

Abstract
Spinocerebellar ataxia type 7 (SCA7) is a dominantly inherited neurodegenerative disorder caused by a CAG – polyglutamine (polyQ) repeat expansion in the ataxin-7 gene. In polyQ disorders, synaptic dysfunction and neurodegeneration may develop prior to symptom onset. However, conditional expression studies of polyQ disease models demonstrate that suppression of gene expression can yield complete reversal of established behavioral abnormalities. To determine if SCA7 neurological and neurodegenerative phenotypes are reversible, we crossed PrP-floxed-SCA7-92Q BAC transgenic mice with a tamoxifen-inducible Cre recombinase transgenic line, CAGGS-Cre-ER™. PrP-floxed-SCA7-92Q BAC;CAGGS-Cre-ER™ bigenic mice were treated with a single dose of tamoxifen 1 month after the onset of detectable ataxia, which resulted in ∼50% reduction of polyQ-ataxin-7 expression. Tamoxifen treatment halted or reversed SCA7 motor symptoms, reduced ataxin-7 aggregation in Purkinje cells (PCs), and prevented loss of climbing fiber (CF)–PC synapses in comparison to vehicle-treated bigenic animals and tamoxifen-treated PrP-floxed-SCA7-92Q BAC single transgenic mice. Despite this phenotype rescue, reduced ataxin-7 expression did not result in full recovery of cerebellar molecular layer thickness or prevent Bergmann glia degeneration. These results demonstrate that suppression of mutant gene expression by only 50% in a polyQ disease model can have a significant impact on disease phenotypes, even when initiated after the onset of detectable behavioral deficits. The findings reported here are consistent with the emerging view that therapies aimed at reducing neurotoxic gene expression hold the potential to halt or reverse disease progression in afflicted patients, even after the onset of neurological disability.
INTRODUCTION
Spinocerebellar ataxia type 7 (SCA7) is a dominantly inherited progressive neurodegenerative disorder caused by a polyglutamine (polyQ) repeat expansion in the ataxin-7 gene. PolyQ-expanded ataxin-7 leads to cell loss in the retina, brainstem and cerebellum, resulting in blindness and progressive loss of motor control (1). Other disorders caused by a CAG repeat expansion encoding an elongated polyglutamine tract in their causative proteins are Huntington's disease (HD), spinal and bulbar muscular atrophy, dentatorubral pallidoluysian atrophy and SCA types 1, 2, 3, 6 and 17. A common feature of polyQ expansion disease is mid-life onset with slowly progressive neurological abnormalities that worsen over years to decades, before culminating in early mortality (2).
Previous studies of polyQ disease mouse models indicate that neuronal dysfunction and neuropathology are reversible up to a certain point, as the affected cells possess the capacity to recover upon termination of mutant gene expression (3,4). In an inducible model of HD, cessation of mutant huntingtin expression after the onset of detectable motor abnormalities permitted a nearly complete reversal of motor symptoms and clearance of protein aggregates from the striatum. Progressive striatal volume loss, reactive astrogliosis and dopamine-1 receptor loss were also prevented (3). In an inducible model of SCA1, motor function and Purkinje cell (PC) histopathology improved when mutant transgene expression was terminated, even at late disease stages, suggesting that SCA1 pathology is reversible. However, recovery from disease phenotypes was greatest in the SCA1 inducible model when termination of disease gene expression occurred early (4). In the case of SCA1, polyQ-ataxin-1 aggregates were cleared from PCs at all stages of disease gene termination. Taken together, these two studies suggest that HD and SCA1 require continued expression of the causative gene to maintain disease progression, raising hopes that phenotype reversibility is a feature of polyQ diseases.
We have previously developed and described the PrP-floxed-SCA7-92Q BAC transgenic model of SCA7, which carries a mutant human ataxin-7 complementary DNA (cDNA) with 92 CAG repeats, flanked by loxP sites within a murine prion protein transgene in the context of a bacterial artificial chromosome (5,6). This mouse model expresses polyQ-ataxin-7 ubiquitously and develops progressive ataxia and cerebellar histopathology, including thinning of the cerebellar molecular layer and degeneration of Bergmann glia (BG) processes (6). We previously reported that cell-type-specific excision of polyQ-ataxin-7 from BG, from neurons of the inferior olive (IO) and cerebellar PCs, or from all three cell types influences both motor behavior and pathological features of the disease phenotype (6). While identifying the specific cell types involved in generating the SCA7 phenotype is important for understanding disease pathogenesis and designing rational therapies, these cell-type-specific excision studies did not address whether the SCA7 disease phenotype can be halted or reversed at later disease stages.
In this study, we employed the same conditional mouse model of SCA7 to determine whether the suppression of mutant ataxin-7 expression after symptom onset would reverse the symptoms or prevent the progression of neuropathology. In order to fully analyze the process of reversibility, we first carefully characterized pathological features of SCA7 disease progression in the PrP-floxed-SCA7-92Q BAC line. We observed that molecular layer thinning occurs at the onset of behavioral abnormalities, but well before BG processes are lost. Similar to findings reported in SCA1 mice (7,8), we documented that climbing fiber (CF) terminals originating from the IO become redistributed in PrP-floxed-SCA7-92Q BAC mice. We crossed the PrP-floxed-SCA7-92Q BAC line with mice expressing tamoxifen-inducible Cre recombinase, and administered tamoxifen after the onset of motor abnormalities and neuropathology. Bigenic mice administered tamoxifen demonstrated ∼50% reduction of polyQ-ataxin-7 transgene expression. Inducing Cre-mediated excision of polyQ-ataxin-7 1 month after symptom onset prevented the progression of the behavioral phenotype. Tamoxifen treatment of bigenic mice also reduced polyQ-ataxin-7 aggregate formation within PC nuclei and prevented CF synapse abnormalities, suggesting that altered IO input contributes to SCA7 motor disease progression. Diminished polyQ-ataxin-7 gene expression after symptom onset did not eliminate PC or BG pathology, despite resolving IO–CF neuropathology and decreasing polyQ-ataxin-7 aggregation. Our findings thus indicate that ∼50% reduction of mutant ataxin-7 gene expression, even after the onset of behavioral abnormalities and PC degeneration, can prevent SCA7 disease progression and ameliorate key features of SCA7 disease. These findings suggest that therapeutic suppression of disease gene expression in SCA7 patients at far less than100% gene knock-down efficiency may still achieve significant alleviation of neurological abnormalities.
RESULTS
Purkinje cell degeneration coincides with the onset of motor dysfunction in PrP-floxed-SCA7-92Q BAC mice
Several studies have shown that polyQ disease mouse models exhibit detectable pathological abnormalities prior to behavioral symptom onset (8–11). Such early pathology may be reversible, if transgene expression is inactivated early in disease natural history (8,11). To determine whether PrP-floxed-SCA7-92Q BAC mice develop presymptomatic PC pathology, sagittal brain sections from 10-week-old PrP-floxed-SCA7-92Q BAC mice and non-transgenic controls were labeled with antibodies against calbindin. The number of PCs and their dendritic arbors appeared similar between the two groups (Fig. 1A and B). Quantification of the cerebellar molecular layer thickness, using a previously described method (6), demonstrated no difference between PrP-floxed-SCA7-92Q BAC and non-transgenic mice (Fig. 1C). Thus, PrP-floxed-SCA7-92Q BAC mice have no histological abnormalities of PCs at 10 weeks of age, confirming normal development of PC dendritic arbors and suggesting that PC pathology may correlate with the onset of behavioral deficits.
Figure 1.
