

Abstract
The cellular inhibitor of apoptosis 1 (cIAP1) protein is an essential regulator of canonical and noncanonical nuclear factor κB (NF-κB) signaling pathways. NF-κB signaling is known to play important roles in myogenesis and degenerative muscle disorders such as Duchenne muscular dystrophy (DMD), but the involvement of cIAP1 in muscle disease has not been studied directly. Here, we asked whether the loss of cIAP1 would influence the pathology of skeletal muscle in the mdx mouse model of DMD. Double-mutant cIAP1−/−;mdx mice exhibited reduced muscle damage and decreased fiber centronucleation in the soleus, compared with single-mutant cIAP1+/+;mdx mice. This improvement in pathology was associated with a reduction in muscle infiltration by macrophages and diminished expression of inflammatory cytokines such as IL-6 and tumor necrosis factor-α. Furthermore, the cIAP1−/−;mdx mice exhibited reduced serum creatine kinase, and improved exercise endurance associated with improved exercise resilience by the diaphragm. Mechanistically, the loss of cIAP1 was sufficient to drive constitutive activation of the noncanonical NF-κB pathway, which led to increased myoblast fusion in vitro and in vivo. Collectively, these results show that the loss of cIAP1 protects skeletal muscle from the degenerative pathology resulting from systemic loss of dystrophin.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a severe and progressive X-linked neuromuscular disorder resulting from mutations causing the loss of dystrophin (1). Dystrophin functions as a major structural protein in skeletal muscle, and its loss renders the muscle highly vulnerable to mechanical damage. In humans, continuing cycles of degeneration and regeneration result in the exhaustion and senescence of the satellite cells, which are responsible for muscle regeneration and eventual replacement of muscle with fat and connective tissue. Affected individuals present with muscle weakness as early as two years of age, and usually die in their late teens or early twenties from pulmonary or cardiac failure. To date, there are no durable treatments or cures for this disorder; it is thus crucial to develop strategies that either improve the regenerative potential or decrease the damage susceptibility of affected muscle.
Although the cause of DMD is well defined, the factors that both perpetuate and aggravate the resulting muscle pathology are poorly understood. A point of convergence for existing corticosteroid therapies, immune system biology and skeletal muscle regeneration is the nuclear factor κB (NF-κB) signaling pathway. In recent years, multiple studies have addressed the role of NF-κB in DMD, using the mdx mouse model in which a nonsense mutation in the DMD gene causes a loss of functional dystrophin (2,3). NF-κB transcription factors function as homo- or heterodimers formed from five proteins: RelA/p65, RelB, c-Rel, p105/p50 and p100/p52. The dimers remain sequestered in the cytoplasm by inhibitor of κB (IκB) proteins until a cascade of signaling events triggers ubiquitination and degradation of IκB, allowing the translocation of NF-κB to the nucleus. Canonical NF-κB activity, mediated through the RelA:p50 dimer, is chronically elevated in dystrophic muscle, and many reports suggest that this elevation contributes to the resulting pathology (4–6). The cellular inhibitors of apoptosis-1 and -2 (cIAP1 and cIAP2) proteins regulate NF-κB signal transduction through various mechanisms. The cIAPs are E3 ubiquitin ligases, and generally function by ubiquitinating key scaffold adaptor proteins and kinases, targeting them for either degradation or formation of signaling scaffolds (7,8). The cIAP1/2 proteins are essential for canonical signaling triggered by tumor necrosis factor (TNF) superfamily members, such as TNFα, CD40L and TWEAK (TNFα-like weak inducer of apoptosis) (7–10). In this case, either cIAP1 or cIAP2 catalyzes the K63 ubiquitination of the kinase RIP1 (receptor interacting kinase 1), which assembles a signaling complex that is required for signal transduction. The cIAP1/2 proteins also function as negative regulators of a noncanonical NF-κB pathway. Here, cIAP1/2, along with the adapter proteins TRAF2 and TRAF3, forms a ubiquitin ligase complex that targets the protein kinase NIK (NF-κB-inducing kinase) for degradation (8,11). In the absence of cIAP1/2, the accumulation of NIK triggers the processing of p100 into its functional p52 subunit, which forms a heterodimer with RelB and translocates to the nucleus. NF-κB signaling through p52:RelB leads to the transcription of target genes that are both shared with and distinct from the canonical pathway (12,13). Although canonical NF-κB signaling is well studied in the context of muscle regeneration, the noncanonical pathway is as yet poorly understood. Recent studies have highlighted the noncanonical pathway as promoting an oxidative muscle fate, at least in part through transcriptional upregulation of the mitochondrial regulator PGC-1β (14–16). Furthermore, we recently showed that the noncanonical pathway is an apical regulator of myoblast fusion, and improves regeneration following cardiotoxin-induced injury (17). Either the loss of cIAP1 or treatment with the cytokine TWEAK (9,18) is sufficient to induce robust activation of the noncanonical pathway and promote myoblast fusion in vitro and in vivo. However, the outcomes of cIAP1/2 loss on muscle function and regeneration in a disease model have not been studied. Notably, only cIAP1 and not cIAP2 is expressed in muscle cells (7), though both cIAP1 and cIAP2 are present in myeloid cells which infiltrate muscle tissue following injury. A cIAP1 knockout animal is thus effectively a cIAP1/2 double-knockout in muscle, whereas most other tissues retain the compensatory function of cIAP2.
Here, we examined the effect of a loss of cIAP1 on the pathology of skeletal muscle in the mdx mouse. We show that the loss of cIAP1 is associated with decreased damage, reduced inflammation and improved contractile responsiveness of the soleus muscle. We also show that in the absence of cIAP1, mdx mice exhibit reduced diaphragm permeability and improved endurance to treadmill exercise. In primary mdx myoblasts, we show that the loss of cIAP1 leads to elevated activation of both canonical and noncanonical NF-κB pathways, leading to delayed (but not impaired) myoblast differentiation and increased myoblast fusion, respectively. These results suggest that the downregulation of cIAP1, or the activation of the noncanonical NF-κB pathway, may have potential therapeutic utility in the treatment of DMD.
RESULTS
cIAP1−/−;mdx double-mutant mice have decreased pathology in the soleus muscle
To determine the role played by cIAP1 in the pathology of mdx muscle, we generated whole-body cIAP1−/−;mdx double-mutant mice, as well as single-mutant cIAP1+/+;mdx controls, by crossing cIAP1−/− males (19) with mdx females. Immunohistochemistry for dystrophin, as well as western blotting for cIAP1 expression (Fig. 1A and B), was used to confirm genotypes determined by PCR and sequencing. We went on to characterize the muscle pathology of the cIAP1−/−;mdx mice. The presence of a nucleus at the center of a muscle fiber is an indicator of recent regeneration, as mature healthy fibers have nuclei at the periphery (20,21). Cross-sections of 12-week-old male soleus and extensor digitorum longus (EDL) muscles were stained with H&E for analysis. The cIAP1−/−;mdx solei had ∼50% fewer centronucleated fibers than their cIAP1+/+;mdx counterparts (28 ± 9.7% versus 57 ± 1.2%, respectively, Fig. 2A and B), which suggests that fewer muscle fibers required repair in the cIAP1−/−;mdx solei. However, there was no difference in the proportion of centronucleated fibers between cIAP1+/+;mdx and cIAP1−/−;mdx EDL muscles (Fig. 2C). There was a modest reduction in the number of fibers expressing embryonic myosin heavy chain (eMHC) in both soleus and EDL muscles (Supplementary Material, Fig. S1). Since a reduction in fiber centronucleation may also suggest a deficit in regeneration, we directly measured muscle damage by quantifying the amount of muscle containing necrotic tissue and dense infiltration by mononuclear cells (monocytes and macrophages). There was a considerable reduction in total size of such regions in cIAP1−/−;mdx solei (Fig. 2D and E). These results suggest that the loss of cIAP1 results in decreased muscle damage and a reduced requirement for regeneration.
Figure 1.

