


Abstract
Replication protein A (RPA), essential for DNA replication, repair and DNA damage signalling, possesses six ssDNA-binding domains (DBDs), including DBD-F on the N-terminus of the largest subunit, RPA70. This domain functions as a binding site for p53 and other DNA damage and repair proteins that contain amphipathic alpha helical domains. Here, we demonstrate direct binding of both ssDNA and the transactivation domain 2 of p53 (p53TAD2) to DBD-F, as well as DBD-F-directed dsDNA strand separation by RPA, all of which are inhibited by fumaropimaric acid (FPA). FPA binds directly to RPA, resulting in a conformational shift as determined through quenching of intrinsic tryptophan fluorescence in full length RPA. Structural analogues of FPA provide insight on chemical properties that are required for inhibition. Finally, we confirm the inability of RPA possessing R41E and R43E mutations to bind to p53, destabilize dsDNA and quench tryptophan fluorescence by FPA, suggesting that protein binding, DNA modulation and inhibitor binding all occur within the same site on DBD-F. The disruption of p53–RPA interactions by FPA may disturb the regulatory functions of p53 and RPA, thereby inhibiting cellular pathways that control the cell cycle and maintain the integrity of the human genome.
INTRODUCTION
Genome stability requires the interplay of many signalling and DNA repair pathways, often requiring the action and regulation of multifunctional proteins that can modulate their activities appropriately during periods of DNA replication stress. Replication protein A (RPA), the major single-stranded DNA (ssDNA)-binding protein in eukaryotic cells, coordinates multiple DNA metabolic functions through interactions with numerous proteins critical to the DNA damage response (DDR) and DNA repair (1). RPA consists of three subunits (RPA70, RPA32 and RPA14) encompassing five ssDNA-binding domains (DBDs) that contribute to the high affinity of RPA binding to ssDNA (Figure 1) (2). RPA also has an affinity for dsDNA. In vitro experiments have shown that RPA binds to dsDNA and destabilizes the double helix, resulting in strand separation and RPA binding to ssDNA (3–5). The sixth identified binding domain, DBD-F, located on the N-terminus of RPA70, has been identified as the DBD primarily responsible for this destabilization activity of dsDNA (4). Although the precise mechanism of helix destabilization is not fully understood, the ability of DBD-F to bind ssDNA independently of the other DBDs with low affinity may be relevant to RPA unwinding activity (6). Additionally, DBD-F is a protein–protein interaction domain that is important in DNA repair and cell cycle checkpoint activities. A DBD-F mutant strain in yeast, rfa-t11, was found to be replication competent, but homologous recombination (HR) and checkpoint defective, suggesting that DBD-F was essential for specific DNA repair and the DDR signalling pathways, but not essential for replication in unstressed cells (7,8). Transient siRNA studies confirmed the importance of this domain in the DDR, as human cells expressing RPA70 with the analogous rfa-t11 mutation resulted in replication comparable with cells expressing wt-RPA70; however, they were sensitive to camptothecin- and etoposide-induced replication stress (9,10).
Figure 1.
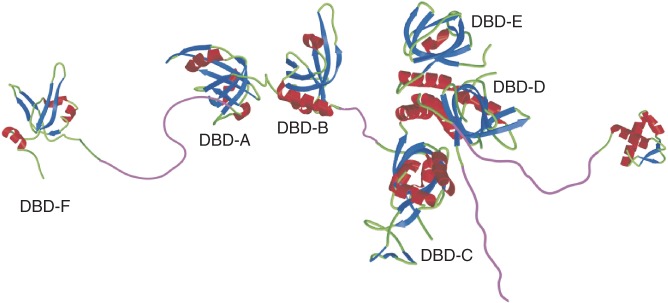
Illustration of the RPA heterotrimer depicting the oligonucleotide/oligosaccharide binding folds DBD-A through DBD-E. Modified from image provided by Dr Marc Wold.