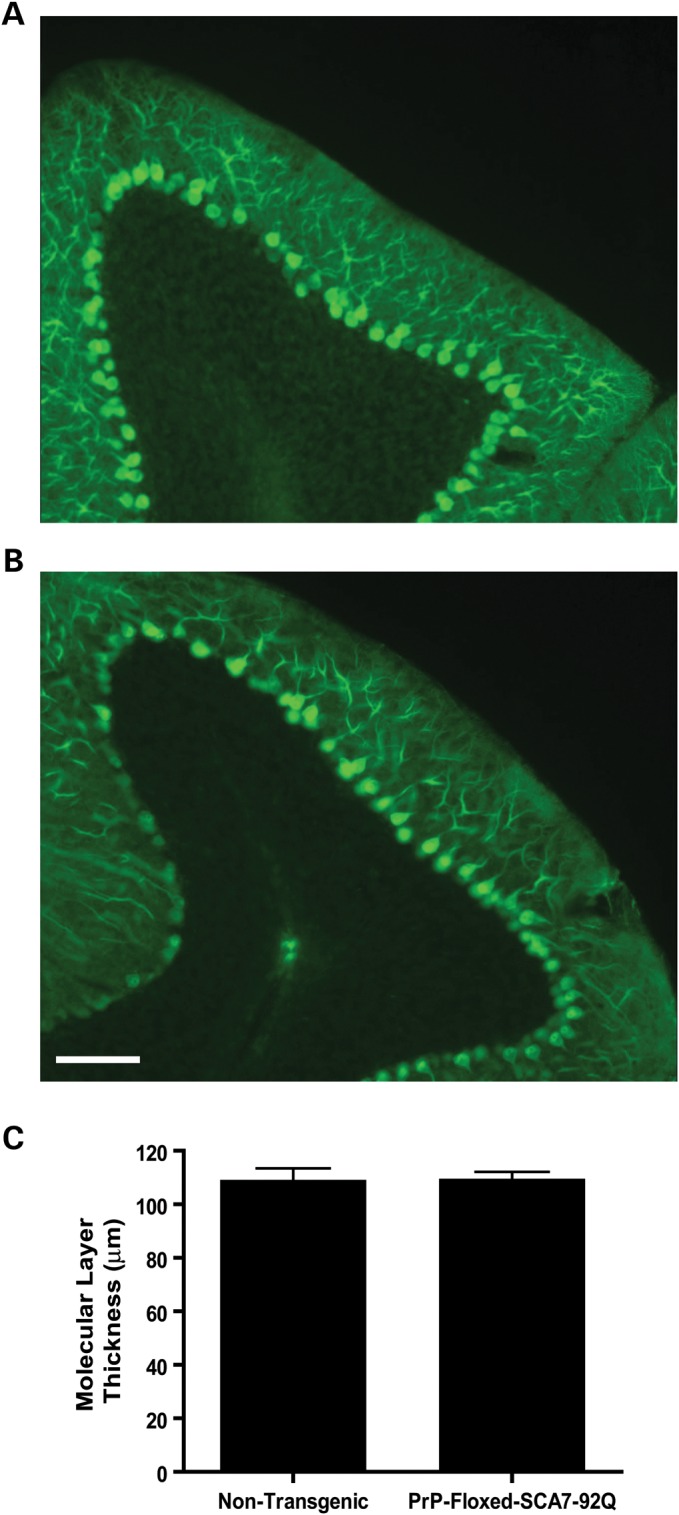
PrP-floxed-SCA7-92Q BAC mice develop normal cerebellar histology. Sagittal cerebellar sections immunostained for calbindin indicate that both (A) non-transgenic and (B) PrP-floxed-SCA7-92Q BAC mice exhibit intense calbindin immunoreactivity for PC bodies, with extensive dendritic arborization. Scale bar, 100 µm. (C) Cerebellar molecular layer thickness is similar for non-transgenic (n = 4) and PrP-floxed-SCA7-92Q BAC (n = 6) mice (two-tailed t-test, unpaired). Error bars = SEM.
We recently reported that PrP-floxed-SCA7-92Q BAC mice have detectable motor deficits at 20 weeks of age (6). To determine whether PC pathology coincides with the onset of the neurological phenotype, we examined sagittal cerebellar sections labeled for calbindin in early symptomatic animals. We observed that 20-week-old PrP-floxed-SCA7-92Q BAC mice have visible degeneration of PC dendrites compared with non-transgenic controls (Fig. 2A and B). Molecular layer thickness measurements revealed that PrP-floxed-SCA7-92Q BAC mice display significantly thinner cerebellar molecular layers than non-transgenic littermates (Fig. 2C). The degree of molecular layer thinning in 25-week-old mice is similar to 20-week-old mice (data not shown). These observations indicate that behavioral deficit onset in PrP-floxed-SCA7-92Q BAC mice coincides with the onset of cerebellar histopathology.
Figure 2.

PrP-floxed-SCA7-92Q BAC mice exhibit PC degeneration, but normal BG at early symptomatic stage. Calbindin immunostaining of sagittal cerebellar sections from 20-week-old mice reveals more extensive PC dendritic branching and a thicker molecular layer in (A) non-transgenic mice in comparison to (B) PrP-floxed-SCA7-92Q BAC mice. Scale bar, 100 µm. (C) Quantification of the cerebellar molecular layer thickness reveals thinning of the molecular layer in PrP-floxed-SCA7-92Q BAC mice (n = 6) compared with non-transgenic mice (n = 4; two-tailed t-test, unpaired; P < 0.01). GFAP immunostaining of sagittal cerebellar sections indicates a similar cellular appearance and staining intensity for (D) non-transgenic mice and (E) PrP-floxed-SCA7-92Q BAC mice at 20 weeks of age. Scale bar, 25 µm. Quantification of BG process numbers demonstrates that the number of GFAP-positive processes is comparable for PrP-floxed-SCA7-92Q BAC mice (n = 6) and non-transgenic mice (n = 4–5) at (F) 20 weeks of age or (G) 25 weeks of age (two-tailed t-test, unpaired). Error bars = SEM.
Previously, we observed that degenerating PCs can cause BG pathology, including the loss of BG processes in the cerebellar molecular layer (6). To determine whether BG pathology develops coincidently with PC pathology in early symptomatic PrP-floxed-SCA7-92Q BAC mice, we immunostained sagittal cerebellar sections for glial fibrillary acidic protein (GFAP) and quantified BG process numbers, using previously reported methods (6). BG in PrP-floxed-SCA7-92Q BAC mice at 20 and 25 weeks of age were not significantly affected compared with non-transgenic mice (Fig. 2D–G). We observed some features previously reported in mice expressing polyQ-expanded ataxin-7 exclusively in BG (12), including excessive branching and fewer BG processes reaching the pial surface (Fig. 2D and E). However, quantification of BG process numbers in the cerebellar molecular layer revealed that PrP-floxed-SCA7-92Q BAC mice do not exhibit significant BG process loss at either time point (Fig. 2F and G). These findings suggest that BG degeneration occurs after PC pathology is present, further supporting the hypothesis that damaged PCs cause injury to BG in a non-cell-autonomous manner.
Tamoxifen-inducible Cre recombinase permits suppression of polyQ-ataxin-7 expression in PrP-floxed-SCA7-92Q BAC;CAGGS-Cre-ERTM mice
Patients with neurodegenerative diseases such as SCA7 may be diagnosed well after symptom onset. Therefore, future treatments, which are effective after neurodegeneration begins, remain desirable. However, it is not currently known whether any intervention can arrest SCA7 neurodegeneration after symptom onset. While earlier studies on HD and SCA1 indicate that certain phenotype features can be halted or reversed by cessation of disease gene expression (3,4), this has not yet been investigated in the context of SCA7. To determine whether SCA7 neurological and neurodegenerative phenotypes are reversible, we crossed a tamoxifen-inducible Cre recombinase transgenic line, CAGGS-Cre-ER™ (13), with PrP-floxed-SCA7-92Q BAC mice. Bigenic mice (PrP-floxed-SCA7-92Q BAC;CAGGS-Cre-ER™) express Cre recombinase ubiquitously, though Cre is sequestered into the cytoplasm due to fusion with a mutant form of the estrogen receptor (ER) ligand-binding domain. Once tamoxifen binds, the Cre-ER™ fusion protein translocates to the nucleus, where Cre recombinase can excise the loxP-flanked CAG-expanded ataxin-7 cDNA (Fig. 3A). Because symptom onset in PrP-floxed-SCA7-92Q BAC mice occurs at ∼20 weeks of age (6), we orally administered a single 20 mg dose of tamoxifen citrate dissolved in corn oil, or corn oil alone, to bigenic (PrP-floxed-SCA7-92Q BAC;CAGGS-Cre-ER™), singly transgenic (CAGGS-Cre-ER™ or PrP-floxed-SCA7-92Q BAC), and non-transgenic littermate controls at 24 weeks of age (Fig. 3A), well after symptom onset. To determine the efficiency of polyQ-ataxin-7 gene excision after tamoxifen administration, cerebella were collected from PrP-floxed-SCA7-92Q BAC; CAGGS-Cre-ER™ bigenic mice administered tamoxifen or vehicle, and from PrP-floxed-SCA7-92Q BAC mice administered tamoxifen. Extracted total cerebellar RNA was analyzed by qRT–PCR for human mutant ataxin-7 mRNA relative to 18S rRNA. Cerebellar RNA from tamoxifen-treated bigenic mice displayed ∼50% reduction in human mutant ataxin-7 mRNA compared with oil-treated bigenic mice and tamoxifen-treated PrP-floxed-SCA7-92Q BAC singly transgenic mice (Fig. 3B). This confirms the efficacy of this system for pronounced suppression of mutant transgene expression.