Generation and confirmation of cIAP1−/−;mdx double-mutant mice. (A) The mice were generated from crossings of cIAP1−/− males with mdx females. The loss of dystrophin expression was confirmed by immunohistochemistry on 10 μm gastrocnemius sections (right panels). Individual muscle fibers were identified by laminin staining (left panels). Gastrocnemii from wild-type mice were used as controls. Scale bar represents 100 μm. (B) Gastrocnemius muscles from wild-type, cIAP1−/−, single-mutant (cIAP1+/+;mdx) and double-mutant (cIAP1−/−;mdx) mice were assayed by western blotting for the expression of cIAP1. Each lane represents an individual animal. GAPDH was used as a loading control.
Figure 2.
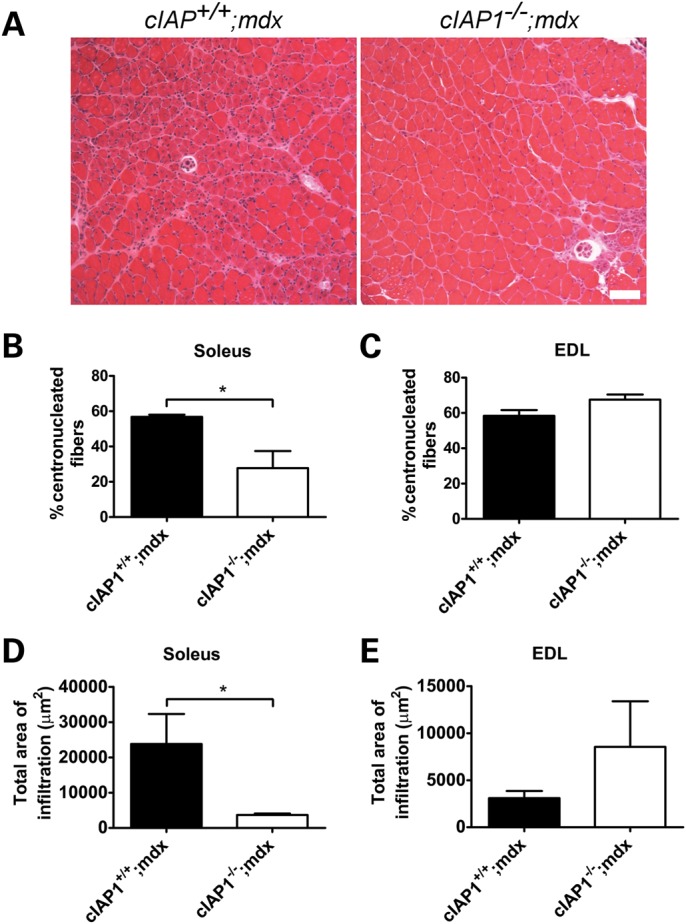
The loss of cIAP1 reduces damage in mdx solei. (A) Cross-sections of soleus muscles were stained with H&E, highlighting centronucleated fibers, and the percentage of fibers with centrally located nuclei was counted. Scale bar represents 100 μm. The percentage of fibers containing centrally located nuclei (B and C) and the total area of necrotic tissue (D and E) were measured from the H&E-stained cross-sections, using the Northern Eclipse software. Data are means ± SEM, n = 5. *P < 0.05 versus cIAP1+/+;mdx controls. EDL, extensor digitorum longus.
A secondary indicator of muscle regeneration is the presence of infiltrating macrophages in damaged muscle (5). We reasoned that if the major outcome of cIAP1 loss was a decrease in muscle damage, this would be reflected by reduced infiltration of pro-inflammatory macrophages into the dystrophic muscle. To test this, we performed quantitative RT–PCR on RNA extracted from soleus and EDL samples and measured the expression of the pro-inflammatory macrophage marker CD68. cIAP1−/−;mdx solei contained about half the CD68 expression of the cIAP1+/+;mdx controls (Fig. 3A and B), whereas CD68 expression in the EDL was unchanged between both genotypes. We further characterized the inflammatory environment in the soleus and EDL muscles by examining the expression of other macrophage and cytokine markers. CD163, which is a marker of anti-inflammatory M2 macrophages (22,23), was markedly increased in cIAP1−/−;mdx soleus, but not in the EDL (Fig. 3C). This outcome was again consistent with the observations of reduced dystrophic damage in the cIAP1−/−;mdx soleus. Furthermore, mRNA expression of cytokines IL6, IL10 and TNF were all decreased in the cIAP1−/−;mdx soleus and EDL (Fig. 3D–F), indicative of a generally reduced inflammatory response in the cIAP1−/−;mdx mice. Taken together, these data suggest that the loss of cIAP1 attenuates the inflammatory response in dystrophic muscle.
Figure 3.
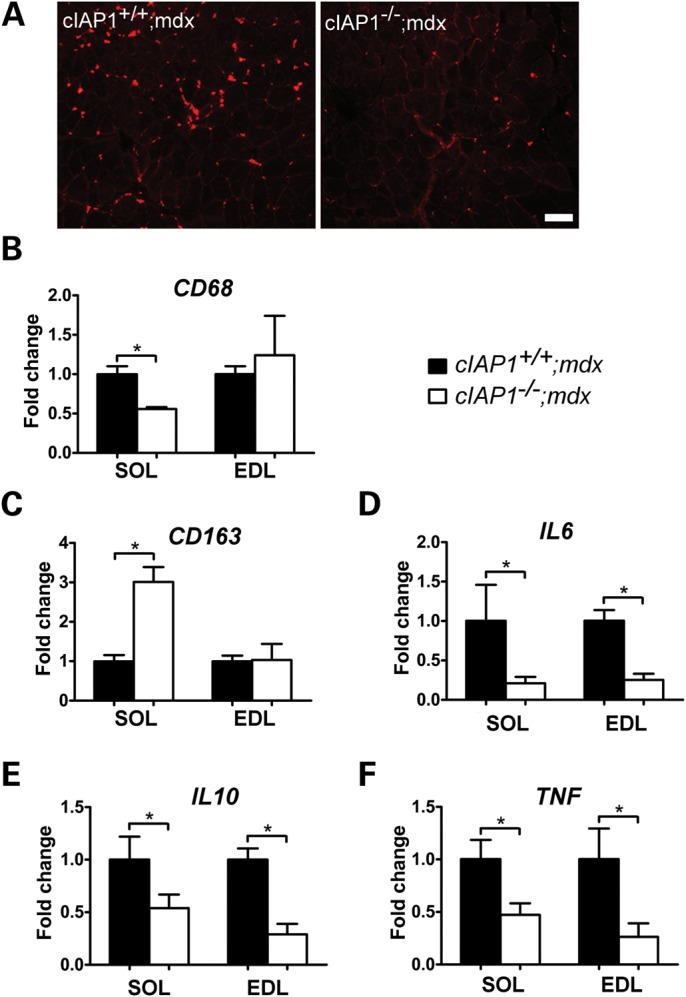
Hallmarks of inflammation and macrophage infiltration are reduced in cIAP1−/−;mdx solei. (A) Cross-sections of soleus muscles from cIAP1+/+;mdx and cIAP1−/−;mdx mice were immunolabeled for CD68 (red) to detect pro-inflammatory macrophages. Scale bar represents 100 μm. (B–F) Total RNA was isolated from soleus and EDL samples for quantitative RT–PCR detection of markers of inflammatory macrophages (CD68, B), noninflammatory macrophages (CD163, C) and cytokines (D–F). Values are expressed relative to cIAP1+/+;mdx data. Graphs represent means ± SEM, n = 4. *P < 0.05 versus cIAP1+/+;mdx controls. SOL, soleus; EDL, extensor digitorum longus.
