

Abstract
Design of non-nucleoside inhibitors of HIV-1 reverse transcriptase with improved activity towards Tyr181Cys containing variants was pursued with the assistance of free energy perturbation (FEP) calculations. Optimization of the 4-R substituent in 1 led to ethyl and isopropyl analogs 1e and 1f with 1 – 7 nM potency towards both the wild-type virus and a Tyr181C variant.
Non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs) are an important component of combination therapies for treatment of HIV infection.1,2 Their further improvement is important to seek safer alternatives that can be given in low dosages and that may be needed to combat newly emerging viral variants. Compounds that can penetrate the blood-brain barrier well might provide an additional advance for addressing latent viral reservoirs.3 A continuing challenge for developing NNRTIs is achievement of activity against clinically relevant viral variants that incorporate single and multiple mutations in the reverse transcriptase enzyme (HIV-RT). A particularly troublesome mutation has been Tyr181Cys (Y181C), which often arises quickly in patients who begin NNRTI therapy.4 The first generation drugs, nevirapine and delavirdine, are inactive towards HIV-1 strains with this mutation, and the second-generation efavirenz is debilitated by Y181C when combined with Lys103Asn.1,4 In contrast, the most recent introductions, etravirine and rilpivirine, show sub-10 nM potency in cell assays towards these variants and many others.5
In our own work, several new classes of NNRTIs have been explored.6–9 The Y181C variant has always been problematic and it has required deliberate efforts to overcome. One solution involved reduction of contact of the inhibitors with Tyr181,8 while another took advantage of a crystal structure with an alternative orientation of Tyr181.9 A third approach was to enhance interactions in a distal region of the NNRTI binding site that might yield general benefits for activity.7c Specifically, 1a (R = X = H) has an EC50 of 13 nM towards wild-type HIV-1, but shows no activity towards a Y181C-containing strain. The X = Cl analog 1b fares only a little better with EC50s of 6 nM for the WT virus and 420 nM for the Y181C variant.7b A remedy was sought by extending the inhibitors to the ‘east’ to occupy a channel between Phe227 and Pro236. Though it was possible to replace the cyano group of 1 with novel alternatives, the best that was found for activity was 2a (R = H) with EC50s of 31 nM and 3 μM, respectively.7b
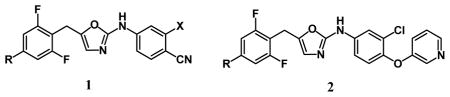
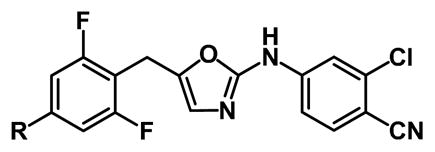
As described here, a new effort for the oxazoles 1 has been made by analyzing the potential for productive modifications of the 4-R group. The strategy was to seek improved Y181C activity, even if there is some loss of WT activity. Structural model building with the BOMB program and OPLS force fields10 suggested that some elaboration of R might be possible, but that substituents much larger than methyl might lead to steric clashes with the WT protein. However, as illustrated for the case with R = ethyl in Figure 1, it was expected that a group such as ethyl or propyl might constructively occupy the space vacated by the Tyr181 to Cys181 change. The problem with such structural visualization is that too many complexes look good. One cannot see if contacts are, in fact, too close, and one cannot visualize potential entropic losses owing to restriction of translational, rotational, or torsional degrees of freedom. Thus, as in the past, we turned to free-energy perturbation (FEP) calculations with configurational sampling at 25 °C using Monte Carlo (MC) simulations to obtain quantitative predictions.6–10
Figure 1.

Snapshots of 1e bound to the NNRTI site for wild-type (top) and Tyr181Cys HIV-RT from MC/FEP simulations. Carbon atoms of 1e are in yellow. Some residues are omitted for clarity.
The MC/FEP calculations followed the same protocols as previously described.7,8 Initial coordinates of the complexes were constructed from the 1S9E PDB file11 using the MCPRO12 and BOMB programs.10 The Y181C variant was generated manually from the WT structure and all complexes were relaxed with short conjugate gradient minimizations. The model included the 178 amino acid residues nearest the ligand. The unbound ligands and complexes were solvated in 25-Å caps with 2000 and 1250 TIP4P water molecules. The FEP calculations utilized 11 windows of simple overlap sampling. Each window covered 10–15 million (M) configurations of equilibration and 20–30 M configurations of averaging. The energetics were evaluated with the OPLS-AA force field for the protein, OPLS/CM1A for the ligands, and TIP4P for water.13 The MC/FEP calculations were carried out for both the WT and Y181C variant of HIV-RT with MCPRO; the resultant relative free energies of binding are summarized in Table 1.
