


Abstract
The E3 ubiquitin ligase Mule/ARF-BP1 plays an important role in the cellular DNA damage response by controlling base excision repair and p53 protein levels. However, how the activity of Mule is regulated in response to DNA damage is currently unknown. Here, we report that the Ser18-containing isoform of the USP7 deubiquitylation enzyme (USP7S) controls Mule stability by preventing its self-ubiquitylation and subsequent proteasomal degradation. We find that in response to DNA damage, downregulation of USP7S leads to self-ubiquitylation and proteasomal degradation of Mule, which eventually leads to p53 accumulation. Cells that are unable to downregulate Mule show reduced ability to upregulate p53 levels in response to DNA damage. We also find that, as Mule inactivation is required for stabilization of base excision repair enzymes, the failure of cells to downregulate Mule after DNA damage results in deficient DNA repair. Our data describe a novel mechanism by which Mule is regulated in response to DNA damage and coordinates cellular DNA damage responses and DNA repair.
INTRODUCTION
The p53 tumour suppressor protein plays a major role in the cellular DNA damage response by initiating either a cell cycle delay, to allow the completion of DNA repair processes before DNA replication, or by inducing apoptotic cell death in the case of excessive DNA damage that cannot be repaired (1,2). Although Mdm2 is widely accepted to be the major E3 ubiquitin ligase involved in the regulation of cellular p53 levels (3,4), other E3 ubiquitin ligases, such as Mule (5), COP1 (6) and Pirh2 (7), have been shown to regulate the stability and activity of p53 in vitro and in living cells. However, their specific role in the cellular DNA damage response is unclear.
Among these E3 ubiquitin ligases, Mule (also known as ARF-BP1, E3Histone, UREB1, HUWE1, HECTH9 and LASU1) has recently attracted a significant amount of attention, as it was discovered to play multiple roles at the various stages of the cellular responses to DNA damage. Mule has been shown to play a key role in the tuning of the capacity of the base excision repair system, and, consequently, its ability to respond to oxidative DNA damage, by regulating the cellular levels of DNA polymerase β (8) and DNA polymerase λ (9). Other substrates of Mule, involved in multiple cellular processes, such as cell proliferation, apoptosis and DNA repair, have also been identified [for review see (10)]. Importantly, it has been demonstrated that Mule ubiquitylates p53 protein directly, as depletion of Mule using siRNA results in a significant increase in the cellular levels of p53, and, consequently, elevates p53-induced apoptosis (5). This observation was further supported by the generation of mule knockout mice, which were found to be embryonic lethal because of the significant accumulation of p53 (11). Taken together, these data indicate that p53 suppression in unstressed cells is one of the major functions of Mule. Therefore, in response to DNA damage, the p53-suppression function of Mule should be downregulated to enable the cellular DNA damage response. Indeed, Mule is inhibited by the ARF tumour suppressor protein that is induced by oncogenic stress (5,8). However, ARF-deficient cells are still able to elevate p53 levels in response to ionizing radiation (12), suggesting the existence of an ARF-independent pathway for Mule downregulation in response to DNA damage.
We have recently identified an important role for a specific isoform of USP7 that is phosphorylated at serine 18 residue (further referred to as USP7S) in regulation of p53 levels in response to DNA damage (13). As it was previously shown that Mule may potentially interact with USP7S (14), we decided to investigate the role of USP7S in the regulation of Mule. Here, we report that USP7S controls the cellular levels of Mule in response to DNA damage, and it regulates the efficiency of DNA repair.
MATERIALS AND METHODS
Antibodies, proteins, plasmids, chemicals and cell lines
Polyclonal pUSP7S and USP7S antibodies were produced by Biomatik as described in (13). Actin (ab6276), ubiquitin (ab7254) and HA (ab9110) antibodies were purchased from Abcam, Flag antibodies (200471) were from Agilent Technologies, p53 (sc-126) antibodies were from Santa Cruz, Mule antibodies used for in vivo and in vitro studies were from Bethyl Laboratories (UREB1, BET-A300-486A) and ProSci (4213), respectively, Pol λ antibodies (A301-640A) were from Bethyl Laboratories and PPM1G antibodies were kindly provided by O. Gruss. Pol β antibodies were produced as described in (15).