cIAP1−/−;mdx solei demonstrate improved contractile properties
Deficits in the contractile properties of mdx muscle are well-documented effects of DMD pathology (24–26). Specifically, the increased membrane permeability of dystrophic muscle permits excessive influx of calcium, which may initiate the cascade of pathology and eventual necrosis (27–29). Such disruptions of calcium homeostasis manifest as excessive force production at low frequencies of stimulation. We asked whether the loss of cIAP1 would restore normal force–frequency characteristics to mdx muscle. To test this, we first determined the maximum isometric force produced by isolated soleus and EDL muscles. As previously reported (30–32), maximum force produced by the dystrophic solei was unaffected by the loss of dystrophin in either cIAP1+/+;mdx or cIAP1−/−;mdx muscle (Fig. 4A). In contrast, force output was reduced by 30% in the cIAP1+/+;mdx EDLs compared with the wild-type controls (Fig. 4B). The cIAP1−/−;mdx EDLs exhibited a slight increase in force compared with the cIAP1+/+;mdx, but the increase was not statistically significant. We next determined the force–frequency relationships of the soleus and EDL muscles by measuring the percentage of maximum force produced at stimulating frequencies between 0 and 140 Hz. The cIAP1+/+;mdx solei produced a force–frequency curve that was significantly shifted toward the left (requiring lower stimulation frequencies to produce the same force) compared with wild-type controls (Fig. 4C), which indicates that the cIAP1+/+;mdx solei were aberrantly responsive to stimulation. In contrast, the force–frequency curve of the cIAP1−/−;mdx solei was essentially identical to the wild-type controls (Fig. 4A), which suggests that there is normalization of calcium homeostasis and responsiveness in the cIAP1−/−;mdx solei. cIAP1+/+;mdx EDLs also had a force–frequency curve shifted toward lower frequencies when compared with wild-type EDL (Fig. 4B). However, the force–frequency responsiveness was not normalized in the cIAP1−/−;mdx EDL muscles. Dystrophic muscles maintain contractile properties by altering the fiber-type composition toward slow/oxidative fibers (33,34). We asked whether the loss of cIAP1 would restore normal proportions of slow fibers to the soleus. To test this, we immunolabeled cross-sections of wild-type, cIAP1+/+;mdx and cIAP1−/−;mdx solei with antibodies to type I (slow) MHC and quantified the percentage of fibers expressing this MHC isoform. The cIAP1+/+;mdx solei had ∼60% more slow fibers than the undamaged wild-type solei (43.5 ± 4 versus 27.4 ± 2%, Supplementary Material, Fig. S2). In contrast, the cIAP1−/−;mdx solei exhibited approximately the same percentage of slow fibers (30.4 ± 2%) as the wild-type muscles. Surprisingly, immunolabeling of EDL sections from cIAP1+/+;mdx and cIAP1−/−;mdx mice identified no slow fibers in either genotype (data not shown). Collectively, these results indicate that the loss of cIAP1 normalizes contractile responsiveness and fiber-type distributions in mdx solei, an outcome that is consistent with our observations of reduced muscle pathology in the cIAP1−/−;mdx mice.
Figure 4.

The loss of cIAP1 restores normal frequency responsiveness to mdx solei. Isometric force maxima of muscles stimulated at 140 Hz were determined from soleus (A) and EDL (B) muscles of wild-type, cIAP1+/+;mdx and cIAP1−/−;mdx mice. Force–frequency curves were obtained from the same soleus (C) and EDL (D) muscles. Data are means ± SEM, n = 5. *P < 0.05 versus wild-type controls. SOL, soleus; EDL, extensor digitorum longus.
Loss of cIAP1 leads to decreased diaphragm membrane permeability and increased exercise endurance in mdx mice
We next asked whether the improvements seen in muscle pathology with the loss of cIAP1 led to functional recovery. Muscle fiber ‘leakiness’ is a hallmark of DMD, and results in the permeation of muscle creatine kinase into the serum (35–37). We assessed the amount of creatine kinase in cIAP1+/+;mdx and cIAP1−/−;mdx sera. The unexercised cIAP1−/−;mdx mice demonstrated a 14% reduction in serum creatine kinase compared with cIAP1+/+;mdx mice (Fig. 5A). This relatively modest but significant decrease was consistent with improvements seen predominantly in slow muscle, such as the soleus. We asked whether this was sufficient to produce a detectable outcome in the animals' exercise endurance. We tested mice on a two-part treadmill-running task: first, they were run uphill at a 15° incline at 10 m/min for 2 min. Subsequently, they were switched to a downhill run (15°) at 10 m/min and their time-to-exhaustion was recorded. The cIAP1−/−;mdx mice ran about three times longer than the cIAP1+/+;mdx controls (Fig. 5B). In spite of the variances in animal run-times, notably 75% (6 of 8) of the cIAP1−/−;mdx mice ran for longer than 10 min, compared with 17% (2 of 12) of the cIAP1+/+;mdx mice. A number of factors, including various endurance and cardiovascular influences, could have accounted for this improvement. The diaphragm is one of the most susceptible muscles to eccentric damage in the mdx mouse, and is also the muscle that most closely mimics the degeneration seen with DMD in humans (38–40). Improved resistance of the diaphragm to damage may explain the increased run-time of the cIAP1−/−;mdx mice. To test this, cIAP1+/+;mdx and cIAP1−/−;mdx mice were treadmill-exercised at 10 m/min for 10 min on a level plane. Subsequently, they were injected with Evans Blue dye, a membrane-impermeable fluorescent dye that labels damaged fibers. Separate cohorts of cIAP1+/+;mdx and cIAP1−/−;mdx animals were injected with Evans Blue, but were not exercised. Twenty-four hours later, the diaphragms were isolated, sectioned and immunolabeled for laminin to identify damaged fibers that contained Evans Blue (which appears red under fluorescence). The cIAP1−/−;mdx diaphragms had 50% fewer Evans Blue-labeled fibers than the cIAP1+/+;mdx controls without exercise, and 75% fewer labeled fibers after exercise (Fig. 5D). Notably, there was no increase in the proportion of Evans Blue-labeled fibers in the exercised cIAP1−/−;mdx diaphragms, compared with unexercised controls. These data suggest that the loss of cIAP1 improves exercise performance at least in part by improving the exercise resilience of the diaphragm.
Figure 5.

The loss of cIAP1 decreases muscle permeability and increases exercise endurance. (A) Creatine kinase levels were measured from cIAP1+/+;mdx and cIAP1−/−;mdx mouse sera. For the latter two bars, mice were first exercised for 10 min by running on a treadmill, without incline. Sera were collected 24 h after exercise. (B) Mice were exercised by running on a treadmill, set at a 15° incline and 10 m/min, for 2min. They were then switched to a 15° decline, and the time-to-exhaustion was recorded. Data plotted are for individual animals tested. Horizontal bar indicates the mean time-to-exhaustion for each group. (C and D) Groups of cIAP1+/+;mdx and cIAP1−/−;mdx mice were exercised on a treadmill at 10 m/min for 10 min, and injected intraperitoneally with 10 mg/kg Evans Blue. Another group of mice was injected with equivalent amounts of Evans Blue without treadmill exercise. Twenty-four hours later, the mice were euthanized, and the diaphragms were sectioned and immunolabeled for laminin to identify muscle fibers (green). Evans Blue-labeled fibers were identified by red fluorescence. The percentage of laminin-labeled fibers co-labeled with Evans Blue was calculated. Values were expressed relative to cIAP1+/+;mdx data. Data are means ± SEM, n = 3–4 for (A), n = 4 for (C and D). *P < 0.05 versus cIAP1+/+;mdx controls. Scale bar represents 250 μm.