Table 1.
MC/FEP Results for Relative Free Energies of Binding (kcal/mol).
| ||||
|---|---|---|---|---|
| R | Wild-Type | Tyr181Cys | ||
| ΔΔ Gb | σa | ΔΔGb | σa | |
| H | 0.0 | 0.0 | 0.0 | 0.0 |
| F | 0.03 | 0.07 | −0.14 | 0.05 |
| Cl | −2.38 | 0.08 | −2.33 | 0.07 |
| Clxb | −3.33 | 0.09 | −3.09 | 0.08 |
| Me | −4.11 | 0.11 | −4.72 | 0.11 |
| Et | −5.71 | 0.19 | −7.62 | 0.18 |
| Pr | −4.34 | 0.24 | −9.18 | 0.25 |
| i-Pr | −4.93 | 0.26 | −10.17 | 0.23 |
| (R)-s-Bu | −3.46 | 0.34 | −8.25 | 0.34 |
| (S)-s-Bu | −2.55 | 0.34 | −7.59 | 0.29 |
| t-Bu | −3.90 | 0.35 | −8.87 | 0.30 |
| OMe | −4.11 | 0.30 | −4.73 | 0.28 |
| OEt | −3.14 | 0.30 | −6.07 | 0.30 |
± σ is the computed uncertainty in ΔΔGb.
Includes an extra point charge on Cl to enable halogen bonding; see ref. 14.
For the WT protein, the prediction is that binding should be enhanced by replacing R = H by a methyl or ethyl group, but further enlargement would be detrimental. Halogen and ether substituents are also predicted to be less effective than small alkyl groups. Then, the results for the Y181C variant are striking with the prediction that there would be significantly greater gain than for the WT enzyme and that peak binding should occur for R = isopropyl. It might be noted that by visualization, the R = s-Bu analogs look very nice with the two carbon branch extending into the Tyr181 cavity (Figure 2). Again, visualization is inadequate. Thus, the expectation was that for R = Et and i-Pr, the WT activity should be good relative to R = H, but there should be significantly greater gains in potency towards the Y181C-bearing virus.
Figure 2.
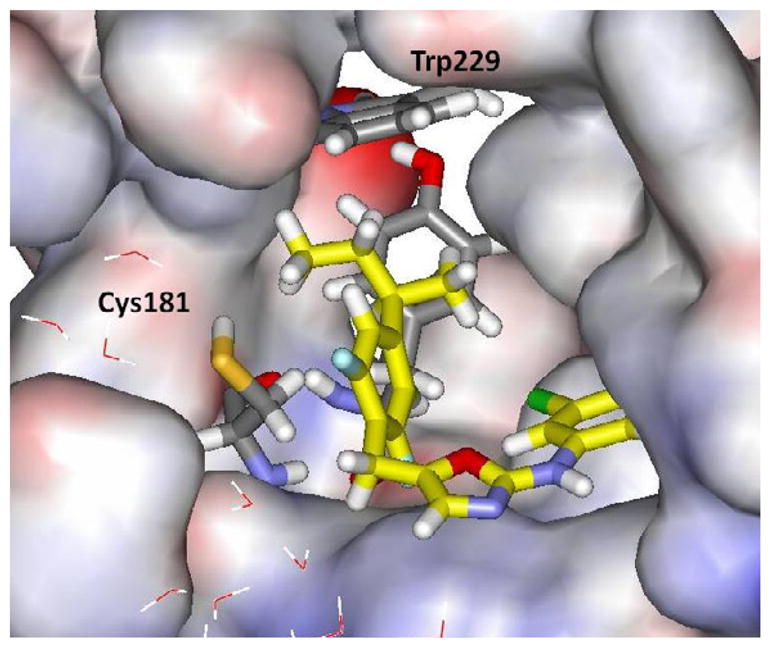
Snapshot of the (R)-s-butyl analog 1i bound to the NNRTI site for Tyr181Cys HIV-RT from MC/FEP simulations. The s-butyl group is likely too constrained to yield improved potency over the i-Pr analog.