Ubiquitin, E1 and E2 enzymes were purchased from Boston Biochemicals. Mammalian expression vectors encoding the usp7s wild-type as well as usp7sC223S mutant genes and production and purification of the corresponding proteins were as described in (13). Bacterial and mammalian expression vectors encoding Mule HECT-domain were kindly provided by Dr M. Eilers. The GST-tagged truncated Mule protein was expressed in Escherichia coli cells and purified using GSTrap FF column chromatography (GE Healthcare).
HeLa (adenocarcinoma) and HCT116p53+/+ (colorectal carcinoma) cell lines were purchased from ATCC.
Whole-cell extracts
Whole-cell extracts were prepared as described in (8).
RNA interference
Cells were transfected using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer’s protocol. The siRNA sequences used to target the Mule, PPM1G and Mdm2 transcripts were as follows: 5′-AAUUGCUAUGUCUCUGGGACA-3′ (5), 5′-AGGCUACCAUGACUAUUGA-3′ (16) and 5′-AAGCCAUUGCUUUUGAAGUUA-3′ (17), respectively. To knockdown Ser18-containing USP7 isoform (USP7S), the 5′-AGCGGGCGAGCAGCAGUUG-3′ sequence was used (13). The GFP siRNA sequence (5′-GCUGACCCUGAAGUUCAUCUU-3′) served as a control.
DNA damaging treatments
Cells were subjected to ionizing radiation using GSR-D1 137Cs γ-irradiator (RPS Services Limited) at a dose rate of 1.8 Gy/min (8-Gy dose). For hydrogen peroxide and etoposide treatments, cells were treated with either 150 µM H2O2 for 15 min or 10 µM etoposide dissolved in DMSO for 1 h. The medium was changed after the treatment, and cells were incubated at 37°C to allow for DNA repair. The Comet assay was performed as described in (8).
In vitro ubiquitylation and deubiquitylation assays
Mule (25 pmol) was self-ubiquitylated in the presence of 7 pmol E1, 65 pmol UbcH7 and 6 pmol ubiquitin in buffer containing 25 mM Tris–HCl, pH 8.0, 4 mM adenosine triphosphate, 5 mM MgCl2, 200 µM CaCl2, 1 mM DTT and 10 µM MG-132 for 1 h at 30°C with shaking. Ubiquitin and E2 were removed from the reaction mixture by filtration and buffer exchange into deubiquitylation buffer containing 50 mM Tris–HCl, pH 8.0, 150 mM KCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 5% glycerol and 2 mM DTT using Amicon Ultra 30K units (Millipore). For the deubiquitylation assay, ubiquitylated Mule (0.6 pmol) was incubated with equal amounts of wild-type USP7S or inactive C223S USP7S mutant (1.2 and 2.4 pmol) in deubiquitylation buffer for 30 min at 30°C with shaking. To stop the reaction, sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer (25 mM Tris–HCl, pH 6.8, 2.5% β-mercaptoethanol, 1% SDS, 5% glycerol, 1 mM EDTA and 0.05 mg/ml bromophenol blue) was added to samples, which were then heated for 5 min at 95°C before separation of the proteins on a 4–20% SDS–PAGE gel, followed by transfer to a PVDF membrane and western blotting with Ubi, Mule and USP7S antibodies.
Western blots
Western blots were performed by standard procedure as recommended by the vendor (Novex). Blots were visualized and quantified using the Odyssey image analysis system (Li-Cor Biosciences). For quantification (below the appropriate figure), the expression levels of proteins were normalized against the actin loading control as calculated from at least three independent experiments.
Real-time polymerase chain reaction
Real-time polymerase chain reaction was performed as described in (13), using the 5′-TGAATGCTCTGGCTGCATAC-3′ and 5′-CCCCAGGTTTAGGATCAGATT-3′ primers for amplification of the Mule transcript. The results are presented as a mean ± standard deviation of three independent biological experiments. Statistical analysis was performed using Student’s paired t-test with a two-tailed distribution.