Loss of cIAP1 increases myoblast fusion
Although the cIAP1−/−;mdx exhibited protection against dystrophic damage, a mechanism linking cIAP1 loss with muscle development or function remained unclear. We previously demonstrated that the loss of cIAP1 leads to constitutive activation of the noncanonical NF-κB pathway and increases myoblast fusion (17). We asked whether a similar mechanism may account for the protective effect of cIAP1 loss in mdx muscle. To test this, we isolated primary myoblasts from wild-type, cIAP1−/−;mdx and cIAP1+/+;mdx muscle and differentiated the cells into myotubes by switching them to reduced-serum media for 3 days. Myotubes formed from cIAP1+/+;mdx myoblasts were normal and indistinguishable from wild-type (Fig. 6A), which is consistent with previous comparisons of wild-type and mdx primary myotubes (41,42). In contrast, myoblast fusion was enhanced in cIAP1−/−;mdx myoblasts, producing myotubes that were 50% larger than either wild-type or cIAP1+/+;mdx cells (Fig. 6A and B). Since cIAP1 is known to regulate both canonical and noncanonical NF-κB signaling, we asked whether the loss of cIAP1 affected the activation of either pathway. Canonical and noncanonical NF-κB activity decreased over the course of differentiation in wild-type myoblasts, and was virtually identical in cIAP1+/+;mdx cells (Fig. 6C and D). In contrast, activity in both pathways was elevated in differentiating cIAP1−/−;mdx cells. To confirm these observations, myoblasts differentiated for between 0 and 3 days were analyzed by western blotting for the phosphorylation of p65, which is an indicator of canonical pathway activation, and for the processing of p100 to p52, which is indicative of noncanonical pathway activation. p65 phosphorylation was elevated in cIAP1−/−;mdx myoblasts compared with wild-type and cIAP1+/+;mdx cells (Fig. 6E). Levels of p52 were also greater in cIAP1−/−;mdx myoblasts compared with the wild-type and cIAP1+/+;mdx cells, though p65 phosphorylation and p52 expression declined over the course of differentiation in every instance. Levels of MHC were reduced in differentiating cIAP1−/−;mdx cells, which suggests that the loss of cIAP1 delays myoblast differentiation. Nevertheless, the timely formation of hypernucleated myotubes from cIAP1−/−;mdx myoblasts suggested that the delay was insufficient to attenuate myogenesis. To confirm these observations in vivo, we measured the cross-sectional areas of regenerating (centronucleated) muscle fibers in cIAP1+/+;mdx and cIAP1−/−;mdx solei. There was a distinct shift in fiber size distributions toward large fibers in the cIAP1−/−;mdx mice (Fig. 6F), though overall muscle mass was unaffected (Supplementary Material, Fig. S3). Notably, 82% of the cIAP1−/−;mdx fibers were >500 µm2 in cross-sectional area, compared with 62% of the cIAP1+/+;mdx controls. Taken together, these results suggest that the loss of cIAP1 leads to increased myogenesis and myoblast fusion, both in vitro and in vivo, in spite of a delay in differentiation observed in cIAP1−/−;mdx myoblasts.
Figure 6.

The loss of cIAP1 leads to increased myoblast fusion. (A) Primary myoblasts of the indicated genotypes were differentiated by serum withdrawal for 3 days. Photomicrographs were taken of the resulting myotubes on a Zeiss PrimoVert microscope. Scale bar represents 50 µm. (B) Myotube diameters were measured from photomicrographs captured as in (A). Bars represent mean ± SEM, n = 3 independent experiments, approximately 200 myotubes per condition. (C and D) Myoblasts were differentiated for between 0 and 3 days; at each time point, cells were harvested for NF-κB activity by DNA-binding ELISA. Antibodies specific to p65 (C) or p100 (D) were used to detect DNA-binding complexes specific to the canonical or noncanonical NF-κB pathways, respectively. Values are relative to wild-type samples at 0 days of differentiation, and are shown as mean ± SEM, n = 3 independent experiments. (E) Myoblasts differentiated as in (C) and (D) were analyzed by western blotting. Diff., days of differentiation. (F) Cross-sections of solei from cIAP1+/+;mdx and cIAP1−/−;mdx mice were stained with H&E to identify centronucleated fibers. These fibers were traced using the ImageJ software to obtain cross-sectional areas. Values represented are means ± SEM of four solei per genotype.
DISCUSSION
Our results demonstrate for the first time that the loss of cIAP1, an essential regulator of NF-κB signaling, can ameliorate muscle pathology and improve muscle function in the mdx mouse. These effects are evidenced by reduced fiber damage, reduced muscle inflammation, improved muscle contractile properties, normalized fiber-type distributions, reduced serum creatine kinase, increased exercise endurance and reduced diaphragm permeability and damage following exercise. Histologically and functionally, the improvements were manifested particularly in the soleus and diaphragm muscles, but not in the EDL. Combined with the reductions in serum creatine kinase that indicate decreased muscle damage in the animals as a whole, the evidence points to a positive effect of the elimination of cIAP1 on muscle pathology in DMD.
A consistent finding of our study was that, at multiple levels of analysis, the loss of cIAP1 protected soleus but not EDL muscle from dystrophic damage. This disparity was evident in the differences in centronucleation, necrotic damage, macrophage infiltration and physiological responsiveness between both muscles. The reason for these differences may lie in the different physical demands placed on fast-twitch fibers, which predominate in the EDL, versus slow-twitch fibers that are abundant in the soleus. The EDL produces considerably more force than the soleus (∼45 N/cm2 peak isometric force in the wild-type EDL, versus ∼17 N/cm2 in the soleus, Fig. 4A and B), rendering fast-twitch muscles more susceptible to activity-induced damage. This is consistent with previous observations that fast fibers are particularly prone to damage associated with DMD (43,44). It is thus likely that the protection accorded by the loss of cIAP1 applies only to ranges of force output routinely exceeded by muscles such as the EDL. In spite of this restriction in scope, the loss of cIAP1 is nevertheless sufficient to produce detectable functional improvements in exercise performance in the mdx mice, in part through increased damage resistance of the diaphragm. Our observations of decreased muscle damage and decreased centronucleation, in response to the loss of an NF-κB regulator, are seemingly in contrast to a previous study by Acharyya et al. (5). This study indicated that the attenuation of canonical NF-κB signaling through p65 promotes regeneration by increasing muscle centronucleation. However, the methodological differences between that study and the present one can be reconciled in the outcomes: although in both studies it is possible to reduce inflammation by blunting canonical NF-κB signaling (exemplified in the current study by reduced necrosis, Fig. 2; and reduced macrophage infiltration, Fig. 3), the additional effect of increased noncanonical NF-κB activation in the cIAP1−/−;mdx mice is to reduce muscle injury and consequently reduce fiber centronucleation. The cIAP1 knockout thus provides information about the differential roles of canonical and noncanonical NF-κB signaling in muscle regeneration and pathology.
The reductions in muscle damage we observed in the cIAP1−/−;mdx mice may reflect the convergence of multiple distinct mechanisms. First, the loss of cIAP1 activates the noncanonical NF-κB pathway to increase myoblast fusion (17), producing larger muscle fibers that may be intrinsically more resistant to damage (45). Recent reports suggest that the noncanonical pathway promotes an oxidative muscle fate (15,16), which may also confer a protective role against DMD (44,46). Second, we and others have shown that cytokine-induced activation of canonical NF-κB signaling is blunted in the absence of cIAP1 (7,8,47). The cIAP1−/−;mdx muscle is thus protected from the effects of chronic NF-κB signaling that normally result from the heavy cytokine load present in regenerating muscle. The loss of cIAP1 also triggers ligand-autonomous activation of canonical NF-κB activity, which in itself can aggravate muscle pathology in mdx mice (5,48). However, our data show that the level of canonical activation triggered by the loss of cIAP1 is insufficient to impair myogenesis either in vitro or in vivo (Fig. 6). This suggests that, following the loss of cIAP1, the pro-myogenic effects resulting from the activation of the noncanonical NF-κB pathway outweigh any consequences of canonical pathway activation, the latter of which can aggravate mdx muscle pathology (5). Finally, a number of other major signaling pathways are dysregulated in mdx muscle, particularly mitogen-activated protein (MAP) kinases (49–51). A recent study showed that cIAP1 and cIAP2 are essential for MAP kinase signaling in response to cytokine triggers (10). The loss of cIAP1 thus provides an avenue by which signaling pathways that aggravate mdx pathology can be attenuated to reduce degeneration and promote a pro-regenerative environment for muscle.