Figure 3.

Cre-mediated excision of mutant ataxin-7 in PrP-floxed-SCA7-92Q BAC mice. (A) Outline of the breeding and treatment paradigm employed in this study. CAGGS-Cre-ER™ and non-transgenic animals were pooled together in one group designated as ‘SCA7-negative’. (B) Quantitative real-time RT–PCR analysis reveals that ataxin-7-92Q transgene expression is reduced by ∼50% in bigenic mice induced to express Cre recombinase in the nucleus (n = 3/group; P < 0.01, ANOVA with Bonferroni post-test). Error bars = SEM.
Suppression of polyQ-ataxin-7 expression restores motor function in SCA7 mice
SCA7-negative (CAGGS-Cre-ER™ or non-transgenic), SCA7-positive (PrP-floxed-SCA7-92Q BAC) and bigenic (PrP-floxed-SCA7-92Q BAC;CAGGS-Cre-ER™) mice were dosed either with tamoxifen or with vehicle 4 weeks after symptom onset at 24 weeks of age. At 23 weeks (pre-treatment), bigenic mice exhibited significant abnormalities in composite phenotype score, similar to SCA7-positive mice. All bigenic and SCA7-positive mice were significantly impaired compared with SCA7-negative mice (Fig. 4A). Singly transgenic SCA7 mice treated with tamoxifen or oil progressively worsened in accordance with a previous report (6). Bigenic mice administered oil at 24 weeks also exhibited a progressive neurological phenotype. Bigenic mice treated with a single dose of tamoxifen worsened slightly at 25 weeks, but their composite phenotype score reached a plateau and remained consistent throughout the remainder of the study period (Fig. 4A). Although the bigenic tamoxifen-treated mice exhibited neurological abnormalities, they no longer had progressive symptoms nor were their symptoms as severe as those exhibited by SCA7-positive mice or bigenic oil-treated mice. Hence, Cre-mediated excision of the ataxin-7 transgene 4 weeks after symptom onset arrested SCA7 disease progression. This finding was corroborated by assessment of performance on an accelerating rotarod. At 23 weeks of age, SCA7-positive and bigenic mice fell off the accelerating rotarod more quickly than SCA7-negative mice, suggesting a trend toward deficiencies in motor coordination (Fig. 4B). At 42 weeks of age (19 weeks post-treatment and end stage), SCA7-positive mice given tamoxifen or oil performed significantly worse on the rotarod compared with SCA7-negative mice. Bigenic mice administered oil also performed poorly. Bigenic tamoxifen-treated mice, however, performed similarly to the SCA7-negative mice (Fig. 4B), indicating that the suppression of polyQ-ataxin-7 expression can reverse the SCA7 ataxia phenotype as measured by the rotarod, even after the onset of visible neurological disease in SCA7 transgenic mice.
Figure 4.
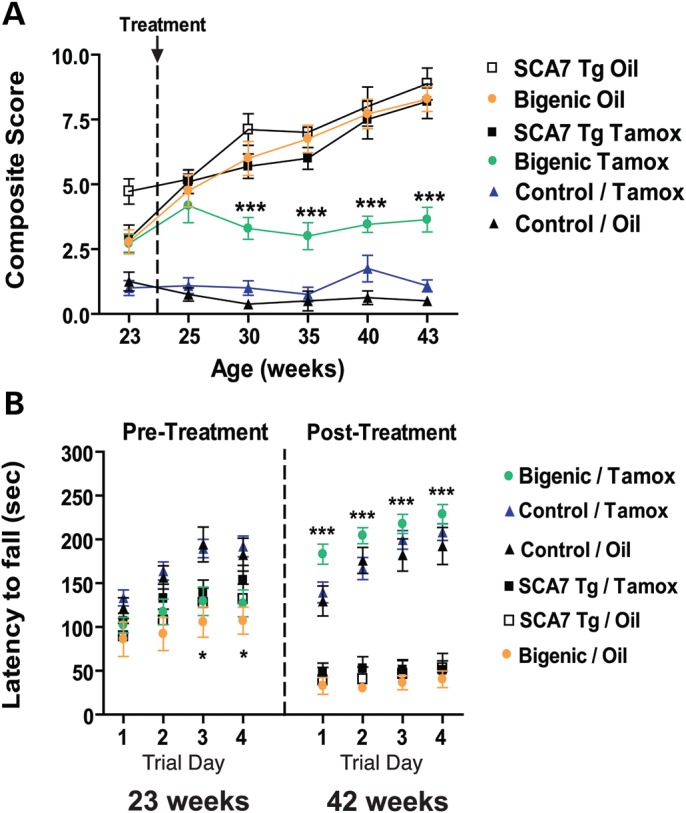
Excision of ataxin-7-92Q expression after symptom onset halts or reverses motor phenotypes in SCA7 transgenic mice. (A) We evaluated the neurological status of cohorts of SCA7 transgenic mice using a composite phenotype score, which consists of four individual tests that exemplify different aspects of the mouse SCA7 phenotype. Each component is scored from 0 (normal) to 3 (severely affected). The tests assess the ability to walk on a cage ledge, hindlimb clasping upon tail suspension, the existence of an abnormal gait and the development of kyphosis (hunched back secondary to neurodegeneration). Scores from all four tests are combined into a single, composite score. Using this testing paradigm, we found that after suppression of mutant ataxin-7 transgene expression at 24 weeks of age, neurological function is preserved in tamoxifen-treated bigenic mice, as they perform significantly better than oil-treated bigenic mice or SCA7 transgenic mice (P < 0.001, ANOVA with Bonferonni post-test). Error bars = SEM. (B) We employed the accelerating rotarod task to evaluate cohorts of SCA7 transgenic mice, including bigenic mice subjected to tamoxifen-induced excision of the ataxin-7-92Q transgene after symptom onset at 24 weeks, and found that treated bigenic mice perform comparably with SCA7-negative mice and significantly better than oil-treated bigenic mice at 42 weeks of age, even though bigenic mice performed significantly worse than control mice prior to treatment (*P < 0.05, ***P < 0.001, ANOVA with Bonferonni post-test). Error bars = SEM.
Suppression of polyQ-ataxin-7 expression does not reverse molecular layer thinning or prevent Bergmann glial pathology in SCA7 mice
We next tested whether the suppression of mutant ataxin-7 transgene expression could prevent the progression of cerebellar histopathology in PrP-floxed-SCA7-92Q BAC transgenic mice. We collected sagittal cerebellar sections from SCA7-negative mice, SCA7-positive mice and bigenic mice, and found that cerebella from tamoxifen-treated SCA7-positive and oil-treated bigenic mice exhibited PCs with shrunken somae and reduced dendritic arborization, while PCs from tamoxifen-treated bigenic mice appeared healthier (Fig. 5A–D). Analysis of the cerebellar molecular layer thickness revealed that tamoxifen-treated bigenic mice had a thicker molecular layer than tamoxifen-treated SCA7-positive and oil-treated bigenic mice, but these differences were not statistically significant (Fig. 5E). Although diminished polyQ-ataxin-7 expression after the onset of molecular layer thinning did not result in a statistically significant increase in the molecular layer thickness, PC pathology appeared improved. These findings suggest that some aspects of PC pathology may be influenced by reduced mutant gene expression, while other features, such as molecular layer thinning (which was already evident long before tamoxifen treatment was administered), remain unaffected by this intervention.