RESULTS AND DISCUSSION
USP7S interacts with Mule and prevents its self-ubiquitylation
To address the role of USP7S in the regulation of Mule protein levels, we firstly followed up the proteomic studies by the Harper laboratory (14) that suggested an interaction of Mule with USP7S and demonstrated that catalytically active Mule HECT-domain efficiently interacts with USP7S, both in vitro (Figure 1A) and in vivo (Figure 1B). As with many other E3 ubiquitin ligases, Mule is self-ubiquitylated when incubated in the presence of ubiquitin [(5); Figure 1C, first lane]. In a cellular environment, such self-ubiquitylation can result in proteasomal degradation of the protein, although this may be prevented by deubiquitylation. Therefore, we analysed whether USP7S can limit Mule self-ubiquitylation through its associated deubiquitylation activity. Indeed, using self-ubiquitylated Mule as a substrate, we demonstrated that USP7S, but not its catalytically inactive mutant (C223S), efficiently deubiquitylates Mule in an in vitro reaction reconstituted with the recombinant proteins (Figure 1C).
Figure 1.
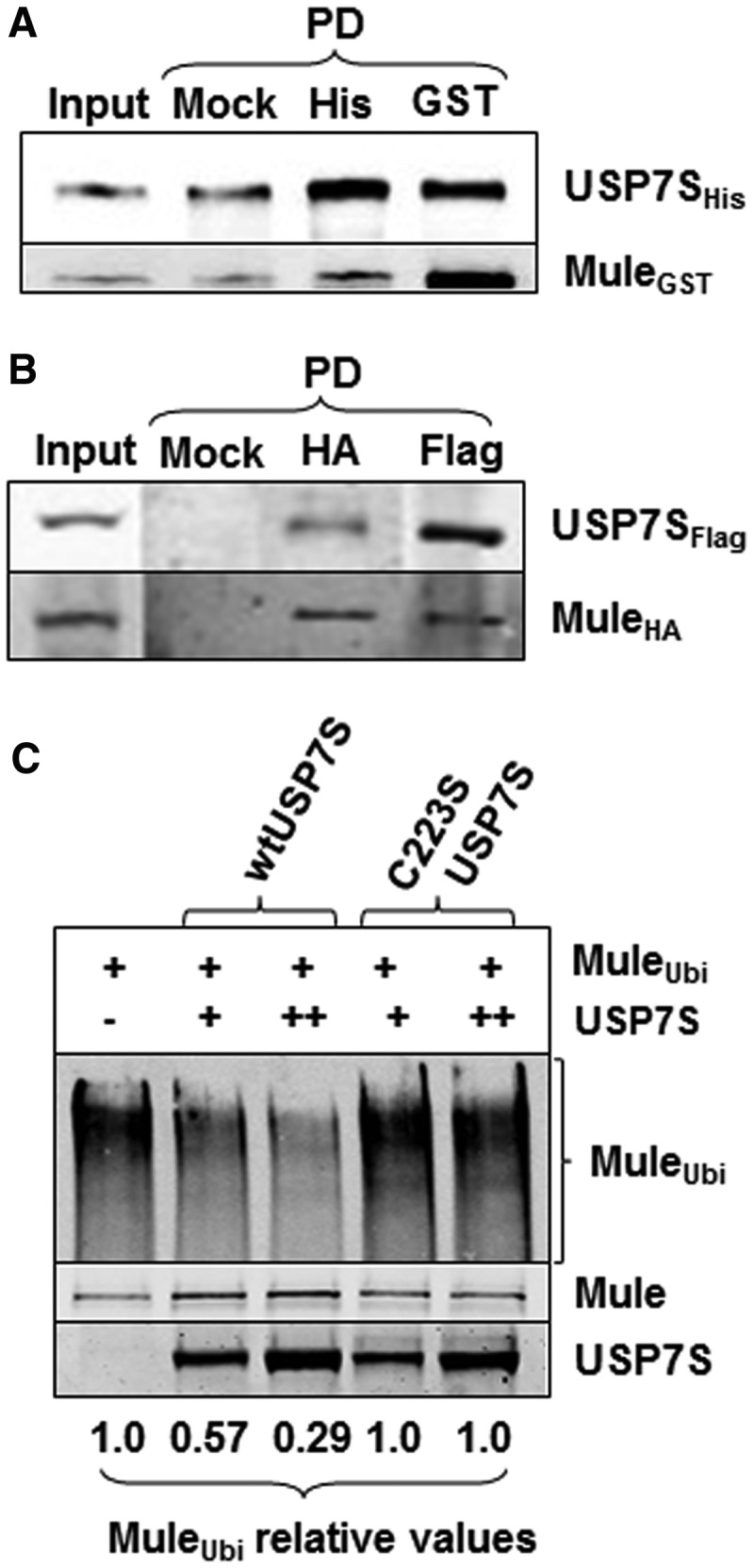
USP7S interacts with Mule and prevents its self-ubiquitylation in vitro. (A) Equal amounts (2.5 pmol) of purified recombinant His-tagged USP7S and GST-tagged Mule were incubated in buffer containing 50 mM Tris–HCl, pH 8.0, 50 mM KCl, 1 mM EDTA, 1 mM DTT for 30 min at room temperature with shaking. USP7S and Mule were then pulled down using either magnetic beads as a control (Mock PD, 100%) or using magnetic Ni-NTA agarose beads (His PD, 100%) or GST agarose beads (GST PD, 100%), respectively. The beads were boiled in SDS–PAGE sample buffer for 5 min, and proteins were separated by 10% SDS–PAGE and analysed using USP7S and Mule antibodies (10% of the input is loaded). (B) HeLa cells were simultaneously transfected with USP7S and Mule (1 pmol each) expression plasmids, whole-cell extracts were prepared and used to pull down HA-tagged Mule or Flag-tagged USP7S using either magnetic beads as a control (Mock PD, 100%), HA- (HA PD, 100%) or Flag-agarose (Flag PD, 100%) beads. Proteins were separated by 4–20% SDS–PAGE and analysed using USP7S and Mule antibodies (15% of the input is loaded). (C) In vitro deubiquitylation of self-ubiquitylated Mule HECT-domain by recombinant wild-type USP7S protein (wtUSP7S) or its inactive mutant (C223S USP7S) analysed by western blotting. Equal loading of the enzyme is demonstrated using USP7S antibodies.
USP7S controls the cellular levels of Mule
Having established that Mule and USP7S form a complex and that USP7S deubiquitylates self-ubiquitylated Mule in vitro, we questioned whether USP7S also controls Mule protein levels in living cells. To answer this question, we suppressed the expression of USP7S in HeLa cells using siRNA and found decreased levels of Mule (Figure 2A). To further prove that downregulation of Mule protein levels after USP7S knockdown is because of a reduced amount of USP7S, rather than an indirect effect of the siRNA, we first depleted USP7S and then transfected these cells with a plasmid encoding siRNA resistant USP7S mRNA and observed a complete restoration of Mule protein levels (Figure 2B). Interestingly, overexpression of USP7S in HeLa cells did not affect the cellular levels of Mule (Figure 2C), suggesting that there is a cellular excess of the USP7S deubiquitylating activity that ensures relatively high stability of Mule, which consequently suppresses the levels of the p53 tumour suppressor under unstressed conditions.
Figure 2.