Evidence highlighting the importance of the inflammatory response in the pathology of DMD may bear critically on the phenomena we observed in this study. A caspase-11 mutation was recently identified in the ES cells used to derive the cIAP1−/− mice (52,53). Caspase-11 is an inducible regulator of IL-1β production and caspase-1 activation from a variety of bacterial pathogens and toxins (52,54,55). The caspase-11 mutation identified in animals and ES cells of the 129 mouse strain is a five base-pair deletion in the splice acceptor region of exon 7, which produces a truncated and nonfunctional transcript. The cIAP1−/− mice were derived from 129 ES cells and, in spite of extensive backcrossing to the C57BL/6 strain (which retains the wild-type caspase-11), the mutation remains in cIAP1−/− mice currently in use. It is thus possible that this mutation, which is also present in the cIAP1−/−;mdx mice (data not shown), may contribute to the attenuated inflammatory response observed in the soleus and EDL muscles of these mice. However, evidence suggests that the resistance to muscle degeneration we observed in these mice is specific to the loss of cIAP1. All reports to date on the involvement of caspase-11 in the inflammatory response indicate that it activates the inflammasome in response to a limited subset of stimuli (52,56), particularly Gram-negative bacteria (55,57). To our knowledge there is no evidence of caspase-11 involvement in the damage-associated inflammatory response. Furthermore, the cIAP1−/−;mdx EDL muscle, which shows no rescue from mdx pathology, also exhibits similar degrees of pro-inflammatory macrophage recruitment (as measured by CD68 expression) to cIAP1+/+;mdx muscle. This suggests that macrophage recruitment is not intrinsically impaired by the caspase-11 mutation. In contrast, the cIAP1−/−;mdx soleus, which shows reduced mdx-associated pathology, reduced CD68 expression and increased CD163 (anti-inflammatory macrophage) expression, suggesting that macrophage recruitment is associated specifically with the extent of muscle injury. We also observed reduced expression of IL6, TNF and IL10 in both soleus and EDL muscles of cIAP1−/−;mdx mice (Fig. 3). These observations are also likely due to the loss of cIAP1, since we previously showed that both cIAP1 and cIAP2 are essential for cytokine production by macrophages (58). Collectively, the results show that cIAP1 loss can function at multiple levels to reduce mdx pathology.
A further finding of this study was that the cIAP1−/−;mdx mice exhibited increased endurance to treadmill exercise. Even accounting for the outliers, the cIAP1−/−;mdx animals' time-to-exhaustion was three times longer than those of the cIAP1+/+;mdx controls. One of the hallmark symptoms of DMD is chronic and progressive impairment of respiratory function (59). The diaphragm, as the muscle most responsible for this function, is also the most reliable indicator of damage or recovery in the mdx mouse (38,60,61). On equivalent treadmill exercise tasks, the cIAP1−/−;mdx diaphragms displayed less exercise-induced damage than the cIAP1+/+;mdx counterparts, suggesting that improved diaphragm resilience partially explained the differences in performance between the groups. The reduced diaphragm damage, which was measured by Evans Blue permeability, is also an indicator of reduced muscle ‘leakiness’. This observation is consistent with the decreased serum creatine kinase in the cIAP1−/−;mdx mice, as well as the normalized force–frequency curves in the cIAP1−/−;mdx solei (indicative of reduced calcium influx to the muscle). Similar phenotypes have been observed in previous studies, in which an increase in fiber size was associated with reduced mdx pathology (45,62,63). These observations indicate that muscle resilience can be augmented either by increasing muscle fiber size during myogenesis (by increasing myoblast fusion) or through hypertrophy of extant fibers.
In summary, we observed that the loss of cIAP1 was associated with decreased soleus damage and increased exercise performance of mdx mice. This opens the way for therapeutic approaches using pharmacological inhibitors of cIAP1. The cIAP1/2 small-molecule antagonists, known as Smac-mimetic compounds, can be administered orally and are well tolerated (64). Their use in mdx animals that normally express cIAP1 would provide a clearer understanding of the role of this protein in the pathology of muscular dystrophy.
MATERIALS AND METHODS
Experimental mice
All experiments were reviewed and approved by the Animal Care Committee of the University of Ottawa. Mice were fed standard laboratory chow ad libitum and housed according to the guidelines of the Canadian Council for Animal Care. Female mdx mice homozygous for the point mutation in the dystrophin gene were mated with male cIAP1−/− mice described elsewhere (19). They were further backcrossed to obtain double-mutant cIAP1−/−;mdx mice, as well as cIAP1+/+;mdx controls. The mdx mice were derived from a C57BL/10 background, and the cIAP1−/− mice were on a C57BL/6 background. Male mice of age 10–12 weeks were used for the experiments described herein. Age-matched C57BL/6 males were used as dystrophin-containing controls where indicated. Genotypes of the mice were determined by PCR screening of tail snip or ear notch DNA for the cIAP1 deletion, and by sequencing the locus in exon 23 of the dystrophin gene containing the nonsense mutation (3). Primers used for sequencing were 5′-CCTTCTTCTTGATATGAATGAAACT (forward) and 5′-CACCAACTGGGAGGAAAGT (reverse).
Treadmill running
To determine time-to-exhaustion on a treadmill-running task, cIAP1+/+;mdx and cIAP1−/−;mdx mice were run for 2 min at 10 m/min on a treadmill with a 15° incline. They were subsequently run at a 15° decline until they could no longer maintain this speed, even with gentle prodding. For the assessment of diaphragm resilience, four mice of each genotype were run for 10 min at 10 m/min on a treadmill with a 0° incline. A second group of four mice remained unexercised; all animals were subsequently injected intraperitoneally with 10 mg/kg of Evans Blue (Sigma-Aldrich, Oakville, Ontario, Canada) in sterile PBS. Diaphragms were excised 24 h after injection for histological examination.
Force measurement
Force–frequency relationships were measured in soleus and EDL muscles. Mice were anesthetized with a single intraperitoneal injection of 2.2 mg of ketamine, 0.44 mg of xylazine and 0.22 mg of acepromazine per 10 g of body mass. Soleus and EDL were excised and tendons tied with surgical silk (#6). One tendon was attached to the force transducer, whereas the other attached to fixed metal pin. Muscles were continuously immersed in a previously described physiological saline solution (65) and maintained at 25°C. Muscle length (Le) was adjusted to obtain maximum isometric tetanic force elicited every 5 min with 200 ms trains of supramaximal 10 V, 0.3 ms square pulses at 200 Hz. Stimulation pulses were delivered using platinum wires on opposite side and near the middle portion of muscles. The wires were connected to a stimulation isolation unit (SIU5, Grass Instruments, USA), which in turn was connected to a S88 Grass stimulator. Force was monitored with a Cambridge ergometer (model 300, Aurora Scientific, Inc., Aurora, Ontario, Canada) or with a Kulite force transducer model BD100 (Kulite Semiconductor Products, Inc., Leonia, NJ, USA). After muscles had equilibrated for 30 min, forces were measured while stimulation frequency was increased from 1 to 140 Hz.
Quantitative RT–PCR
Total RNA was extracted from soleus and EDL samples, using the Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA, USA), following manufacturer's instructions. One-step real-time SYBR Green RT–PCR reactions were carried out with each sample in triplicate, using 100 ng of RNA per reaction. RT–PCR reactions were performed in a 50 μl of reaction volume, using the QuantiTect RT–PCR (Qiagen). GAPDH was used as the housekeeping gene. Primers used were as listed in Table 1.
Table 1.