The significance of DBD-F as a domain for protein–protein interactions was first described through an association with p53 (11–13). More recently, studies revealed that checkpoint activation, in part, is mediated through the recruitment of checkpoint proteins Rad9, ATR interacting protein (ATRIP) and Mre11 by DBD-F, as these proteins contain an amphipathic alpha helical domain that binds to the basic cleft of DBD-F (14–16). With the emergence of DBD-F as a recruiting scaffold for the assembly of DDR proteins, we have been interested in this domain as a novel target for cancer therapy, leading to our previous discovery of fumaropimaric acid (FPA) as an inhibitor of RPA protein interactions (17).
Tumour suppressor p53, the most commonly mutated gene in human cancers, primarily regulates the transcription of numerous genes involved in cell cycle control, apoptosis and DNA repair (18,19). p53 functions as a homotetramer and consists of DNA-binding and tetramerization domains that are flanked by two intrinsically disordered regions at both the N- and C-termini, the N-terminal transactivation and C-terminal regulatory domains, respectively (20). The N-terminal transactivation domain can be further divided into two subdomains, TAD1 (amino acids 1–40) and TAD2 (amino acids 41–61) (21). As TAD2 comes in contact with proteins containing DNA-binding domains, this intrinsically disordered region conforms to an amphipathic α-helix upon binding to proteins such as and RPA (13,22). The p53TAD2 behaves as a ssDNA mimetic competing with ssDNA for binding to the DNA binding oligonucleotide/ oligosaccharide-binding (OB) folds located within BRCA2 and RPA (23,24). Sequestration of p53 by BRCA2 and RPA has been suggested to inhibit the transcriptional activity of p53 with consequent down-regulation of apoptosis (25,26). Evidence for this model was demonstrated by overexpression of BRCA2 or a BRCA2 peptide that binds p53 and significantly reduced p53-mediated apoptosis (25). Conversely, the direct association of p53 with BRCA2 and RPA may interfere with HR independent of p53 transcriptional activity. This is supported by evidence that p53-mediated downregulation of replicative stress-dependent HR required p53 interaction with RPA (27).
Here, we show that DBD-F directly binds p53TAD2 and ssDNA, and that both of these interactions are inhibited by FPA. FPA binding results in a conformational shift in RPA occurring at a distant region from the binding surface. These results denote a more interactive relationship between DBD-F and other RPA domains than previously thought (28). We provide evidence that a double mutation (R41E and R43E) within DBD-F prevents both p53 binding and helix destabilization activity, suggesting that DBD-F may operate as a true protein-DNA binding domain in vivo. Molecular modelling simulations predict that DBD-F is highly flexible and is therefore susceptible to structural change due to ligand binding or subunit interactions. Finally, using a set of structurally related compounds, we have determined that both hydrophobic and electrostatic interactions play a role in the stability of the FPA–DBD-F complex formation.
MATERIALS AND METHODS
Reagents
5′ IRDye® 700 labelled polyT-30mer and unlabelled polyA-30mer oligonucleotides were purchased from Integrated DNA Technologies, Inc. The N-terminal X-rhodamine labelled p53TAD2, containing amino acids 40–57 (Xr-NH2-MDDLMLSPDDIEQWFTED), was produced by AnaSpec. A p53TAD2 peptide containing an hemagglutinin (HA) tag (NH2-MDDLMLSPDDIEQWFTEDYPYDVPDYA) that does not contain an X-rhodamine label, referred to as p53TAD2-HA, was produced by Thermo Scientific. FPA and structurally related compounds were received from the Developmental Therapeutics Program of the National Cancer Institute.