Cellular levels of Mule are regulated by USP7S. (A) HeLa cells were transfected with USP7S-specific siRNA (200 pmol) for 72 h. (B) HeLa cells were first transfected with 200 pmol USP7S-specific siRNA for 48 h and then transfected with a plasmid encoding siRNA resistant USP7S (0.3 pmol) for further 24 h. (C) HeLa cells were transfected with USP7S (0.6 pmol) expressing plasmid for 24 h. (A–C) Whole-cell extracts were prepared, proteins were separated by 4–20% SDS–PAGE and analysed by western blotting using the indicated antibodies. (D) HeLa cells were transfected with USP7S specific siRNA as previously described, total RNA was prepared and the levels of Mule and GAPDH mRNA were analysed by quantitative real-time polymerase chain reaction in three independent experiments. The knockdown efficiency was confirmed by western blotting. (E) HeLa cells were subjected to the USP7S knockdown as previously described; after 48 h, cells were treated with 2 µM MG-132 for 14 h. Whole-cell extracts were prepared, and proteins were separated by 4–20% SDS–PAGE and analysed by western blotting using the indicated antibodies.
To prove that reduced Mule protein levels in USP7S depleted cells were a result of its protein degradation, rather than downregulation of mule gene transcription, we measured Mule mRNA levels in USP7S-knockdown HeLa cells. Indeed, we detected an increased, rather than a decreased, level of mule mRNA in these cells compared with control cells (Figure 2D). An elevation of mule mRNA levels involves, most probably, a compensatory mechanism that is intended to restore cellular Mule protein levels, so that the steady-state equilibrium of the E3 ubiquitin ligase, and its substrates, can be maintained. Having proved that the regulation of cellular Mule levels by USP7S occurs at a protein level, we further aimed to demonstrate that this downregulation is accomplished through proteasomal degradation. To this end, after a USP7S knockdown that leads to reduction in Mule cellular protein levels (Figure 2E, lane 2), we treated cells with the proteasome inhibitor MG-132 and were able to restore Mule protein levels (Figure 2E, lane 3). Thus, we concluded that USP7S prevents Mule self-ubiquitylation and subsequent proteasomal degradation.
Downregulation of Mule in response to DNA damage is controlled by USP7S
Taken together, our data suggest that the steady-state protein levels of Mule are tightly regulated by USP7S-dependent deubiquitylation that balances Mule self-ubiquitylation; therefore, it is controlled mainly by the activity of the USP7S protein. We have recently demonstrated that under acute DNA damage stress, USP7S is downregulated at the early stages of the cellular DNA damage response (13). As we have linked the cellular steady-state protein levels of Mule to the level and activity of USP7S protein, we hypothesized that Mule may also be downregulated during the early stages of the DNA damage response. Indeed, when we monitored the effect of various DNA damaging agents on Mule protein levels, we found that within 15–60 min after treatment with hydrogen peroxide (Figure 3A), etoposide (Figure 3B) or ionizing radiation (Figure 3C), both pUSP7S and Mule were simultaneously reduced. This was followed by a restoration in their protein levels at later time points post-treatment and highlights a direct correlation between the protein levels of USP7S and Mule. As we previously demonstrated that proteasomal degradation of USP7S in response to DNA damage is triggered by its dephosphorylation by the ATM-dependent phosphatase PPM1G (13), we prompted to analyse whether deregulation of Mule protein levels in response to DNA damage is also dependent on PPM1G activity. To this end, we treated PPM1G siRNA-depleted HCT116p53+/+ cells with etoposide and observed that there was no decrease in Mule, as well as pUSP7S protein levels, in response to DNA damage in cells lacking PPM1G in comparison with control-treated cells (Figure 3D). This suggests that PPM1G is required for downregulation of Mule protein levels, in response to DNA damage.
Figure 3.

Mule is downregulated after DNA damage, and this is controlled by USP7S. (A–C) HCT116 p53+/+ cells were either left untreated (Contr) or exposed to (A) 150 µM hydrogen peroxide for 15 min, (B) 10 µM etoposide (Eto) for 1 h or (C) 8-Gy ionizing radiation (IR) and harvested at the indicated time points post-treatment. (D) HCT116 p53+/+ cells were transfected with PPM1G siRNA (200 pmol) for 72 h, cells were then harvested either without any additional treatment (−) or 1 h post-treatment with etoposide (+) as previously described. Whole-cell extracts were prepared, proteins were separated by 4–20% SDS–PAGE and analysed by western blotting using the indicated antibodies.