List of primers used for qPCR experiments
| Primer | Sequence | Primer length | Product size |
|---|---|---|---|
| CD68 forward | TTCCAAGAGAAGGCAAAG | 18 | 271 |
| CD68 reverse | GCTGGTGTGAACTGTGAC | 18 | |
| CD163 forward | TTG CTA TTG GTT TGC TTG TTC | 21 | 76 |
| CD163 reverse | GCT TGC CTG CTC TCT ATC G | 19 | |
| IL6 forward | GAG TTG TGC AAT GGC AAT TCT G | 22 | 50 |
| IL6 reverse | GCA AGT GCA TCA TCG TTG TTC AT | 23 | |
| IL10 forward | GCA CTA CCA AAG CCA CAA GG | 20 | 83 |
| IL10 reverse | TAA GAG CAG GCA GCA TAG CA | 20 | |
| TNF forward | TAT GGC TCA GGG TCC AAC TC | 20 | 75 |
| TNF reverse | GGT CAC TGT CCC AGC ATC TT | 20 | |
| GAPDH forward | CAT GTT CCA GTA TGA CTC CAC TC | 23 | 136 |
| GAPDH reverse | GGC CTC ACC CCA TTT GAT GT | 20 |
Creatine kinase assay
Serum creatine kinase was performed using an assay kit (BioAssay Systems, Hayward, CA, USA), following manufacturer's instructions. Blood was collected by cardiac puncture from animals anesthetized with pentobarbital. Immediately after bleeding, animals were killed by cervical dislocation. Sera were separated by centrifugation at 300g for 5 min and were stored at −70°C until use.
Histology and immunohistochemistry
Ten-micron sections were cut on a cryostat from fresh frozen samples embedded in OCT (Tissue Tek II), and frozen in isopentane precooled in liquid nitrogen. Hematoxylin and eosin (H&E) staining was used to identify centronucleated fibers and regions of degeneration. For immunohistochemistry, antibodies used were rabbit anti-laminin (catalog #L9393, Sigma), mouse anti-dystrophin (MANDYS1, Developmental Studies Hybridoma Bank, IA, USA), mouse anti-type I myosin (A4.840, Developmental Studies Hybridoma Bank) and mouse anti-embryonic myosin (F1.652, Developmental Studies Hybridoma Bank). Slide-mounted cross-sections were incubated overnight with primary antibodies diluted in PBS containing 0.3% Triton X-100 (Sigma) and 0.5% bovine serum albumin (Invitrogen). After washing, cover slips were incubated with the appropriate secondary antibody, diluted in PBS with Hoechst 33258 (Sigma) as a nuclear counterstain and incubated for 1 h at room temperature. This was followed by washing, mounting of cover slips and visualization. Sections were photographed using a Zeiss Axiophot microscope equipped with a digital camera and the Northern Eclipse image analysis software, version 6.0 (Empix Imaging, Mississauga, Ontario, Canada). Areas of degeneration were measured in Northern Eclipse from the entire area of each analyzed muscle section. For the soleus and EDL muscles, data were collected from all fibers in a cross-section; for the diaphragm, 400–500 fibers from randomly selected fields were counted for the indicated analyses.
Cell culture
Primary myoblasts were prepared from 3–6-week-old female mice killed by cervical dislocation, and lower limb muscles were carefully dissected away from the bone. Muscle samples were digested with collagenase-dispase solution (Roche) for 2–3 h, before being triturated, washed by centrifugation twice at 300g for 5 min and resuspended in DMEM (ATCC) containing 10% equine serum and 5 ng/ml basic FGF (Peprotech). Fibroblasts were separated out by ‘preplating’ on a regular tissue culture coated-plate for 1 h before the cell suspension was seeded into a six-well plate coated with Matrigel (BD Biosciences). Two days later, the myoblasts were switched to and maintained in a growth medium containing 20 FBS, 10% equine serum, 10 ng/ml bFGF and 2 ng/ml HGF (Peprotech), and also on Matrigel.
NF-κB activity assay
Binding of p65- and p52-containing complexes to DNA was measured using an NF-κB DNA binding ELISA (Active Motif, Carlsbad, CA, USA), following the manufacturer's instructions. Analysis was performed using a FLUOstar Optima fluorescence microplate reader (BMG Labtech, Offenburg, Germany).
Western blotting
Western blotting was performed as described previously (7). Rabbit polyclonal antibodies to p65, phospho-p65 (S536) and p100 were obtained from Cell Signaling Technologies. Antibodies to total MHC were obtained from the Developmental Studies Hybridoma Bank, and antibodies to cIAP1 were from Enzo Life Sciences.
Statistical analyses
All quantitative data are represented as mean ± SEM. Statistical analyses were performed in GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA), using a t-test with significance set at P < 0.05. Split-plot ANOVA was used to determine statistical differences for the force–frequency curves. The mouse and muscle treatments were part of the whole plot because they were independent from one another, whereas the frequency treatment was in the split-plot because forces at all frequencies were obtained from the same muscles. The ANOVA calculations were made using version 9.0GLM (general linear model) procedures of the Statistical Analysis Software (SAS Institute, Inc., Cary, NC, USA). Least significant difference was used to locate the significant differences (66).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by funds from the Canadian Institutes of Health Research, the Howard Hughes Medical Institute (HHMI), and the Muscular Dystrophy Association USA.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Drs Michael Rudnicki, Luc Sabourin, Cathy Tsilfidis (all at the Ottawa Hospital Research Institute) and Ilona Skerjanc (University of Ottawa) for their helpful advice and support. The authors also thank Dr. Tak Mak (University of Toronto) for providing the cIAP1−/− mice. R. G. K. is an HHMI International Research Scholar, a Fellow of the Royal Society of Canada and a Distinguished Professor of the University of Ottawa.
Conflict of interest statement. None declared.
REFERENCES
- 1.Hoffman E.P., Brown R.H., Jr, Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Ryder-Cook A.S., Sicinski P., Thomas K., Davies K.E., Worton R.G., Barnard E.A., Darlison M.G., Barnard P.J. Localization of the mdx mutation within the mouse dystrophin gene. EMBO J. 1988;7:3017–3021. doi: 10.1002/j.1460-2075.1988.tb03165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sicinski P., Geng Y., Ryder-Cook A.S., Barnard E.A., Darlison M.G., Barnard P.J. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 4.Messina S., Bitto A., Aguennouz M., Minutoli L., Monici M.C., Altavilla D., Squadrito F., Vita G. Nuclear factor kappa-B blockade reduces skeletal muscle degeneration and enhances muscle function in mdx mice. Exp. Neurol. 2006;198:234–241. doi: 10.1016/j.expneurol.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 5.Acharyya S., Villalta S.A., Bakkar N., Bupha-Intr T., Janssen P.M., Carathers M., Li Z.W., Beg A.A., Ghosh S., Sahenk Z., et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Invest. 2007;117:889–901. doi: 10.1172/JCI30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Acharyya S., Sharma S.M., Cheng A.S., Ladner K.J., He W., Kline W., Wang H., Ostrowski M.C., Huang T.H., Guttridge D.C. TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: implications in Duchenne muscular dystrophy. PloS One. 2010;5:e12479. doi: 10.1371/journal.pone.0012479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahoney D.J., Cheung H.H., Mrad R.L., Plenchette S., Simard C., Enwere E., Arora V., Mak T.W., Lacasse E.C., Waring J., et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc. Natl Acad. Sci. USA. 2008;105:11778–11783. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zarnegar B.J., Wang Y., Mahoney D.J., Dempsey P.W., Cheung H.H., He J., Shiba T., Yang X., Yeh W.C., Mak T.W., et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008;9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vince J.E., Chau D., Callus B., Wong W.W., Hawkins C.J., Schneider P., McKinlay M., Benetatos C.A., Condon S.M., Chunduru S.K., et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J. Cell Biol. 2008;182:171–184. doi: 10.1083/jcb.200801010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Varfolomeev E., Goncharov T., Maecker H., Zobel K., Komuves L.G., Deshayes K., Vucic D. Cellular inhibitors of apoptosis are global regulators of NF-kappaB and MAPK activation by members of the TNF family of receptors. Sci. Signal. 2012;5:ra22. doi: 10.1126/scisignal.2001878. [DOI] [PubMed] [Google Scholar]