Cloning, expression and purification of RPA, p53 and DBD-F fusion proteins
The human recombinant RPA and RPAΔF were purified from Escherichia coli as described previously (29). RPA containing the RPA70 mutations R41E and R43E, RPA (R41E, R43E), was generated using the Phusion® site-directed mutagenesis kit (NEB), sequenced, and the corresponding protein purified accordingly. The maltose-binding protein (MBP) fusion protein containing amino acids 1–168 of DBD-F, referred to as DBD-FMBP, was created by cutting out a NcoI/PstI RPA70 fragment from the RPA pET-11d vector, and ligating it into a pMBP-parallel2 expression vector (30). The resulting vector was expressed in E. coli, strain BL21, induced with isopropyl β-D-1-thiogalactopyranoside (IPTG), and purified using a gravity flow MBP-binding column (New England Biolabs). The concentrations of purified proteins were quantified using a Bradford assay. For expression of p53–GST fusion protein and unmodified GST, Addgene plasmid 10852 and pGEX-4T-1 were expressed in BL21 cells (31). After centrifugation, cells were sonicated in PBS in the presence of 1 mM PMSF and 1 mM DTT, clarified by centrifugation, and used directly in the binding assay. Characterization of the purified proteins, p53 and GST lysates, is shown in Supplementary Figure S4.
Electrophoretic mobility shift assays and helix destabilization assays
Electrophoretic mobility shift assays (EMSAs) for RPA, RPAΔF and RPA (R41E, R43E) were performed in binding buffer B (25 mM HEPES, 10% (v/v) glycerol, 10 mM NaCl, pH 8.0). The ssDNA probe is a 5′ IRDye® 700 labelled polyT-30mer oligonucleotide. To make the dsDNA probe, the labelled polyT-30mer was annealed to a polyA-30mer and treated with mung bean nuclease as reported previously (17). Concentrations of ssDNA and dsDNA probes were 20 nM. Concentrations of RPA were 10 nM and 40 nM for ssDNA and dsDNA binding, respectively. The concentrations of the RPA mutants were normalized relative to RPA ssDNA-binding activity. EMSAs for the DBD-FMBP fusion protein were performed in binding buffer A (25 mM HEPES, 10% (v/v) glycerol, 100 mM NaCl, pH 8.0) with 2 µM DBD-FMBP and electrophoresed at 4°C. Gels were scanned on a near infrared scanner, (LI-COR Biosciences).
Fluorescence anisotropy association curves
Data points were collected using a model A-1010 Alphascan photon counting fluorometer (Photon Technologies) in which the emission of the X-rhodamine-labelled p53TAD2 was monitored in binding buffer A at 25°C upon titration with RPA or DBD-FMBP. The dye was excited using a xenon short arc lamp (Ushio, UXL-75XE, No. ME0282) followed by the excitation monochromator set at 585 nm (bandpass of ∼5 nm). The exciting beam passed through a rotatable polarizer containing a mounted KN-36 Polaroid sheet (Polaroid) before reaching the sample cuvet. Emitted light was collected at 630 nm (bandpass of ∼5 nm) after passing through a similar second polarizer (analyzer) placed between the sample cuvet and emission monochromator and photomultiplier (R928 PMT, Hamamatsu).