Downregulation of Mule after DNA damage is required for efficient DNA repair and p53 upregulation
It was earlier demonstrated that a Mule knockdown, in a similar way as an Mdm2 knockdown, stabilizes p53 and, therefore, reduces the rate of cell proliferation and induces p53-dependent apoptosis (5,11). We now demonstrate that treatment of cells with a variety of DNA damaging agents results in a substantial reduction of cellular levels of Mule (Figure 3). We further demonstrate that both downregulation of Mule by siRNA or in response to ionizing radiation results in elevated levels of the cellular p53 protein (Figure 4A). We, therefore, hypothesized that downregulation of Mule in response to DNA damage is required for the p53 response to be activated. In support of this hypothesis, we found that overexpression of Mule reduces the upregulation of p53 in response to ionizing radiation (Figure 4B, compare lanes 2 and 4; Figure 4C). It should be noted that, as the p53 regulation in response to DNA damage is accomplished via multiple pathways, the restriction of p53 upregulation by DNA damage imposed by Mule overexpression is partially rescued through other mechanisms. Most probably this happens via downregulation of Mdm2, which is also accomplished through the same ATM–PPM1G–USP7S pathway that controls Mule levels (13).
Figure 4.

Mule downregulation after DNA damage is required to coordinate the p53 DNA damage response with DNA repair. (A) HCT116 p53+/+ cells were transfected with Mule-specific siRNA (200 pmol) for 72 h (left panel). Alternatively (right panel), cells were either left untreated (−) or exposed to 8-Gy ionizing radiation (+) and harvested 1 h post-treatment (IR). (B) HCT116 p53+/+ cells were transfected with HA-tagged Mule expressing plasmid (1 pmol) for 24 h and then treated with IR as described in (A). (C) Graphical analysis of the data presented in (B). Statistical data are presented as a mean ± standard deviation, *P < 0.001 as analysed by Student’s t-test from three independent experiments. (D, E) HeLa cells were transfected with HA-tagged Mule HECT-domain expressing plasmid (1 pmol) for 24 h. (E) The cells were then either left untreated (UN) or treated with 25 µM hydrogen peroxide for 5 min, allowed to repair for 0–120 min, and the levels of single strand breaks and alkali labile sites were analysed by the alkaline single-cell gel electrophoresis (Comet) assay. Shown are the mean tail DNA values (%) with standard deviations from three independent experiments. Statistically significant results comparing untreated cells (Control) and Mule plasmid-treated cells (Mule) are represented by *P < 0.01 and **P < 0.001, as analyzed by Student’s t-test. (A, B, D) Whole-cell extracts were prepared, proteins were separated by 4–20% SDS–PAGE and analysed by western blotting using the indicated antibodies.
Mule also controls a number of proteins involved in the cellular DNA damage response that are not regulated by Mdm2, in particular DNA polymerases β and λ (8,9), which are important players in base excision repair. Indeed, we have previously demonstrated that inactivation of Mule results in more efficient DNA repair (13). Correspondingly, if Mule downregulation in response to DNA damage is required for DNA repair, then overexpression of Mule should reduce the cellular capacity for DNA repair. In fact, we found that overexpression of Mule in HeLa cells results in a reduction of Pol β and Pol λ levels (Figure 4D), and a reduced rate of DNA repair as measured by the alkaline single-cell gel electrophoresis (comet) assay (Figure 4E).
In our previous work, we demonstrated that the regulation of Mdm2, and consequently p53 protein levels, is accomplished through the ATM–PPM1G–USP7S pathway (13). We now demonstrate that Mule is also downregulated in response to DNA damage via the same signalling pathway, and this downregulation is required for p53 accumulation and efficient DNA repair. As we have previously shown, in unstressed cells, the majority of USP7S is phosphorylated by the CK2 protein kinase, and thus is stabilized (13). This ensures stabilization of USP7S substrates, such as Mule and Mdm2, which mediate proteasomal degradation of p53 in unstressed cells. In response to DNA damage, the protein phosphatase PPM1G is activated by ATM that results in dephosphorylation, and therefore destabilization, of USP7S. This, in turn, leads to DNA damage-dependent downregulation of both Mdm2 (13) and Mule, as we demonstrated in this study (Figure 5).