- 11.Vallabhapurapu S., Matsuzawa A., Zhang W., Tseng P.H., Keats J.J., Wang H., Vignali D.A., Bergsagel P.L., Karin M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat. Immunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonizzi G., Bebien M., Otero D.C., Johnson-Vroom K.E., Cao Y., Vu D., Jegga A.G., Aronow B.J., Ghosh G., Rickert R.C., et al. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. EMBO J. 2004;23:4202–4210. doi: 10.1038/sj.emboj.7600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smale S.T. Dimer-specific regulatory mechanisms within the NF-kappaB family of transcription factors. Immunol. Rev. 2012;246:193–204. doi: 10.1111/j.1600-065X.2011.01091.x. [DOI] [PubMed] [Google Scholar]
- 14.Arany Z., Lebrasseur N., Morris C., Smith E., Yang W., Ma Y., Chin S., Spiegelman B.M. The transcriptional coactivator PGC-1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab. 2007;5:35–46. doi: 10.1016/j.cmet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Bakkar N., Wang J., Ladner K.J., Wang H., Dahlman J.M., Carathers M., Acharyya S., Rudnicki M.A., Hollenbach A.D., Guttridge D.C. IKK/NF-kappaB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. J. Cell Biol. 2008;180:787–802. doi: 10.1083/jcb.200707179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bakkar N., Ladner K., Canan B.D., Liyanarachchi S., Bal N.C., Pant M., Periasamy M., Li Q., Janssen P.M., Guttridge D.C. IKKalpha and alternative NF-kappaB regulate PGC-1beta to promote oxidative muscle metabolism. J. Cell Biol. 2012;196:497–511. doi: 10.1083/jcb.201108118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Enwere E.K., Holbrook J., Lejmi-Mrad R., Vineham J., Timusk K., Sivaraj B., Isaac M., Uehling D., Al-awar R., LaCasse E., et al. TWEAK and cIAP1 regulate myoblast fusion through the noncanonical NF-kappaB signaling pathway. Sci. Signal. 2012;5:ra75. doi: 10.1126/scisignal.2003086. [DOI] [PubMed] [Google Scholar]
- 18.Saitoh T., Nakayama M., Nakano H., Yagita H., Yamamoto N., Yamaoka S. TWEAK induces NF-kappaB2 p100 processing and long lasting NF-kappaB activation. J. Biol. Chem. 2003;278:36005–36012. doi: 10.1074/jbc.M304266200. [DOI] [PubMed] [Google Scholar]
- 19.Conze D.B., Albert L., Ferrick D.A., Goeddel D.V., Yeh W.C., Mak T., Ashwell J.D. Posttranscriptional downregulation of c-IAP2 by the ubiquitin protein ligase c-IAP1 in vivo. Mol. Cell. Biol. 2005;25:3348–3356. doi: 10.1128/MCB.25.8.3348-3356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng L., Gu X., Sanderson J.E., Wang X., Lee K., Yao X., Liu H., Cheung W.L., Li M. A new function of a previously isolated compound that stimulates activation and differentiation of myogenic precursor cells leading to efficient myofiber regeneration and muscle repair. Int. J. Biochem. Cell Biol. 2006;38:1123–1133. doi: 10.1016/j.biocel.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 21.Stupka N., Plant D.R., Schertzer J.D., Emerson T.M., Bassel-Duby R., Olson E.N., Lynch G.S. Activated calcineurin ameliorates contraction-induced injury to skeletal muscles of mdx dystrophic mice. J. Physiol. 2006;575:645–656. doi: 10.1113/jphysiol.2006.108472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Villalta S.A., Nguyen H.X., Deng B., Gotoh T., Tidball J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2009;18:482–496. doi: 10.1093/hmg/ddn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chihara T., Hashimoto M., Osman A., Hiyoshi-Yoshidomi Y., Suzu I., Chutiwitoonchai N., Hiyoshi M., Okada S., Suzu S. HIV-1 proteins preferentially activate anti-inflammatory M2-type macrophages. J. Immunol. 2012;188:3620–3627. doi: 10.4049/jimmunol.1101593. [DOI] [PubMed] [Google Scholar]
- 24.Sacco P., Jones D.A., Dick J.R., Vrbova G. Contractile properties and susceptibility to exercise-induced damage of normal and mdx mouse tibialis anterior muscle. Clin. Sci. (Lond.) 1992;82:227–236. doi: 10.1042/cs0820227. [DOI] [PubMed] [Google Scholar]
- 25.Lynch G.S. Role of contraction-induced injury in the mechanisms of muscle damage in muscular dystrophy. Clin. Exp. Pharmacol. Physiol. 2004;31:557–561. doi: 10.1111/j.1440-1681.2004.04026.x. [DOI] [PubMed] [Google Scholar]
- 26.Chan S., Head S.I., Morley J.W. Branched fibers in dystrophic mdx muscle are associated with a loss of force following lengthening contractions. Am. J. Physiol. Cell Physiol. 2007;293:C985–C992. doi: 10.1152/ajpcell.00128.2007. [DOI] [PubMed] [Google Scholar]
- 27.Wang X., Weisleder N., Collet C., Zhou J., Chu Y., Hirata Y., Zhao X., Pan Z., Brotto M., Cheng H., et al. Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat. Cell Biol. 2005;7:525–530. doi: 10.1038/ncb1254. [DOI] [PubMed] [Google Scholar]
- 28.Constantin B., Sebille S., Cognard C. New insights in the regulation of calcium transfers by muscle dystrophin-based cytoskeleton: implications in DMD. J. Muscle Res. Cell Motil. 2006;27:375–386. doi: 10.1007/s10974-006-9085-2. [DOI] [PubMed] [Google Scholar]
- 29.Turner P.R., Westwood T., Regen C.M., Steinhardt R.A. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature. 1988;335:735–738. doi: 10.1038/335735a0. [DOI] [PubMed] [Google Scholar]
- 30.Coulton G.R., Curtin N.A., Morgan J.E., Partridge T.A. The mdx mouse skeletal muscle myopathy: II. Contractile properties. Neuropathol. Appl. Neurobiol. 1988;14:299–314. doi: 10.1111/j.1365-2990.1988.tb00890.x. [DOI] [PubMed] [Google Scholar]
- 31.Consolino C.M., Brooks S.V. Susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of mdx mice. J. Appl. Physiol. 2004;96:633–638. doi: 10.1152/japplphysiol.00587.2003. [DOI] [PubMed] [Google Scholar]
- 32.Gumerson J.D., Kabaeva Z.T., Davis C.S., Faulkner J.A., Michele D.E. Soleus muscle in glycosylation-deficient muscular dystrophy is protected from contraction-induced injury. Am. J. Physiol. Cell Physiol. 2010;299:C1430–C1440. doi: 10.1152/ajpcell.00192.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spassov A., Gredes T., Gedrange T., Lucke S., Morgenstern S., Pavlovic D., Kunert-Keil C. Differential expression of myosin heavy chain isoforms in the masticatory muscles of dystrophin-deficient mice. Eur. J. Orthod. 2011;33:613–619. doi: 10.1093/ejo/cjq113. [DOI] [PubMed] [Google Scholar]
- 34.Joanne P., Hourde C., Ochala J., Cauderan Y., Medja F., Vignaud A., Mouisel E., Hadj-Said W., Arandel L., Garcia L., et al. Impaired adaptive response to mechanical overloading in dystrophic skeletal muscle. PloS One. 2012;7:e35346. doi: 10.1371/journal.pone.0035346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quinlan J.G., Johnson S.R., Samaha F.J. Dantrolene normalizes serum creatine kinase in MDX mice. Muscle Nerve. 1990;13:268–269. doi: 10.1002/mus.880130316. [DOI] [PubMed] [Google Scholar]