Each pair of intensity measurements, parallel (IVV) and perpendicular (IVH), was collected after the reaction reached equilibrium. The anisotropy values (<r> = (IVV − G·IVH) / (IVV + 2·G·IVH), where G is the grating factor (0.69 for X-rhodamine-labelled p53TAD2), reported are averages of three independent values at each point in the titration. For each intensity measurement, 30 data points were collected over a 1-s time period, fit by linear regression, and the fitted value at the midpoint, 0.5 s, was the recorded value for that time interval and used in determining the average over three such measurements. When there was no change in fluorescence, only anisotropy (<r>) was reported. Otherwise, the change in total fluorescence intensity (F, where F = IVV + 2·G·IVH) or anisotropy multiplied by fluorescence intensity (rF, where rF = <r> · F) was used to determine a binding constant (Ka) because these two functions are linear in concentrations of fluorescent species. The fraction (Y) of p53TAD2 bound to RPA or DBD-FMBP was determined using the appropriate average signal (<r>, F or rF), and the best fit to a single binding site model (Equation 1) was obtained by a least squares minimization where Ka and<r> max are parameters. For instance,
| (1) |
where <r>obs(i) is the average anisotropy at iteration i, <r>min and <r>max are the minimum and maximum anisotropies, RT is the total concentration of protein (RPA or DBD-FMBP) and PT is the total concentration of p53TAD2. Equation 1 can be rearranged into a quadratic form (Equation 2):
| (2) |
where the quadratic coefficients are: a = −Ka·PT, b = (1+ PT·Ka + RT·Ka), c = −RT·Ka and Y can then be solved using the familiar quadratic equation with the positive sign proceeding the radical. Kd is the concentration of free ligand at which Y = 0.5 and Kd = 1/Ka. When the binding is weak and the concentration of fluorescent ligand is low, Equation 1 can be simplified as shown in Equation 3, also with two parameters, Ka and <r>max:
| (3) |
Parameter errors reported are one standard deviation. For data sets with fewer than 20 points, the confidence region was obtained by applying the Student’s t-test. The concentrations were adjusted for any dilution effects.
Tryptophan fluorescence quenching
Quenching of tryptophan fluorescence was used to determine direct binding of FPA to RPA and aid in determining whether both ligands could simultaneously be bound to RPA. The fluorescence of the sample, 3 μM RPA upon addition of FPA in binding buffer A at 25°C, was collected using the same apparatus as for the fluorescence anisotropy data, with an excitation wavelength of 292 nm. For the emission wavelengths, a single value at every 1 nm was collected from 325–375 nm and data were smoothed using a Savitzky–Golay quadratic smoothing function with a 7-point filter. The average of 5–8 smoothed buffer fluorescence intensity values at 348 nm was subtracted from the average of 5–8 similarly smoothed sample values and the difference for each data point recorded (Fobs(i)). The data points were fit according to the single binding site model (Equation 3) with two parameters (Fmin and Ka), where Y(i) = 1 − [(Fobs(i) − Fmin) / (Fmax − Fmin)], Fobs(i) is the total observed fluorescence at iteration i, Fmin and Fmax are the minimum and maximum fluorescence respectively and Y(i) is the fraction of RPA bound to FPA. The reported Kd is calculated from Ka (Kd = 1/Ka). The concentrations were adjusted for any dilution effects.
Fluorescence anisotropy competition curves
Data points were collected with the same apparatus and method as described for the association curves where X-rhodamine-labelled p53TAD2 is the reporter. Samples were incubated after each addition of the competing ligand at 25°C until equilibrium was reached.
The data set was fit assuming the simplest model: binding of p53TAD2 and FPA to the DBD-FMBP in the full-length RPA is competitive and exclusive. A Newton–Raphson matrix routine (32) for two nonlinear binding equations and the associated Jacobian (J) embedded in a two-dimensional least squares grid search was used to determine two optimal and correlated Ka’s and the joint 68% confidence region for the competition experiment involving RPA, p53TAD2 and FPA. Subsequently the same procedure was followed for the DBD-FMBP using both FPA and p53TAD2-HA as the competing ligands. The Newton–Raphson was constrained by the conservation of the total concentrations of the three species (Equations 4–6):
| (4) |
| (5) |
| (6) |
where RT, PT and FPAT are the total concentrations and R, P and FPA are the free concentrations of RPA, p53TAD2 and FPA, respectively, for each iteration (i), L is the binding polynomial based on the simplest model (1 + Ka1 · P(i) + Ka2 · FPA(i)), and Ka1and Ka2 are the binding constants of p53TAD2 binding to RPA and FPA binding to RPA, respectively.