Figure 5.
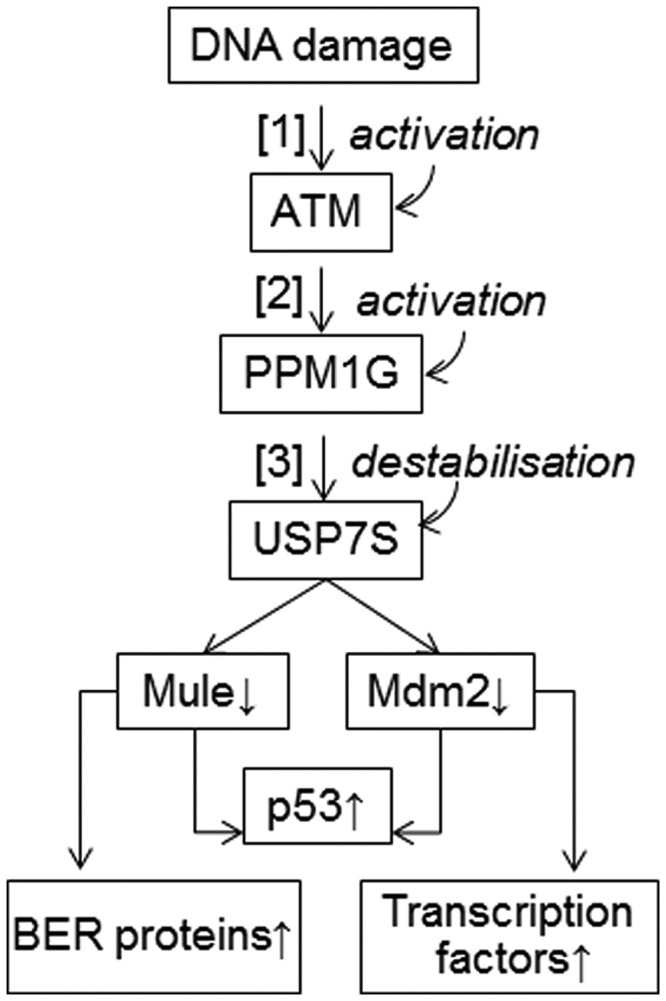
Proposed mechanism for the USP7S-dependent regulation of Mule and Mdm2 protein levels after DNA damage. In response to DNA damage, ATM is activated (1) and stimulates activity of PPM1G (2). PPM1G dephosphorylates and destabilises USP7S (3) that leads to both Mdm2 and Mule self-ubiquitylation and degradation, with the consequent stabilization of p53 and other substrates of Mdm2 and Mule. In unstressed cells, the deubiquitylation enzyme USP7S is stabilized by phosphorylation, and thus stabilizes both Mdm2 and Mule that results in proteasomal degradation of p53.
Therefore, we propose that the cellular levels of p53 are regulated by Mule and Mdm2 via the same DNA damage signalling pathway that involves ATM, PPM1G and USP7S as the upstream regulators. In addition to the common function of p53 regulation, Mule and Mdm2 E3 ubiquitin ligases control a number of distinct proteins such as Pol β, Pol λ and forkhead homeobox type O transcription factors that orchestrate different aspects of the cellular response to DNA damage (Figure 5).
Our findings provide the mechanism of how DNA damage is reported to Mule, which consequently contributes to activation of the p53-dependent DNA damage response, as well as allowing for DNA repair to be accomplished within a required time frame through the regulation of the levels of DNA Pol β and Pol λ. Additionally, it should be noted that Mule has been shown to be overexpressed in a high percentage of tumour cell lines (18,19). As we demonstrated (Figure 4B and C), elevated levels of Mule may attenuate, at least partially, the upregulation of the p53 protein in response to DNA damage and inhibit prompt DNA repair via the base excision repair pathway; therefore, it may possibly be one of the major reasons for genetic instability in these cancer cells.