Figure 5.
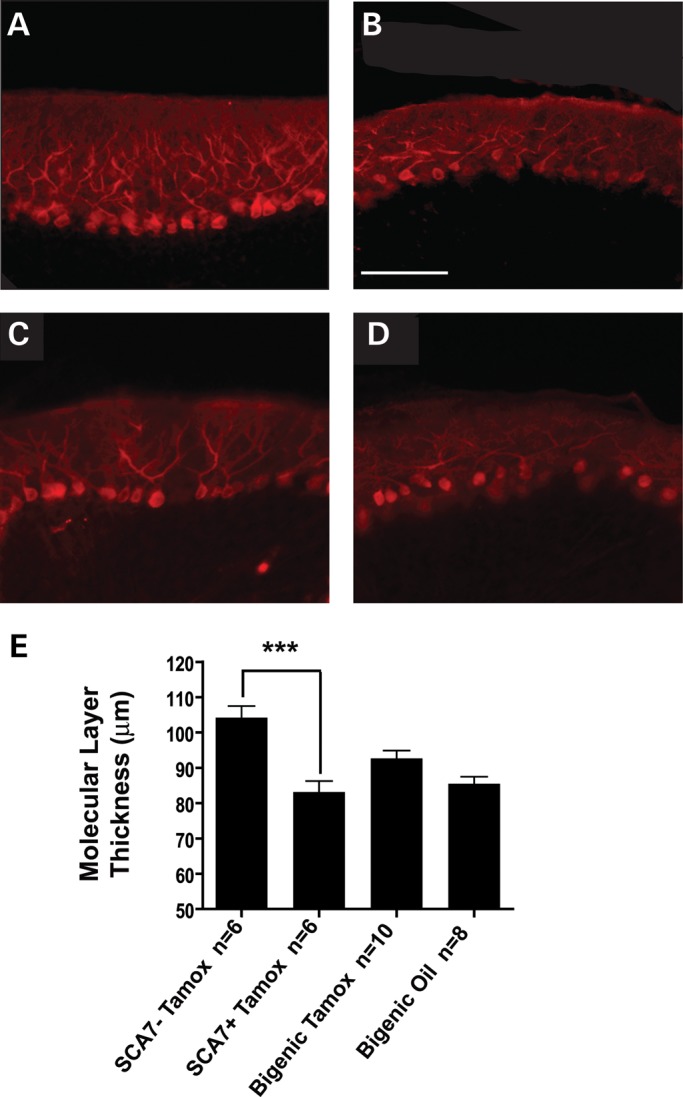
Suppression of ataxin-7-92Q transgene expression in SCA7 mice after symptom onset does not fully rectify cerebellar pathology. Calbindin immunostaining of sagittal cerebellar sections from (A) 43-week-old SCA7-negative mice treated with tamoxifen reveals extensive PC dendrite branching, while (B) age-matched tamoxifen-treated SCA7 transgenic mice display reduced calbindin immunoreactivity and a shrunken molecular layer. (C) In contrast, PCs in tamoxifen-treated bigenic mice had a morphology more similar to non-transgenic mice, unlike (D) oil-treated bigenic mice that exhibit an intermediate morphology. Scale bar, 100 µm. (E) When we quantified the cerebellar molecular layer thickness, we found that singly transgenic SCA7 mice and oil-treated bigenic mice display dramatically reduced cerebellar molecular layer thickness in comparison to non-SCA7 transgenic controls (P < 0.001, ANOVA with Bonferonni post-test), and tamoxifen-treated bigenic mice have a thicker cerebellar molecular layer compared with tamoxifen-treated singly transgenic SCA7 mice, suggesting some improvement, but with a difference that is not statistically significant. Error bars = SEM.
We then evaluated whether SCA7 disease gene excision could prevent the progressive loss of BG processes, which becomes detectable at a later time point. Cerebellar sections from SCA7-negative, SCA7-positive and bigenic mice were immunostained with antibodies against GFAP to visualize BG. Tamoxifen-treated SCA7-positive (singly transgenic) mice displayed cerebella with disorganized and stunted BG processes compared with tamoxifen-treated SCA7-negative mice (Fig. 6A and B). BG from tamoxifen-treated bigenic mice appeared healthier than tamoxifen-treated SCA7-positive BG, while oil-treated bigenic mice harbored BG more similar in appearance to SCA7-positive tamoxifen-treated BG (Fig. 6A–D). Quantification of BG process numbers revealed that both tamoxifen-treated and oil-treated bigenic mice exhibited BG pathology similar to tamoxifen-treated SCA7-positive mice (Fig. 6E), indicating that excision of the ataxin-7 transgene at 24 weeks does not prevent BG process loss, even though BG degeneration occurs after gene excision in PrP-floxed-SCA7-92Q BAC mice (Fig. 2D–G).
Figure 6.

Suppression of ataxin-7-92Q transgene expression in SCA7 mice after symptom onset does not prevent BG process loss. We immunostained cohorts of 43-week-old mice for GFAP, and noted that in comparison to tamoxifen-treated SCA7-negative mice (A), tamoxifen-treated SCA7 singly transgenic mice (B) have fewer, less organized and more stunted BG processes, and that this BG pathology is also apparent in both (C) tamoxifen-treated bigenic mice and (D) oil-treated bigenic mice. Scale bar, 25 µm. (E) Quantification of BG process numbers revealed that tamoxifen-treated bigenic mice display a significant loss of BG processes (P < 0.001, ANOVA with Bonferonni post-test), akin to tamoxifen-treated SCA7 singly transgenic mice or oil-treated bigenic mice. Error bars = SEM.
Suppression of polyQ-ataxin-7 expression after symptom onset prevents inferior olive climbing fiber–Purkinje cell synapse pathology in SCA7 mice
Our recently published findings demonstrated that PCs, BG and neurons of the IO nucleus are important contributors to SCA7 disease onset and pathogenesis (6). Climbing fibers (CFs) originating from IO neurons synapse onto PC dendrites. Antibodies against vesicular glutamate transporter 2 (VGLUT2) specifically label CF synaptic terminals (14). In a mouse model of SCA1, CF synapses, labeled with antibodies to VGLUT2, are decreased near the pial surface in the cerebellar molecular layer of symptomatic mice (7). Because the IO degenerates in SCA7 patients and is likely involved in disease pathogenesis in SCA7 and related cerebellar degenerative disorders (6,7,8,15,16), we investigated whether loss of CF–PC synapses also occurs in our SCA7 mouse model. After immunostaining sagittal cerebellar sections from non-transgenic and PrP-floxed-SCA7-92Q BAC mice for VGLUT2, we documented that VGLUT2-positive CF terminals are evenly distributed throughout the molecular layer along PC dendrites in 20-week-old non-transgenic and PrP-floxed-SCA7-92Q BAC mice, and in 40-week-old non-transgenic mice (Fig. 7A–C). In 40-week-old PrP-floxed-SCA7-92Q BAC mice, however, the CF–PC synapses appeared aberrantly distributed, with a greater number of CF terminals located adjacent to PC soma (Fig. 7A–D). Oil-treated bigenic mice appeared to have VGLUT2 CF terminals arranged in a pattern similar to PrP-floxed-SCA7-92Q BAC mice (Fig. 7C–E). CF terminal distribution in tamoxifen-treated bigenic mice appeared normal, resembling 40-week-old non-transgenic mice, instead of PrP-floxed-SCA7-92Q BAC mice (Fig. 7C–F).
Figure 7.

