergence time for each branch of interest. The NEXUS formatted file was used to generate the BEAST XML input file for the BEAST version 1.7.2 program. To obtain the TMRCA and the time for the branches, four taxon subsets were set up (AR, DR, FR, and R1b). We chose GTR as the substitution model, gamma as the site heterogeneity model, lognormal relaxed clock as the clock model, exponential growth as the tree prior, μ = 1 × 10-9 mutations per nucleotide per year, and 100 million as the MCMC chain length. The log output files were obtained by running the BEAST software. We carried out two independent BEAST runs from the same XML input file and combined the two log output files using LogCombiner, as recommended to increase the ESS (effective sample size) of the analysis and also to allow us to determine whether or not the two independent runs were converging on the same distribution (Supplemental Table S8). Tracer version 1.5 (http://beast.bio.ed.ac.uk/Tracer) was used to analyze the output file and times (Table 1, BEAST), and TreeAnnotator version 1.7.2 to obtain an estimate of the phylogenetic tree. Finally, we viewed the tree in FigTree (http://beast.bio.ed.ac.uk/FigTree) (Supplemental Fig. 4).
Rho
In the second broad approach, we used the phylogeny-based rho statistic (Forster et al. 1996; Saillard et al. 2000), applied both to the same set of 2004 SNPs (tree 3, Table 1, Rho-1) and also to the complete set of 5865 SNPs, including recurrent SNPs (tree 2, Table 1, Rho-2).
In all five estimates (two GENETREE, one BEAST, two rho), calibration was achieved using the directly measured SNP mutation rate of 1.0 × 10−9 mutations per nucleotide per year or 3.0 × 10−8 mutations per nucleotide per generation (Xue et al. 2009). Where necessary, we converted estimates in generations to estimates in years using 30 yr per generation.
Annotation
Each variable site was annotated (where relevant) with its type (SNP, MNP, or indel), location in hg36 and hg37, the reference, ancestral, and derived alleles, dbSNP identification, name in the existing phylogenetic tree, haplogroup in which it is variable, presence as part of a CpG dinucleotide, location in a gene, and consequences for protein structure (Supplemental Table S2). The predicted consequences of missense variants for protein function were taken from the modified Condel scores (Gonzalez-Perez and Lopez-Bigas 2011) in Ensembl.
Data access
The NA21313 sequencing data from this study have been deposited in the European Nucleotide Archive (ENA) (http://www.ebi.ac.uk/ena/) under accession nos. ERS037274 (low coverage) and ERS006694 (high coverage).
Acknowledgments
We thank Richard Durbin for comments, Christophe Dessimoz and Kevin Gori for advice about BEAST, Bob Griffiths for helpful suggestions about running GENETREE, and the Sanger library and sequencing teams for generating the haplogroup A data. This work was supported by grant no. 098051 from The Wellcome Trust and a China Scholarship Council (CSC) fellowship to W.W.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.143198.112.
Freely available online through the Genome Research Open Access option.
References
- The 1000 Genomes Project Consortium 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Bonnen PE, de Bakker PI, Deloukas P, Gabriel SB, et al. 2010. Integrating common and rare genetic variation in diverse human populations. Nature 467: 52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahlo M, Griffiths RC 2000. Inference from gene trees in a subdivided population. Theor Popul Biol 57: 79–95 [DOI] [PubMed] [Google Scholar]
- Balaresque P, Bowden GR, Adams SM, Leung HY, King TE, Rosser ZH, Goodwin J, Moisan JP, Richard C, Millward A, et al. 2010. A predominantly Neolithic origin for European paternal lineages. PLoS Biol 8: e1000285 doi: 10.1371/journal.pbio.1000285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar DM, Villems R, Soodyall H, Blue-Smith J, Pereira L, Metspalu E, Scozzari R, Makkan H, Tzur S, Comas D, et al. 2008. The dawn of human matrilineal diversity. Am J Hum Genet 82: 1130–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berniell-Lee G, Calafell F, Bosch E, Heyer E, Sica L, Mouguiama-Daouda P, van der Veen L, Hombert JM, Quintana-Murci L, Comas D 2009. Genetic and demographic implications of the Bantu expansion: Insights from human paternal lineages. Mol Biol Evol 26: 1581–1589 [DOI] [PubMed] [Google Scholar]
- Busby GB, Brisighelli F, Sanchez-Diz P, Ramos-Luis E, Martinez-Cadenas C, Thomas MG, Bradley DG, Gusmao L, Winney B, Bodmer W, et al. 2011. The peopling of Europe and the cautionary tale of Y chromosome lineage R-M269. Proc Biol Sci 279: 884–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann RL, Stoneking M, Wilson AC 1987. Mitochondrial DNA and human evolution. Nature 325: 31–36 [DOI] [PubMed] [Google Scholar]