The synthesis of analogs of 1 then proceeded featuring the previously utilized cyclization of α-azidoketones and phenyl isothiocyanates (Scheme 1).7 The azides were prepared from substituted 2,6-difluorophenylacetic acids via the α-bromo analogs, as previously described (Scheme 2).7 In turn, the desired acids were prepared starting with aldehydes, which were reduced to alcohols using NaBH4. The alcohols were converted to the bromomethyl derivatives with CBr4 and PPh3, and then treated with TMSCN using K2CO3 as an additive in MeCN to provide the corresponding nitriles in good yield. The nitriles were finally hydrolyzed to the requisite carboxylic acids under acidic or basic conditions (Scheme 2).
Scheme 1.
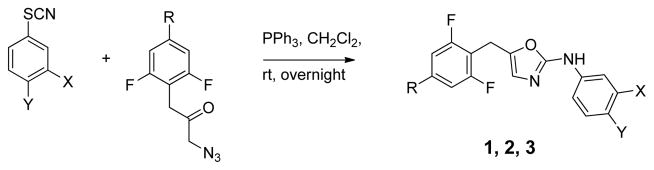
Synthesis of 2-anilinyl-5-benzyloxazoles.
Scheme 2.
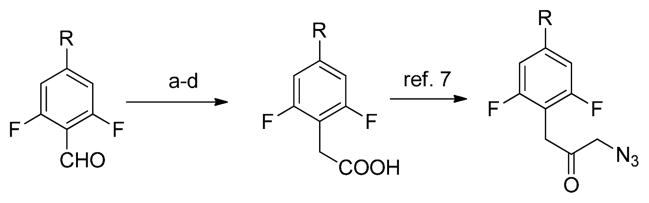
Synthesis of α-azidoacetophenones.
Installation of the 4-R group in the benzyl ring of the aldehydes was generally straightforward starting with the 4-bromo, hydroxyl, or hydroxymethyl precursor.15–20 The one problematic case was for R = t-Bu (Scheme 3). 4-t-Butylbenzoate methyl ester underwent selective dinitration at the positions meta to the t-Bu group. Both nitro groups were reduced, but various conditions for a Balz-Schiemann reaction all failed to effect replacement of the diazonium groups by fluorine. However, it was possible to prepare the 2,6-dichloro derivative from the diamine via a Sandmeyer reaction, which was followed by reduction of the ester to the alcohol and conversion to the carboxylic acid via steps b–d in Scheme 2.
Scheme 3.
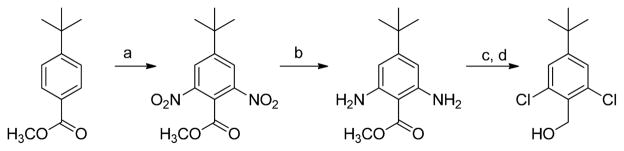
Synthesis of a 4-t-butylbenzyl alcohol.
The identities of all assayed compounds were confirmed by 1H and 13C NMR and high-resolution mass spectrometry; purity was >95% as judged by high-performance liquid chromatography. Activities against the IIIB and variant strains of HIV-1 were measured using MT-2 human T-cells; EC50 values are obtained as the dose required to achieve 50% protection of the infected cells by the MTT colorimetric method. CC50 values for inhibition of MT-2 cell growth by 50% are obtained simultaneously.6–9,21,22 The results are summarized in Table 2.
Table 2.
Anti-HIV-1 Activity (EC50) and cytotoxicity (CC50), μMa
| Compound | R | X | WT | EC50
|
CC50 | |
|---|---|---|---|---|---|---|
| Y181C | K103N/Y181C | |||||
| 1a | H | H | 0.013 | NA | NA | 8 |
| 1b | H | Cl | 0.0062 | 0.420 | NA | 10 |
| 1c | F | Cl | 0.0091 | 0.910 | NA | 7 |
| 1d | Me | Cl | 0.011 | 0.210 | 7.2 | 31 |
| 1e | Et | Cl | 0.0013 | 0.0069 | 0.21 | 10 |
| 1f | i-Pr | Cl | 0.0052 | 0.0072 | 0.12 | 5 |
| 1g | c-Pr | Cl | 0.024 | 1.5 | 10.7 | 65 |
| 1h | CH2c-Pr | Cl | 0.068 | 1.2 | 15.0 | 29 |
| 1ib | s-Bu | Cl | 0.120 | 0.15 | 2.8 | 40 |
| 1j | OEt | Cl | 0.028 | 0.048 | 0.75 | 15 |
| 1k | CH2OMe | Cl | 0.0036 | 0.690 | 6.3 | 15 |
| 2a | H | 0.031 | 3.2 | 4.5 | 16 | |
| 2b | Et | 0.005 | 0.060 | 0.80 | 5 | |
| 3c | t-Bu | Cl | 1.3 | NA | NA | 6 |
| nevirapine | 0.11 | NA | NA | >100 | ||
| efavirenz | 0.002 | 0.010 | 0.030 | 15 | ||
| etravirine | 0.001 | 0.008 | 0.005 | 11 | ||
| rilpivirine | 0.00067 | 0.00065 | 0.002 | 8 | ||
Results using human MT-2 cells. Antiviral and toxicity curves used triplicate samples at each concentration.