VGLUT2 immuno-analysis reveals normal CF–PC synapse distributions in PrP-floxed-SCA7-92Q BAC mice subjected to ataxin-7-92Q transgene suppression. (A–F) We immunostained sagittal cerebellar sections for calbindin (red) and for VGLUT2 (green), a marker of CF–PC synapses. VGLUT2-positive puncta are evenly distributed throughout the cerebellar molecular layer in (A) non-transgenic and (B) PrP-floxed-SCA7-92Q BAC mice at 20 weeks of age. Normal distributions of VGLUT2 puncta were also noted in (C) 40-week-old non-transgenic control mice; however, in (D) 40-week-old PrP-floxed-SCA7-92Q BAC mice, the VGLUT2-labeled CF–PC synapses aggregate proximal to PC bodies and are no longer evenly distributed. (E) VGLUT2 distribution in 43-week-old oil-treated bigenic mice was abnormal, while (F) tamoxifen-treated bigenic mice exhibited CF–PC synapses with more normally distributed VGLUT2 immunostaining. Scale bar, 20 µm. (G) To measure the volume of VGLUT2 immunostaining in each 3-D image, a line was drawn from the PC layer to the pial surface (white line). A second intersecting line (white) was drawn perpendicular to the first line at the midpoint of the molecular layer to divide the proximal region from the distal region. The volume for total VGLUT2 CF–PC synapse immunolabeling was then calculated for the distal and proximal regions (colored boxes). (H) At 20 weeks of age, the distribution of VGLUT2-positive CF–PC synapses in the molecular layer is similar in non-transgenic and PrP-floxed-SCA7-92Q BAC mice (n = 6–7/group). (I) At 40 weeks of age, the distribution of CF–PC synapses is significantly altered with increased VGLUT2 staining observed in the proximal region and lessVGLUT2 staining present in the distal region for PrP-floxed-SCA7-92Q BAC mice compared with non-transgenic mice (P < 0.01, two-tailed t-test, unpaired; n = 6/group). (J) Tamoxifen-treated bigenic mice, however, display a significantly improved VGLUT2 staining distribution at CF–PC synapses, when compared with oil-treated bigenic mice (P < 0.05, two-tailed t-test, unpaired; n = 6/group). Error bars = SEM.
To further quantify the distribution of CF terminals labeled with VGLUT2 immunostaining, 3-D images of sagittal cerebellar sections were collected, and a line separating the distal and proximal halves of the cerebellar molecular layer in relation to PC somae was then constructed. After creating a 3-D box in each of the two regions of the molecular layer and generating a mask to identify areas of intense VGLUT2 immunolabeling of CF terminals, we measured the total volume of VGLUT2 immunolabeling in the distal and proximal regions of the molecular layer (Fig. 7G). Quantification of VGLUT2 immunofluorescence, as a ratio of the volume in the distal molecular layer to the volume in the proximal molecular layer, revealed similar distal-to-proximal VGLUT2 volume ratios in non-transgenic and PrP-floxed-SCA7-92Q BAC mice at 20 weeks of age, and indicated a greater CF terminal volume in proximity to PC somae (ratio <1; Fig. 7H). At 40 weeks of age, this ratio is significantly decreased in PrP-floxed-SCA7-92Q BAC mice compared with non-transgenic controls (Fig. 7I), indicating a redistribution of CF terminals in SCA7 disease. At 43 weeks of age, however, tamoxifen-treated bigenic mice have a significantly increased ratio of distal-to-proximal VGLUT2 volume compared with oil-treated bigenic mice (Fig. 7J). Thus, suppression of polyQ-ataxin-7 expression at 24 weeks prevents SCA7-induced redistribution of CF–PC synapses in the cerebellar molecular layer.
Reduced polyQ-ataxin-7 expression prevents progression of PC aggregate formation
Previous studies of HD and SCA1 transgenic mice documented rapid clearance of intranuclear inclusions containing the aggregated form of the mutant protein upon cessation of mutant gene expression (3,4). We monitored the progressive accumulation of polyQ-ataxin-7 aggregates in PCs in cerebella collected from PrP-floxed-SCA7-92Q BAC mice at 10, 20 and 25 weeks of age (Fig. 8A and B). Using an anti-ataxin-7 antibody, we quantified the number of aggregates as a function of the total PC number. PrP-floxed-SCA7-92Q BAC mice do not exhibit a robust loss of PCs (6). We found that less than 10% of PCs display an ataxin-7 nuclear inclusion at 10 weeks of age, but almost 30% of PCs display such aggregates by 25 weeks of age (Fig. 8B). When we examined the number of polyQ-ataxin-7 aggregates in tamoxifen-treated SCA7-positive and bigenic mice, we noted that ∼50% of PCs in 43-week-old tamoxifen-treated SCA7-positive mice and oil-treated bigenic mice contain inclusions, while only ∼23% of PCs in tamoxifen-treated bigenic mice contain ataxin-7-positive inclusions, similar to the extent of aggregate pathology in 25-week-old PrP-floxed-SCA7-92Q BAC mice (Fig. 8B).
Figure 8.
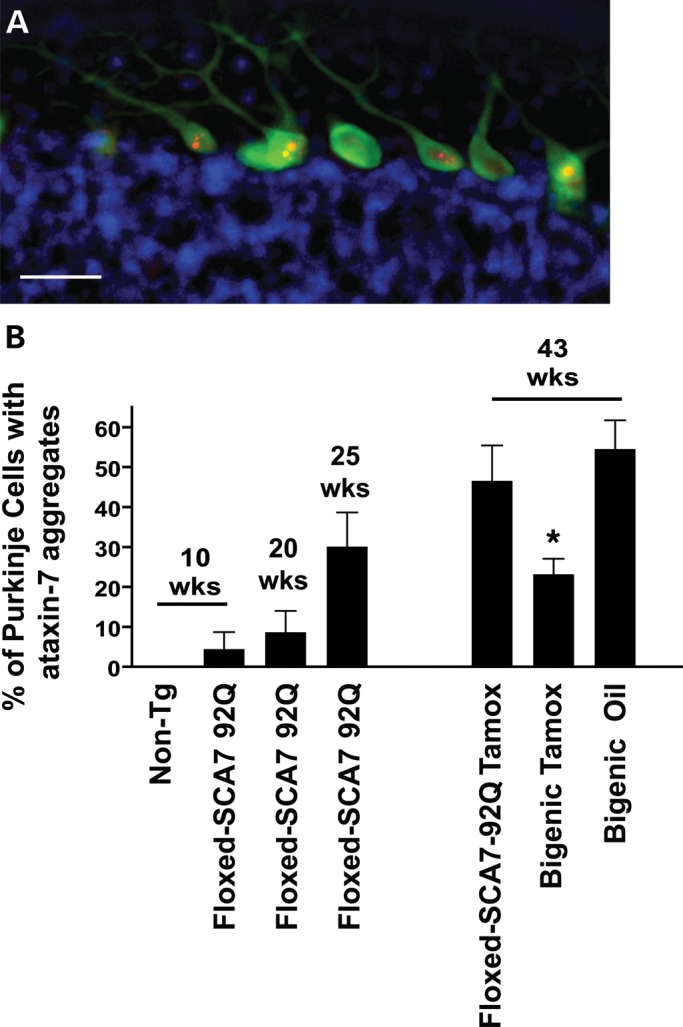
Suppression of ataxin-7-92Q transgene expression in SCA7 mice after symptom onset prevents further aggregate pathology. (A) We immunostained sagittal cerebellar sections for ataxin-7 (red), calbindin (green) and 4′,6-diamidino-2-phenylindole (DAPI) (blue), and as shown here, could readily detect PCs with ataxin-7-positive aggregates. Scale bar, 25 µm. (B) To quantify ataxin-7 aggregate burden, we counted the number of PCs with ataxin-7-positive nuclear inclusions, and divided by the total number of PCs present in the section (n = 3–8 mice/group). PrP-floxed-SCA7-92Q BAC mice accumulate ataxin-7 aggregates beginning at 10 weeks of age, and by 43 weeks of age, ∼50% of PCs display aggregates. Note that only ∼23% of PCs from 43-week-old tamoxifen-treated bigenic animals, however, exhibit ataxin-7 aggregates, which is a significant reduction in comparison to age- and littermate-matched tamoxifen-treated SCA7 singly transgenic mice or oil-treated bigenic mice (P < 0.05, one-way ANOVA with Bonferonni post-test). Error bars = SEM.