- Charchar FJ, Bloomer LD, Barnes TA, Cowley MJ, Nelson CP, Wang Y, Denniff M, Debiec R, Christofidou P, Nankervis S, et al. 2012. Inheritance of coronary artery disease in men: An analysis of the role of the Y chromosome. Lancet 379: 915–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaroni J, Underhill PA, Cavalli-Sforza LL 2009. Y chromosome diversity, human expansion, drift, and cultural evolution. Proc Natl Acad Sci 106: 20174–20179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani F, Trombetta B, Novelletto A, Scozzari R 2008. Recurrent mutation in SNPs within Y chromosome E3b (E-M215) haplogroup: A rebuttal. Am J Hum Biol 20: 614–616 [DOI] [PubMed] [Google Scholar]
- Cruciani F, Trombetta B, Sellitto D, Massaia A, Destro-Bisol G, Watson E, Beraud Colomb E, Dugoujon JM, Moral P, Scozzari R 2010. Human Y chromosome haplogroup R-V88: A paternal genetic record of early mid Holocene trans-Saharan connections and the spread of Chadic languages. Eur J Hum Genet 18: 800–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani F, Trombetta B, Massaia A, Destro-Bisol G, Sellitto D, Scozzari R 2011. A revised root for the human Y chromosomal phylogenetic tree: The origin of patrilineal diversity in Africa. Am J Hum Genet 88: 814–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath M, Palanichamy MG, Mitra B, Jin JQ, Chaudhuri TK, Zhang YP 2011. Y-chromosome haplogroup diversity in the sub-Himalayan Terai and Duars populations of East India. J Hum Genet 56: 765–771 [DOI] [PubMed] [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81 [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29: 1969–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster P, Harding R, Torroni A, Bandelt HJ 1996. Origin and evolution of Native American mtDNA variation: A reappraisal. Am J Hum Genet 59: 935–945 [PMC free article] [PubMed] [Google Scholar]
- Foster EA, Jobling MA, Taylor PG, Donnelly P, de Knijff P, Mieremet R, Zerjal T, Tyler-Smith C 1998. Jefferson fathered slave's last child. Nature 396: 27–28 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Lopez-Bigas N 2011. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 88: 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnarsdottir ED, Li M, Bauchet M, Finstermeier K, Stoneking M 2011. High-throughput sequencing of complete human mtDNA genomes from the Philippines. Genome Res 21: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer MF 1995. A recent common ancestry for human Y chromosomes. Nature 378: 376–378 [DOI] [PubMed] [Google Scholar]
- Hu M, Ayub Q, Guerra-Assuncao JA, Long Q, Ning Z, Huang N, Romero IG, Mamanova L, Akan P, Liu X, et al. 2012. Exploration of signals of positive selection derived from genotype-based human genome scans using re-sequencing data. Hum Genet 131: 665–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling MA, Tyler-Smith C 2003. The human Y chromosome: An evolutionary marker comes of age. Nat Rev Genet 4: 598–612 [DOI] [PubMed] [Google Scholar]
- Jota MS, Lacerda DR, Sandoval JR, Vieira PP, Santos-Lopes SS, Bisso-Machado R, Paixao-Cortes VR, Revollo S, Paz YMC, Fujita R, et al. 2011. A new subhaplogroup of native American Y-Chromosomes from the Andes. Am J Phys Anthropol 146: 553–559 [DOI] [PubMed] [Google Scholar]
- Karafet TM, Mendez FL, Meilerman MB, Underhill PA, Zegura SL, Hammer MF 2008. New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res 18: 830–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ruan J, Durbin R 2008. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18: 1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009. The Sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez FL, Karafet TM, Krahn T, Ostrer H, Soodyall H, Hammer MF 2011. Increased resolution of Y chromosome haplogroup T defines relationships among populations of the Near East, Europe, and Africa. Hum Biol 83: 39–53 [DOI] [PubMed] [Google Scholar]
- Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, et al. 2010. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328: 636–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RG, Jones R, Smith MA 1990. Thermoluminescence dating of a 50,000-year-old human occupation site in northern Australia. Nature 345: 153–156 [Google Scholar]