NA = not active.
Racemic.
The 4-t-butyl-2,6-dichlorobenzyl analog of 1b.
For the WT virus, the ethyl analog 1e did turn out to be the most potent with an EC50 of 1 nM. The gain of a factor of 5 over 1b is less than what might have been expected from Table 1. However, computed free energies of binding are being compared to activities in a cell-based assay and a compression of the computed results is normal.6–10,23 Additional elaboration of the R group was unproductive, as expected, and the 4-t-butyl-2,6-dichloro analog 3 is only a 1 μM NNRTI. For the Y181C HIV-1 variant, it was gratifying that the qualitative predictions from the MC/FEP calculations were confirmed. Indeed, there is pronounced improvement in the activity towards the Y181C-containing virus peaking with the ethyl and isopropyl analogs 1e and 1f with EC50s of 7 nM. As from the computations, the s-Bu analog 1i is significantly less active. For 1f, the potency towards the WT and Y181C variant form of HIV-1 are essentially the same, in sharp contrast to the case for the parent 1b. There is also improvement in potency towards the challenging K103N/Y181C double mutant. Though 1f has about the same potency towards HIV-1 bearing the two mutations as the drug nevirapine has towards the WT virus, the potencies of etravirine and rilpivirine towards the variant are 24- and 60-fold greater still.
Another interesting comparison is for the isopropyl and cyclopropyl analogs 1f and 1g. The cyclopropyl compound is less active against the WT virus by a factor of 5 and 200-fold less active towards the Y181C variant. In the MC/FEP simulations, the isopropyl group is oriented much as in Figure 1 (bottom) with significant projection of one methyl group towards the Tyr181 cavity. Though FEP calculations were not done for the cyclopropyl analog, model building does show the expected reduced projection owing to the ca. 60° internal CCC angles. A cyclopropyl group is decidedly more compact than an isopropyl group, which is disadvantageous in the present case. A cyclopropyl group is also somewhat less lipophilic as apparent in observed octanol/water log P values of 3.3 for cyclopropylbenzene and 3.7 for isopropylbenzene.24
Finally, to integrate the investigation of the ‘eastern extensions’7b with the present work, 2b was synthesized. In comparison to 2a, addition of the ethyl group provides a 5-fold activity boost against the WT virus and a 50-fold gain against the Y181C-bearing variant. The fold gains are almost the same as for the 1b and 1e pair.
In summary, further study of oxazoles 1 as NNRTIs has focused on optimization of the 4-R group to yield greater potency against strains of HIV-1 containing the Tyr181Cys mutation in the reverse transcriptase enzyme. Results of MC/FEP calculations were highly provocative in predicting that substantial differential gains would be obtained for the viral variant over the wild-type virus for R = ethyl and isopropyl. This was confirmed experimentally through synthesis and assaying of the analogs in Table 2. 1e and 1f emerged as NNRTIs with sub-10 nM potency towards both viral strains.