DISCUSSION
A nagging question in therapy development for neurodegenerative disease is the optimal stage at which a therapeutic intervention should be offered. For rare diseases such as SCA7, determining the extent of reversibility for neurological phenotypes and neuronal dysfunction is especially crucial, as opportunities for performing clinical trials will be limited by the small size of affected patient populations. With these issues in mind, we developed an inducible mouse model for SCA7, based upon our PrP-floxed-SCA7-92Q BAC transgenic model that features a floxed human ataxin-7 cDNA carrying 92 CAG repeats, inserted at the first coding exon of the PrP gene contained within a BAC (6). Unlike earlier polyQ disease inducible/conditional models (3,4), these SCA7 transgenic mice express mutant ataxin-7 proteins in neurons, glia and other cell types in the central nervous system (CNS), thereby allowing for an evaluation of the effect of expression suppression upon a network of interconnected neurons and glia. Using this model, we have previously shown that excising polyQ-ataxin-7 from PCs, BG and IO neurons delays neurological disease onset by 20 weeks, extending the age of onset by a factor of two (6). In the present study, we crossed the PrP-floxed-SCA7-92Q BAC transgenic mice with a CAGGS-Cre-ER™ line (13), and then achieved termination of ∼50% of polyQ-ataxin-7 expression in bigenic mice by oral administration of tamoxifen at 24 weeks of age, which is roughly a month after the onset of visible neurological abnormalities and detectable motor incoordination. By comparing PrP-floxed-SCA7-92Q BAC;CAGGS-Cre-ER™ bigenic mice treated with tamoxifen versus vehicle, and including singly transgenic PrP-floxed-SCA7-92Q BAC mice as well as non-SCA7 control mice, we were able to assess the effect of excising the polyQ-ataxin-7 transgene on a range of neurological, histological and neurodegenerative phenotypes. Our findings indicate that disease gene suppression improves motor incoordination, maintains normal CF–PC synapses and improves PC proteostasis, but that other aspects of SCA7 neuropathology do not respond to abated mutant ataxin-7 protein expression.
Motor incoordination is a hallmark feature of SCA7 and related cerebellar degenerations, and stems from dysfunction of neuronal pathways involving cerebellar granule cells, PCs and brainstem neurons in the IO. This circuitry has been extensively studied, and lesions of PCs are sufficient to produce profound ataxia in mouse models (17), confirming the importance of PC afferent and efferent pathways in coordinated movement. In this study, we waited until SCA7 mice exhibited significant impairment in motor coordination before oral administration of tamoxifen to decrease polyQ-ataxin-7 expression. We found that bigenic mice receiving tamoxifen performed comparably with non-SCA7 controls on the accelerating rotarod. This is similar to our previous finding that elimination of polyQ-ataxin-7 in ∼50% of both PCs and IO neurons from birth allows SCA7 transgenic mice to perform normally on the rotarod, based upon comparison to non-transgenic littermates (6). Here, we suppressed polyQ-ataxin-7 transgene expression by ∼50% throughout the cerebellum after 24 weeks of age, and noted a complete normalization of rotarod performance. This recovery of motor function occurred despite an apparent persistence of neuropathological findings typically considered indicative of cerebellar dysfunction, including thinning of the cerebellar molecular layer and BG process loss. Although the molecular layer thickness and BG process number in bigenic mice undergoing polyQ-ataxin-7 expression suppression were comparable with vehicle-treated bigenic mice and with singly transgenic SCA7 mice, a careful examination of these measurements did reveal noticeable improvements, though these observations did not achieve statistical significance (Fig. 5 and 6). Nonetheless, the extent of cerebellar histopathology in bigenic mice subjected to tamoxifen-mediated suppression of expression was significant. These findings underscore that pathology does not always equate with disease, and that additional measures of neural function are required to fully evaluate the status of the cerebellar network.
In the current study, we demonstrated that late-stage PrP-floxed-SCA7-92Q BAC mice have an abnormal distribution of CF–PC synapses. The distribution of CF–PC synapses is normal in SCA7 mice at 20 weeks of age, and inducible Cre recombinase-mediated excision of the ataxin-7-92Q cDNA at 24 weeks of age prevented the development of aberrant CF–PC synapse distribution in 43-week-old SCA7 mice (Fig. 7). CF–PC synapse dislocation occurs after molecular layer thinning and onset of behavioral symptoms, yet the preservation of the CF–PC circuitry in SCA7 animals, with polyQ-ataxin-7 expression decreased by 50% after symptom onset, correlates with improved motor behavior. This suggests that PC function is maintained despite the presence of significant PC and BG pathology. SCA1 is a related neurodegenerative disorder characterized by PC and IO degeneration (16). In a transgenic SCA1 mouse model, PCs displayed a reduced responsiveness to excitatory stimulation from CFs at 6 weeks of age, the age at which rotarod deficits are first detectable (18). Additionally, when disease gene expression was initiated after cerebellar development is complete (at 5 weeks), the CF–PC synaptic inputs were preserved (8). These findings suggest that very early changes in CF–PC synaptic function can contribute to motor deficits. The CF–PC synapse findings in this SCA1 model raise two interesting issues. First, it is possible that functional abnormalities in CF–PC synapses in SCA7 may develop prior to anatomic abnormalities, but that anatomic methods of detection are not sufficiently sensitive to identify the reversible component of this finding. Second, in SCA1 mice, changes in CF–PC synapses are likely initiated by changes in PCs, because this model expresses the disease gene only in PCs (8). It is, therefore, possible that preservation of the CF–PC synapse distribution in the cerebellar molecular layer upon suppression of mutant gene expression in our mouse model of SCA7 indicates a preservation of normal CF–PC synaptic connectivity and function.
In addition to improved motor function and maintenance of CF–PC synapses, reduced polyQ-ataxin-7 protein expression also prevented further formation of polyQ-containing nuclear aggregates in PCs. Tamoxifen-treated bigenic mice, sacrificed at 43 weeks, exhibited roughly the same percentage of PCs with ataxin-7 aggregates as 25-week-old PrP-floxed-SCA7-92Q BAC mice. Because the bigenic mice received tamoxifen at 24 weeks of age, this suggests that suppression of polyQ-ataxin-7 expression by 50% prevented further ataxin-7 nuclear aggregate formation. The impact of disease gene excision on aggregate formation mirrors the effect of disease gene suppression on motor behavior in this cohort of mice. Perhaps PCs spared from forming nuclear aggregates are more capable of effectively maintaining physiological function in spite of dendritic arbor retraction. Proteotoxic stress imposes a heavy burden on energy utilization (19). Reduced expression of polyQ-expanded ataxin-7 likely permits PCs to achieve a more favorable bioenergetics profile, consistent with their ability to maintain behavioral function. Therefore, nuclear aggregates may serve as a surrogate marker for cellular stress, potentially correlating with high levels of toxic oligomers and protofibrils present in the cell (20,21), and their continued presence in PCs would be expected to prevent complete restoration of neural function. We evaluated the extent of decreased mutant ataxin-7 expression on a global rather than a cell-type-specific level (Fig. 3B); hence, the current findings do not differentiate between complete transgene excision in 50% of cells, and a 50% cumulative reduction in expression due to partial transgene excision in a majority of cells. It is possible that PCs harboring a persistent aggregate may not have undergone sufficient polyQ-ataxin-7 gene excision to permit the protein degradation machinery to effectively remove the aggregated protein. Incomplete transgene excision may thus account for the observed lack of reversal of molecular layer thinning and non-cell-autonomous BG degeneration. Failure to eliminate all ataxin-7 protein aggregates in this study also differs from prior investigations of polyQ disease reversibility in HD and SCA1, where the termination of disease gene expression allowed for the eventual removal of polyQ-containing protein aggregates in these models (3,4). One prior study of mice expressing ataxin-7-90Q under rhodopsin promoter control revealed that the proportion of rods in the retina with ataxin-7-positive nuclear aggregates declined from >95% at 26 weeks to ∼50% by 1 year of age. However, the rods never completely cleared the nuclear aggregates even at 2 years of age, despite the fact that polyQ-ataxin-7 expression became negligible at 9 weeks of age in this model (22). These findings suggest that aggregates containing mutant ataxin-7, unlike polyQ-expanded huntingtin and ataxin-1, may not be easily cleared from cells. Nevertheless, since macroaggregates are no longer believed to be the toxic species (23), further studies geared towards the detection of polyQ-ataxin-7 oligomers and other toxic conformers will be required to assess the true extent of proteotoxic stress in such situations.