FUNDING
Medical Research Council and Cancer Research UK (to G.L.D.); Oxford Cancer Research Centre Development Fund (to S.V.K.). Funding for open access charge: Medical Research Council and Cancer Research UK.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Dr Jason Parsons for critically reading the manuscript and Dr Irina Dianova for technical help.
REFERENCES
- 1.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Rozieres S, Maya R, Oren M, Lozano G. The loss of mdm2 induces p53-mediated apoptosis. Oncogene. 2000;19:1691–1697. doi: 10.1038/sj.onc.1203468. [DOI] [PubMed] [Google Scholar]
- 4.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 5.Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121:1071–1083. doi: 10.1016/j.cell.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 6.Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–791. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 7.Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O’Rourke K, Koeppen H, Dixit VM. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429:86–92. doi: 10.1038/nature02514. [DOI] [PubMed] [Google Scholar]
- 8.Parsons JL, Tait PS, Finch D, Dianova II, Edelmann MJ, Khoronenkova SV, Kessler BM, Sharma RA, McKenna WG, Dianov GL. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J. 2009;28:3207–3215. doi: 10.1038/emboj.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Markkanen E, van Loon B, Ferrari E, Parsons JL, Dianov GL, Hubscher U. Regulation of oxidative DNA damage repair by DNA polymerase lambda and MutYH by cross-talk of phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA. 2012;109:437–442. doi: 10.1073/pnas.1110449109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khoronenkova SV, Dianov GL. The emerging role of Mule and ARF in the regulation of base excision repair. FEBS Lett. 2011;585:2831–2835. doi: 10.1016/j.febslet.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 11.Kon N, Zhong J, Qiang L, Accili D, Gu W. Inactivation of arf-bp1 induces p53 activation and diabetic phenotypes in mice. J. Biol. Chem. 2012;287:5102–5111. doi: 10.1074/jbc.M111.322867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamijo T, van de Kamp E, Chong MJ, Zindy F, Diehl JA, Sherr CJ, McKinnon PJ. Loss of the ARF tumor suppressor reverses premature replicative arrest but not radiation hypersensitivity arising from disabled atm function. Cancer Res. 1999;59:2464–2469. [PubMed] [Google Scholar]
- 13.Khoronenkova SV, Dianova II, Ternette N, Kessler BM, Parsons JL, Dianov GL. ATM-dependent downregulation of USP7/HAUSP by PPM1G activates p53 response to DNA damage. Mol. Cell. 2012;45:801–813. doi: 10.1016/j.molcel.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dianova II, Bohr VA, Dianov GL. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry. 2001;40:12639–12644. doi: 10.1021/bi011117i. [DOI] [PubMed] [Google Scholar]
- 16.Petri S, Grimmler M, Over S, Fischer U, Gruss OJ. Dephosphorylation of survival motor neurons (SMN) by PPM1G/PP2Cgamma governs Cajal body localization and stability of the SMN complex. J. Cell. Biol. 2007;179:451–465. doi: 10.1083/jcb.200704163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin Y, Lee H, Zeng SX, Dai MS, Lu H. MDM2 promotes p21waf1/cip1 proteasomal turnover independently of ubiquitylation. EMBO J. 2003;22:6365–6377. doi: 10.1093/emboj/cdg600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, Bernard S, Quarto M, Capra M, Goettig S, et al. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell. 2005;123:409–421. doi: 10.1016/j.cell.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 19.Yoon SY, Lee Y, Kim JH, Chung AS, Joo JH, Kim CN, Kim NS, Choe IS, Kim JW. Over-expression of human UREB1 in colorectal cancer: HECT domain of human UREB1 inhibits the activity of tumor suppressor p53 protein. Biochem. Biophys. Res. Commun. 2005;326:7–17. doi: 10.1016/j.bbrc.2004.11.004. [DOI] [PubMed] [Google Scholar]