Acknowledgments
Gratitude is expressed to the National Institutes of Health (AI44616, GM32136, GM49551) for support. Receipt of reagents through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH is also greatly appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prajapati DG, Ramajayam R, Yadav MR, Giridhar R. Bioorg Med Chem. 2009;17:5744. doi: 10.1016/j.bmc.2009.06.060. [DOI] [PubMed] [Google Scholar]
- 2.de Bethune MP. Antiviral Res. 2010;85:75. doi: 10.1016/j.antiviral.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 3.(a) Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. Science. 2009;323:1304. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]; (b) Richman DD. Curr Opin HIV AIDS. 2011;6:1. doi: 10.1097/COH.0b013e328340ffa6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Clerq E. Nature Rev Drug Disc. 2007;6:1001. doi: 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- 5.Janssen PAJ, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, Heeres J, Koymans L, Vinkers M, Guillemont J, Pasquier E, Kukla M, Ludovici D, Andries K, de Bethune MP, Pauwels R, Das K, Clark AD, Jr, Frenkel YV, Hughes SH, Medaer B, De Knaep F, Bohets H, De Clerck F, Lampo A, Williams P, Stoffels P. J Med Chem. 2005;48:1901. doi: 10.1021/jm049534r. [DOI] [PubMed] [Google Scholar]
- 6.Jorgensen WL, Ruiz-Caro J, Tirado-Rives J, Basavapathruni A, Anderson KS, Hamilton AD. Bioorg Med Chem Lett. 2006;16:663. doi: 10.1016/j.bmcl.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 7.(a) Zeevaart JG, Wang L, Thakur VV, Leung CS, Tirado-Rives J, Bailey CM, Domaoal RA, Anderson KS, Jorgensen WL. J Am Chem Soc. 2008;130:9492. doi: 10.1021/ja8019214. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Leung CS, Zeevaart JG, Domaoal RA, Bollini M, Thakur VV, Spasov K, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2010;20:2485–2488. doi: 10.1016/j.bmcl.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jorgensen WL, Bollini M, Thakur VV, Domaoal RA, Spasov K, Anderson KS. J Am Chem Soc. 2011;133:15686. doi: 10.1021/ja2058583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bollini M, Domaoal RA, Thakur VV, Gallardo-Macias R, Spasov KA, Anderson KA, Jorgensen WL. J Med Chem. 2011;54:8582–8591. doi: 10.1021/jm201134m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jorgensen WL. Acc Chem Res. 2009;42:724–733. doi: 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das K, Clark AD, Jr, Lewi PJ, Heeres J, de Jonge MR, Koymans LMH, Vinkers HM, Daeyaert F, Ludovici DW, Kukla MJ, De Corte B, Kavash RW, Ho CY, Ye H, Lichtenstein MA, Andries K, Pauwels R, de Béthune MP, Boyer PL, Clark P, Hughes SH, Janssen PAJ, Arnold E. J Med Chem. 2004;47:2550. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 12.Jorgensen WL, Tirado-Rives J. J Comput Chem. 2005;26:1689. doi: 10.1002/jcc.20297. [DOI] [PubMed] [Google Scholar]
- 13.Jorgensen WL, Tirado-Rives J. Proc Natl Acad Sci USA. 2005;102:6665. doi: 10.1073/pnas.0408037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jorgensen WL, Schyman P. J Chem Theory Comput. 2012;8:1689. doi: 10.1021/ct300180w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallace DJ, Cheng CY. Tetrahedron Lett. 2002;43:6987. [Google Scholar]
- 16.Kumada M. Pure Appl Chem. 1980;52:669. [Google Scholar]
- 17.Doherty S, Knight JC, Robins EG, Scanlan TH, Champkin PA, Clegg W. J Am Chem Soc. 2001;123:5110. doi: 10.1021/ja010090n. [DOI] [PubMed] [Google Scholar]
- 18.Liang X, Bols MJ. Chem Soc, Perkin Trans 1. 2002:503. [Google Scholar]
- 19.Zhang D, Chen Z, Cai H, Zou X. J Fluor Chem. 2009;130:938. [Google Scholar]
- 20.Massah AR, Mosharafian M, Momeni AR, Aliyan H, Naghash H, Adibnejad M. Synth Commun. 2007;37:1807. [Google Scholar]
- 21.Lin TS, Luo MZ, Liu MC, Pai SB, Dutschman GE, Cheng YC. Biochem Pharmacol. 1994;47:171. doi: 10.1016/0006-2952(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 22.Ray AS, Yang Z, Chu CK, Anderson KS. Antimicrob Agents Chemother. 2002;46:887. doi: 10.1128/AAC.46.3.887-891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guimarães CRW. J Chem Theory Comput. 2011;7:2296. doi: 10.1021/ct200244p. [DOI] [PubMed] [Google Scholar]
- 24.Hansch C, Leo A, Hoekman D. Exploring QSAR – Hydrophobic, Electronic, and Steric Constants. American Chemical Society; Washington: 1995. [Google Scholar]