- 36.Rooney J.E., Gurpur P.B., Burkin D.J. Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy. Proc. Natl Acad. Sci. USA. 2009;106:7991–7996. doi: 10.1073/pnas.0811599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spurney C.F., Gordish-Dressman H., Guerron A.D., Sali A., Pandey G.S., Rawat R., Van Der Meulen J.H., Cha H.J., Pistilli E.E., Partridge T.A., et al. Preclinical drug trials in the mdx mouse: assessment of reliable and sensitive outcome measures. Muscle Nerve. 2009;39:591–602. doi: 10.1002/mus.21211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stedman H.H., Sweeney H.L., Shrager J.B., Maguire H.C., Panettieri R.A., Petrof B., Narusawa M., Leferovich J.M., Sladky J.T., Kelly A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 39.Matecki S., Guibinga G.H., Petrof B.J. Regenerative capacity of the dystrophic (mdx) diaphragm after induced injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004;287:R961–R968. doi: 10.1152/ajpregu.00146.2004. [DOI] [PubMed] [Google Scholar]
- 40.Hnia K., Tuffery-Giraud S., Vermaelen M., Hugon G., Chazalette D., Masmoudi A., Rivier F., Mornet D. Pathological pattern of Mdx mice diaphragm correlates with gradual expression of the short utrophin isoform Up71. Biochim. Biophys. Acta. 2006;1762:362–372. doi: 10.1016/j.bbadis.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rando T.A., Disatnik M.H., Yu Y., Franco A. Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul. Disord. 1998;8:14–21. doi: 10.1016/s0960-8966(97)00124-7. [DOI] [PubMed] [Google Scholar]
- 42.Sandri M., Massimino M.L., Cantini M., Giurisato E., Sandri C., Arslan P., Carraro U. Dystrophin deficient myotubes undergo apoptosis in mouse primary muscle cell culture after DNA damage. Neurosci. Lett. 1998;252:123–126. doi: 10.1016/s0304-3940(98)00563-1. [DOI] [PubMed] [Google Scholar]
- 43.Webster C., Silberstein L., Hays A.P., Blau H.M. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell. 1988;52:503–513. doi: 10.1016/0092-8674(88)90463-1. [DOI] [PubMed] [Google Scholar]
- 44.Selsby J.T., Morine K.J., Pendrak K., Barton E.R., Sweeney H.L. Rescue of dystrophic skeletal muscle by PGC-1alpha involves a fast to slow fiber type shift in the mdx mouse. PloS One. 2012;7:e30063. doi: 10.1371/journal.pone.0030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gehrig S.M., Koopman R., Naim T., Tjoakarfa C., Lynch G.S. Making fast-twitch dystrophic muscles bigger protects them from contraction injury and attenuates the dystrophic pathology. Am. J. Pathol. 2010;176:29–33. doi: 10.2353/ajpath.2010.090760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ljubicic V., Miura P., Burt M., Boudreault L., Khogali S., Lunde J.A., Renaud J.M., Jasmin B.J. Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum. Mol. Genet. 2011;20:3478–3493. doi: 10.1093/hmg/ddr265. [DOI] [PubMed] [Google Scholar]
- 47.Cheung H.H., Beug S.T., St Jean M., Brewster A., Kelly N.L., Wang S., Korneluk R.G. Smac mimetic compounds potentiate interleukin-1beta-mediated cell death. J. Biol. Chem. 2010;285:40612–40623. doi: 10.1074/jbc.M110.183616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reay D.P., Yang M., Watchko J.F., Daood M., O'Day T.L., Rehman K.K., Guttridge D.C., Robbins P.D., Clemens P.R. Systemic delivery of NEMO binding domain/IKKgamma inhibitory peptide to young mdx mice improves dystrophic skeletal muscle histopathology. Neurobiol. Dis. 2011;43:598–608. doi: 10.1016/j.nbd.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura A., Yoshida K., Takeda S., Dohi N., Ikeda S. Progression of dystrophic features and activation of mitogen-activated protein kinases and calcineurin by physical exercise, in hearts of mdx mice. FEBS Lett. 2002;520:18–24. doi: 10.1016/s0014-5793(02)02739-4. [DOI] [PubMed] [Google Scholar]
- 50.Hnia K., Hugon G., Rivier F., Masmoudi A., Mercier J., Mornet D. Modulation of p38 mitogen-activated protein kinase cascade and metalloproteinase activity in diaphragm muscle in response to free radical scavenger administration in dystrophin-deficient Mdx mice. Am. J. Pathol. 2007;170:633–643. doi: 10.2353/ajpath.2007.060344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi H., Boadu E., Mercan F., Le A.M., Flach R.J., Zhang L., Tyner K.J., Olwin B.B., Bennett A.M. MAP kinase phosphatase-1 deficiency impairs skeletal muscle regeneration and exacerbates muscular dystrophy. FASEB J. 2010;24:2985–2997. doi: 10.1096/fj.09-150045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kayagaki N., Warming S., Lamkanfi M., Vande Walle L., Louie S., Dong J., Newton K., Qu Y., Liu J., Heldens S., et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 53.Kenneth N.S., Younger J.M., Hughes E.D., Marcotte D., Barker P.A., Saunders T.L., Duckett C.S. An inactivating caspase 11 passenger mutation originating from the 129 murine strain in mice targeted for c-IAP1. Biochem. J. 2012;443:355–359. doi: 10.1042/BJ20120249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akhter A., Caution K., Abu Khweek A., Tazi M., Abdulrahman B.A., Abdelaziz D.H., Voss O.H., Doseff A.I., Hassan H., Azad A.K., et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity. 2012;37:35–47. doi: 10.1016/j.immuni.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rathinam V.A., Vanaja S.K., Waggoner L., Sokolovska A., Becker C., Stuart L.M., Leong J.M., Fitzgerald K.A. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by Gram-negative bacteria. Cell. 2012;150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mueller N.J., Wilkinson R.A., Fishman J.A. Listeria monocytogenes infection in caspase-11-deficient mice. Infect. Immun. 2002;70:2657–2664. doi: 10.1128/IAI.70.5.2657-2664.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Broz P., Ruby T., Belhocine K., Bouley D.M., Kayagaki N., Dixit V.M., Monack D.M. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. 2012;490:288–291. doi: 10.1038/nature11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bertrand M.J., Doiron K., Labbe K., Korneluk R.G., Barker P.A., Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. 2009;30:789–801. doi: 10.1016/j.immuni.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 59.Toussaint M., Chatwin M., Soudon P. Mechanical ventilation in Duchenne patients with chronic respiratory insufficiency: clinical implications of 20 years published experience. Chronic Respir. Dis. 2007;4:167–177. doi: 10.1177/1479972307080697. [DOI] [PubMed] [Google Scholar]
- 60.Dupont-Versteegden E.E., McCarter R.J. Differential expression of muscular dystrophy in diaphragm versus hindlimb muscles of mdx mice. Muscle Nerve. 1992;15:1105–1110. doi: 10.1002/mus.880151008. [DOI] [PubMed] [Google Scholar]
- 61.Louboutin J.P., Fichter-Gagnepain V., Thaon E., Fardeau M. Morphometric analysis of mdx diaphragm muscle fibres. Comparison with hindlimb muscles. Neuromuscul. Disord. 1993;3:463–469. doi: 10.1016/0960-8966(93)90098-5. [DOI] [PubMed] [Google Scholar]
- 62.Bogdanovich S., Krag T.O., Barton E.R., Morris L.D., Whittemore L.A., Ahima R.S., Khurana T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 63.Minetti G.C., Colussi C., Adami R., Serra C., Mozzetta C., Parente V., Fortuni S., Straino S., Sampaolesi M., Di Padova M., et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat. Med. 2006;12:1147–1150. doi: 10.1038/nm1479. [DOI] [PubMed] [Google Scholar]
- 64.Weisberg E., Ray A., Barrett R., Nelson E., Christie A.L., Porter D., Straub C., Zawel L., Daley J.F., Lazo-Kallanian S., et al. Smac mimetics: implications for enhancement of targeted therapies in leukemia. Leukemia. 2010;24:2100–2109. doi: 10.1038/leu.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cifelli C., Bourassa F., Gariepy L., Banas K., Benkhalti M., Renaud J.M. KATP channel deficiency in mouse flexor digitorum brevis causes fibre damage and impairs Ca2+ release and force development during fatigue in vitro. J. Physiol. 2007;582:843–857. doi: 10.1113/jphysiol.2007.130955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steel R.G., Torrie J.H. Principles and Procedures of Statistics: A Biometrical Approach. New York, NY: McGraw-Hill; 1980. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.