One of the most compelling challenges remaining for the neurodegenerative disease field is to develop meaningful therapies to treat these devastating disorders. Here, we examined the degree of reversibility for SCA7 behavioral, histological and neurodegenerative phenotypes in an inducible mouse model that features widespread expression of the polyQ-expanded ataxin-7 disease protein. Akin to prior studies of HD and SCA1 (3,4), two closely related polyQ diseases, we found that several SCA7 disease phenotypes are reversed, halted or prevented. Our findings thus add to a growing literature suggesting that even in the face of established anatomical and physiological dysfunction, interventions aimed at extinguishing expression of a proteotoxic disease protein hold great promise. That recovery of neurological function is possible in other neurodegenerative diseases, in addition to polyQ disorders, is supported by studies of tau transgenic mice, where termination of expression of the toxic tau isoform yielded dramatic improvements in memory function and synaptic long-term potentiation (24). Although the suppression of polyQ-ataxin-7 gene expression by ∼50% in the inducible SCA7 disease model permitted normalization of rotarod performance and CF–PC synapses, other features of the SCA7 disease process displayed only partial recovery. While the reasons for these differences may reflect a variety of factors, one likely explanation is that our inducible SCA7 model features widespread expression of the disease protein in neurons, glia and other interconnected cell types within the CNS, suggesting that in contrast to mouse model systems where disease gene expression is restricted to specific neuronal populations, the effects of suppression of polyQ disease protein expression are considerably more complicated under these circumstances. Hence, our work underscores the nuances of polyQ disease phenotype reversibility and suggests that a more definitive analysis of the effects of disease protein suppression on disease-associated phenotypes may be better achieved in models featuring widespread disease protein expression. Understanding how disease protein termination therapies impact disease progression in such model systems may yield a more representative picture of expected outcomes in human patients, who, for a growing subset of neurodegenerative disorders, may soon be recruited to receive experimental RNA interference or antisense oligonucleotide gene knock-down therapies. Perhaps, the most encouraging aspect of this work is that a dramatic amelioration of motor dysfunction can be achieved only with 50% knock-down of the disease gene, suggesting that gene silencing approaches in neurodegenerative proteinopathies need not be 100% efficient to yield a meaningful therapeutic outcome.
MATERIALS AND METHODS
Animals
Mice were maintained in a temperature- and humidity-controlled environment on a 12 h light/dark cycle with light onset at 6:00 am. All studies and procedures were approved by an Institutional Animal Care and Use Committee at the University of Washington or the University of California, San Diego. Non-transgenic, PrP-floxed-SCA7-92Q BAC, CAGGS-Cre-ER™ and bigenic littermates were orally administered tamoxifen citrate either as a single 20 mg dose (80 mg/ml in 0.25 ml) dissolved in corn oil or received corn oil alone (0.25 ml) at 24 weeks of age. Mice dosed with tamoxifen were housed separately from those dosed with corn oil.
Behavioral phenotyping
Females and males were used in all studies. An examiner blinded to genotype performed all behavioral measurements. The composite phenotype score and the accelerating rotarod assessment were performed as previously described (5,6). The composite phenotype score consists of four individual tests that exemplify different aspects of the mouse SCA7 phenotype. Each component is scored from 0 (normal) to 3 (severely affected). The tests assess the ability to walk on a cage ledge, hindlimb clasping upon tail suspension, the existence of an abnormal gait and the development of kyphosis (hunched back secondary to neurodegeneration). Scores from all four tests are combined into a single, composite score. More extensive description of this scoring system and demonstration of the abnormalities associated with a specific score can be viewed online at http://www.jove.com/video/1787/a-simple-composite-phenotype-scoring-system-for-evaluating-mouse?ID=1787. The accelerating rotarod assessment consists of 1 day for training followed by four consecutive days of testing. Each mouse runs five trials per day with a 5min break between trials. The worst trial of the day is excluded. Each trial does not exceed 5min.
PCR genotyping
Mice tissue processing, DNA isolation and PCR genotyping were performed as previously described (6). Primer sequences are as follows:
.
| Transgene | Primer sequence | Product (bp) | |
|---|---|---|---|
| PrP-floxed- SCA7-92Q BAC | Forward | CATTTTAGGCCC CACGTATCAC | 587 |
| Reverse | GGCCCGCTCCGACAT | ||
| CAGGS- Cre-ER™ | Forward | CAAAATTTGCCTGC ATTACCG | 553 |
| Reverse | CATTCTCCCACCG TCAGTACG |
Quantitative RT–PCR analysis
Total RNA was isolated using an RNeasy isolation kit (Qiagen), and reverse-transcribed using SuperScript II reverse transcriptase according to the manufacturer's instructions (Invitrogen). The cDNAs were subjected to real-time PCR using primers and probes for human ataxin-7 (assay HsDD165660_m1, Applied Biosystems, Inc.) and mouse 18S RNA (assay Hs99999901_s1, Applied Biosystems, Inc.). Samples were analyzed on a StepOnePlus Real-Time PCR system. Ataxin-7 mRNA levels were normalized to 18S rRNA levels. Average ataxin-7 mRNA/18S rRNA levels in cerebella from tamoxifen-treated PrP-floxed-SCA7-92Q BAC (n = 3), tamoxifen-treated bigenic (n = 3) and oil-treated bigenic (n = 3) mice were calculated using Microsoft Excel and plotted and statistically analyzed in Graphpad Prism 4.0.
Immunohistochemistry
Deeply anesthetized mice were transcardially perfused with phosphate-buffered saline (PBS) followed by 4.0% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB), pH 7.4. Brains were removed and post-fixed in 4.0% PFA in 0.1 m PB overnight, then moved into 0.4% PFA in 0.1 m PB for storage at 4°C. Free-floating 40 µm sagittal brain sections were cut on the Leica Vibratome 1000S and stored in 0.03% PFA in PBS. Free-floating sections were immunostained as previously described (6), except for VGLUT2 immunostaining. Primary antibodies used were anti-calbindin antibody (Swant, 300) at 1:1000 dilution; anti-GFAP antibody (DAKO, Z0334) at 1:1000 dilution or anti-ataxin-7 antibody (Affinity Bioreagents, PA1-749) at 1:500 dilution. For VGLUT2 immunostaining, floating sections were incubated in 0.1 m urea at 85°C for 30 min, followed by cooling to room temperature for 30 min. After a 5 min incubation with 1× PBS, the sections were blocked overnight in 5% normal goat serum (NGS), 1.0% Triton X-100 and 0.2% bovine serum albumin (BSA) at 4°C with shaking. The sections were then incubated with anti-VGLUT2 primary antibody (Millipore, MAB5504) at 1:500 dilution in antibody buffer (5% NGS, 0.3% Triton X-100 and 0.2% BSA) at 4°C with shaking overnight. After the sections were washed 3× in 1× PBS for 10 min, they were incubated with secondary antibody in antibody buffer without BSA at 4°C with shaking overnight. After three 10 min washes in 1× PBS, the sections were blocked overnight a second time before adding a second primary antibody (anti-calbindin) in antibody buffer and incubating overnight at 4°C with shaking. After an additional secondary antibody incubation to detect the anti-calbindin antibody, the sections were washed in 1× PBS, mounted on glass slides and allowed to dry. Fluoro-Gel with Tris buffer mounting media (Electron Microscopy Sciences) was added to the sections before coverslips were applied. For all immunostaining, the secondary antibodies used were AlexaFluor 488 anti-rabbit or anti-mouse IgG (Molecular Probes, Invitrogen) at 1:500 dilution, and/or AlexaFluor 594 anti-rabbit or anti-mouse IgG (Molecular Probes, Invitrogen) at 1:500 dilution. 4′,6-diamidino-2-phenylindole (DAPI) was used at a final concentration of 1:10 000. A Zeiss Axiovert 200 m inverted microscope and Slidebook 5.0 software (Intelligent Imaging Innovations) was used to capture, process and analyze images.