- Rozen S, Marszalek JD, Alagappan RK, Skaletsky H, Page DC 2009. Remarkably little variation in proteins encoded by the Y chromosome's single-copy genes, implying effective purifying selection. Am J Hum Genet 85: 923–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saillard J, Forster P, Lynnerup N, Bandelt HJ, Norby S 2000. mtDNA variation among Greenland Eskimos: The edge of the Beringian expansion. Am J Hum Genet 67: 718–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scally A, Dutheil JY, Hillier LW, Jordan GE, Goodhead I, Herrero J, Hobolth A, Lappalainen T, Mailund T, Marques-Bonet T, et al. 2012. Insights into hominid evolution from the gorilla genome sequence. Nature 483: 169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner SF, Foo C, Gabriel S, Reich D, Daly MJ, Altshuler D 2005. Calibrating a coalescent simulation of human genome sequence variation. Genome Res 15: 1576–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scozzari R, Cruciani F, Santolamazza P, Malaspina P, Torroni A, Sellitto D, Arredi B, Destro-Bisol G, De Stefano G, Rickards O, et al. 1999. Combined use of biallelic and microsatellite Y-chromosome polymorphisms to infer affinities among African populations. Am J Hum Genet 65: 829–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semino O, Passarino G, Oefner PJ, Lin AA, Arbuzova S, Beckman LE, De Benedictis G, Francalacci P, Kouvatsi A, Limborska S, et al. 2000. The genetic legacy of Paleolithic Homo sapiens sapiens in extant Europeans: A Y chromosome perspective. Science 290: 1155–1159 [DOI] [PubMed] [Google Scholar]
- Sezgin E, Lind JM, Shrestha S, Hendrickson S, Goedert JJ, Donfield S, Kirk GD, Phair JP, Troyer JL, O'Brien SJ, et al. 2009. Association of Y chromosome haplogroup I with HIV progression, and HAART outcome. Hum Genet 125: 281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Ayub Q, Vermeulen M, Shao RG, Zuniga S, van der Gaag K, de Knijff P, Kayser M, Xue Y, Tyler-Smith C 2010. A worldwide survey of human male demographic history based on Y-SNP and Y-STR data from the HGDP-CEPH populations. Mol Biol Evol 27: 385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims LM, Garvey D, Ballantyne J 2009. Improved resolution haplogroup G phylogeny in the Y chromosome, revealed by a set of newly characterized SNPs. PLoS ONE 4: e5792 doi: 10.1371/journal.pone.0005792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, Pyntikova T, Ali J, Bieri T, et al. 2003. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423: 825–837 [DOI] [PubMed] [Google Scholar]
- Trombetta B, Cruciani F, Underhill PA, Sellitto D, Scozzari R 2010. Footprints of X-to-Y gene conversion in recent human evolution. Mol Biol Evol 27: 714–725 [DOI] [PubMed] [Google Scholar]
- Trombetta B, Cruciani F, Sellitto D, Scozzari R 2011. A new topology of the human Y chromosome haplogroup E1b1 (E-P2) revealed through the use of newly characterized binary polymorphisms. PLoS ONE 6: e16073 doi: 10.1371/journal.pone.0016073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler-Smith C, Krausz C 2009. The will-o'-the-wisp of genetics: Hunting for the azoospermia factor gene. N Engl J Med 360: 925–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill PA, Shen P, Lin AA, Jin L, Passarino G, Yang WH, Kauffman E, Bonne-Tamir B, Bertranpetit J, Francalacci P, et al. 2000. Y chromosome sequence variation and the history of human populations. Nat Genet 26: 358–361 [DOI] [PubMed] [Google Scholar]
- Underhill PA, Myres NM, Rootsi S, Metspalu M, Zhivotovsky LA, King RJ, Lin AA, Chow CE, Semino O, Battaglia V, et al. 2010. Separating the post-Glacial coancestry of European and Asian Y chromosomes within haplogroup R1a. Eur J Hum Genet 18: 479–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson AC 1991. African populations and the evolution of human mitochondrial DNA. Science 253: 1503–1507 [DOI] [PubMed] [Google Scholar]
- Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E 2009. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res 19: 327–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Wang Q, Long Q, Ng BL, Swerdlow H, Burton J, Skuce C, Taylor R, Abdellah Z, Zhao Y, et al. 2009. Human Y chromosome base-substitution mutation rate measured by direct sequencing in a deep-rooting pedigree. Curr Biol 19: 1453–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalloua PA, Platt DE, El Sibai M, Khalife J, Makhoul N, Haber M, Xue Y, Izaabel H, Bosch E, Adams SM, et al. 2008. Identifying genetic traces of historical expansions: Phoenician footprints in the Mediterranean. Am J Hum Genet 83: 633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerjal T, Xue Y, Bertorelle G, Wells RS, Bao W, Zhu S, Qamar R, Ayub Q, Mohyuddin A, Fu S, et al. 2003. The genetic legacy of the Mongols. Am J Hum Genet 72: 717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]