Cerebellar molecular layer thickness and BG process numbers were quantified as previously described (6). Molecular layer measurements were averaged for each genotype, plotted and analyzed statistically in Graphpad Prism 4.0. Average number of processes for each genotype was calculated, plotted and analyzed statistically in Graphpad Prism 4.0.
To quantify the volume of VGLUT2 immunofluorescence, 3-D image Z-stacks were collected from the three most dorsal folia in each section, three sections per animal (n = 6/group). A blinded observer deconvolved the images using the Nearest Neighbors algorithm of Slidebook 5.0, and then rotated the images using bicubic interpolation to position the PC layer parallel to the horizontal axis. A line was drawn from the PC layer to the pial surface. A second line was drawn perpendicular to, and at the midpoint of the first line to separate the molecular layer into two halves: one ‘distal’ to the PC layer and one ‘proximal’ to the PC layer. A segment mask was generated to encompass VGLUT2 immunofluorescence, and a rectangular box measuring 450 × 60 pixels extending through the Z-stack was drawn in the distal region, with a second box drawn in the proximal region. The total volume of VGLUT2 immunofluorescence was calculated within each distal and proximal box and the data were plotted, graphed and analyzed in Graphpad Prism 4.0.
Ataxin-7 positive nuclear inclusions were quantified using calbindin, ataxin-7 and DAPI triple-labeled cerebellar sections. Two-dimensional images were created for the three most dorsal folia per section, three sections per individual, except at 43 weeks (two dorsal folia persection and one sectionper/individual). We used group sizes of n = 3 at 10, 20 and 25 weeks, and n = 5–8 at 43 weeks. The number of ataxin-7-positive nuclear inclusions and the total number of PC somae were quantified in each image, and the average number of ataxin-7 nuclear inclusions/10 PCs was plotted in Graphpad Prism 4.0.
FUNDING
This work was supported by the National Institutes of Health (R01 EY14061 and R01 NS41648 to A.R.L., R01 NS52535 to G.A.G., GM07108 to S.A.F., with facilities support from P30-HD02274 to the UW Center on Human Development and Disability).
ACKNOWLEDGEMENTS
We would like to thank C. Chang, S. Greenwald, J. Song, D.E. Possin and J. Huang for research support.
Conflict of Interest statement. None declared.
References
- 1.Garden G.A., La Spada A.R. Molecular pathogenesis and cellular pathology of spinocerebellar ataxia type 7 neurodegeneration. Cerebellum. 2008;7:138–149. doi: 10.1007/s12311-008-0027-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 3.Yamamoto A., Lucas J.J., Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 4.Zu T., Duvick L.A., Kaytor M.D., Berlinger M.S., Zoghbi H.Y., Clark H.B., Orr H.T. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J. Neurosci. 2004;24:8853–8861. doi: 10.1523/JNEUROSCI.2978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guyenet S.J., Furrer S.A., Damian V.M., Baughan T.D., La Spada A.R., Garden G.A. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J. Vis. Exp. 2010 doi: 10.3791/1787. pii: 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furrer S.A., Mohanachandran M.S., Waldherr S.M., Chang C., Damian V.A., Sopher B.L., Garden G.A., La Spada A.R. Spinocerebellar ataxia type 7 cerebellar disease requires the coordinated action of mutant ataxin-7 in neurons and glia, and displays non-cell-autonomous Bergmann glia degeneration. J. Neurosci. 2011;31:16269–16278. doi: 10.1523/JNEUROSCI.4000-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duvick L., Barnes J., Ebner B., Agrawal S., Andresen M., Lim J., Giesler G.J., Zoghbi H.Y., Orr H.T. SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron. 2010;67:929–935. doi: 10.1016/j.neuron.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnes J.A., Ebner B.A., Duvick L.A., Gao W., Chen G., Orr H.T., Ebner T.J. Abnormalities in the climbing fiber–Purkinje cell circuitry contribute to neuronal dysfunction in ATXN1[82Q] mice. J. Neurosci. 2011;31:12778–12789. doi: 10.1523/JNEUROSCI.2579-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chou A.H., Chen C.Y., Chen S.Y., Chen W.J., Chen Y.L., Weng Y.S., Wang H.L. Polyglutamine-expanded ataxin-7 causes cerebellar dysfunction by inducing transcriptional dysregulation. Neurochem. Int. 2010;56:329–339. doi: 10.1016/j.neuint.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Vig P.J., Hearst S., Shao Q., Lopez M.E., Murphy H.A., 2nd, Safaya E. Glial S100B protein modulates mutant ataxin-1 aggregation and toxicity: TRTK12 peptide, a potential candidate for SCA1 therapy. Cerebellum. 2011;10:254–266. doi: 10.1007/s12311-011-0262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiang Z., Valenza M., Cui L., Leoni V., Jeong H.K., Brilli E., Zhang J., Peng Q., Duan W., Reeves S.A., et al. Peroxisome-proliferator-activated receptor gamma coactivator 1 alpha contributes to dysmyelination in experimental models of Huntington's disease. J. Neurosci. 2011;31:9544–9553. doi: 10.1523/JNEUROSCI.1291-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Custer S.K., Garden G.A., Gill N., Rueb U., Libby R.T., Schultz C., Guyenet S.J., Deller T., Westrum L.E., Sopher B.L., La Spada A.R. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat. Neurosci. 2006;9:1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi S., McMahon A.P. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 14.Fremeau R.T., Jr., Troyer M.D., Pahner I., Nygaard G.O., Tran C.H., Reimer R.J., Bellocchio E.E., Fortin D., Storm-Mathisen J., Edwards R.H. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- 15.Wang H.L., Chou A.H., Lin A.C., Chen S.Y., Weng Y.H., Yeh T.H. Polyglutamine-expanded ataxin-7 upregulates Bax expression by activating p53 in cerebellar and inferior olivary neurons. Exp. Neurol. 2010;224:486–494. doi: 10.1016/j.expneurol.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 16.Ingram M.A., Orr H.T., Clark H.B. Genetically engineered mouse models of the trinucleotide-repeat spinocerebellar ataxias. Brain. Res. Bull. 2011;88:33–42. doi: 10.1016/j.brainresbull.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chakrabarti L., Zahra R., Jackson S.M., Kazemi-Esfarjani P., Sopher B.L., Mason A.G., Toneff T., Ryu S., Shaffer S., Kansy J.W., et al. Mitochondrial dysfunction in NnaD mutant flies and Purkinje cell degeneration mice reveals a role for Nna proteins in neuronal bioenergetics. Neuron. 2010;66:835–847. doi: 10.1016/j.neuron.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark H.B., Burright E.N., Yunis W.S., Larson S., Wilcox C., Hartman B., Matilla A., Zoghbi H.Y., Orr H.T. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J. Neurosci. 1997;17:7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsunemi T., Ashe T.D., Morrison B.E., Soriano K.R., Au J., Roque R.A., Lazarowski E.R., Damian V.A., Masliah E., La Spada A.R. PGC-1α rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3003799. 142ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poirier M.A., Li H., Macosko J., Cai S., Amzel M., Ross C.A. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 2002;277:41032–41037. doi: 10.1074/jbc.M205809200. [DOI] [PubMed] [Google Scholar]
- 21.Wacker J.L., Zareie M.H., Fong H., Sarikaya M., Muchowski P.J. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat. Struct. Mol. Biol. 2004;11:1215–1222. doi: 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- 22.Helmlinger D., Abou-Sleymane G., Yvert G., Rousseau S., Weber C., Trottier Y., Mandel J.L., Devys D. Disease progression despite early loss of polyglutamine protein expression in SCA7 mouse model. J. Neurosci. 2004;24:1881–1887. doi: 10.1523/JNEUROSCI.4407-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hands S.L., Wyttenbach A. Neurotoxic protein oligomerisation associated with polyglutamine diseases. Acta Neuropathol. 2010;120:419–437. doi: 10.1007/s00401-010-0703-0. [DOI] [PubMed] [Google Scholar]
- 24.Sydow A., Van der Jeugd A., Zheng F., Ahmed T., Balschun D., Petrova O., Drexler D., Zhou L., Rune G., Mandelkow E., et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J. Neurosci. 2011;31:2511–2525. doi: 10.1523/JNEUROSCI.5245-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]