The above three conservation conditions reduce to two equations (Equations 5 and 6) that generate the two Newton–Raphson equations (Equations 7 and 8). F1 and F2, the concentrations of free p53TAD2 and FPA respectively, equal zero when the concentrations are correctly determined in the Newton–Raphson routine. For instance,
| (7) |
| (8) |
The Jacobian matrix (Ji,j = ∂Fi/∂Xj, X1 = (P), X2 = (FPA)) is defined by J1,1 = a, J1,2 = b, J2,1 = c and J2,2 = d. The four quantities are identified as: a = −G1 + P(i) ·(Ka12·RT(i)) / L2, b = P(i) ·Ka1· RT(i) ·Ka2 / L2, c = FPA(i) ·Ka2 ·RT(i) ·Ka1 / L2, d = −G2+ FPA(i) ·Ka22 ·RT(i) / L2. The inverse Jacobian matrix (J−1) is then: J−11,1 = d / Δ, J−11,2 = −b / Δ, J−12,1 = −c / Δ and J−12,2 = a / Δ, where delta is the determinant of J (Δ = a·d − b·c).
The Newton–Raphson algorithm (Equation 9) uses known values of PT(i), FPAT(i) and RT(i) and chosen values of Ka1 and Ka2 where iterations (i) proceed to convergence where F1 = F2 = 0.
| (9) |
where V is a column vector with rows P and FPA, and F is a column vector with rows F1 and F2.
The optimal Ka1 and Ka2 values were used to calculate the fraction of p53TAD2 bound to RPA and then converted to a response function (Equation 10) in order to determine the sum of squared residuals with respect to <r>obs:
| (10) |
The maximum or minimum signal in the absence of the competing ligand was determined before titration. Excellent endpoints were obtained using a conventional double-reciprocal plot and refined using a one-dimensional grid search added to the Newton–Raphson routine. The square root of the minimum sum of the squared residuals (σbest) was thus determined. Subsequent grid searches were carried out varying the binding constants until Equation 11 (33) was satisfied for the 68% confidence boundary (σ68%):
| (11) |
with n data points, p parameters, and the critical F value was calculated using an online F distribution calculator (http://www.danielsoper.com/statcalc3/calc.aspx?id=4) with p degrees of freedom in the numerator and (n−p) degrees of freedom in the denominator. Significant overlap between the 68% confidence regions of the individually determined Kd values and the Kd values from the competition experiment validates the assumption of the competitive and exclusive binding model. Therefore, the same program was used to analyse the competitive binding data involving the DBD-FMBP. As the DBD-FMBP does not have a tryptophan, direct binding of FPA cannot be determined using tryptophan fluorescence quenching, and a competition experiment must be used to determine the Kd for the binding of FPA to the DBD-FMBP. The 68% ellipsoidal confidence region for the Kd values of p53TAD2/ DBD-FMBP and FPA/ DBD-FMBP was truncated by the 68% confidence region of the individually determined p53TAD2 and DBD-FMBP Kd, leaving part of an ellipsoid as our final 68% confidence region. The same analysis was used to determine the Kd and 95% confidence region (σ95% = σbest * (1 + (p/(n−p)) *Fp, n−p, 0.05)0.5) for the competitive binding between X-rhodamine-labelled p53TAD2 and p53TAD2-HA.
Protein modelling and docking
The protein structure (PDB ID: 2B29), composed of residues 1–121 of RPA70, was relaxed using all-atom molecular dynamics (MD) simulations. All MD simulations were performed using the GROMACS 4.5.1 package (34). The AMBER96 force field (35) was used to describe the protein structure and the tip3p water model to describe the solvent (36). Protein charge states were set in accordance with a pH of 7, and no counter ions were added (37). The protein was solvated in a periodic dodecahedron-shaped box with ∼5300 water molecules and energy minimized for 200 steps using the steepest descent algorithm. A 0.1-ns-long position restrained MD simulation was followed by a 1-ns-long MD simulation in the moles (N), volume (V) and temperature (T) (NVT) ensemble. The temperature was maintained close to 300 K by weak coupling to an external temperature bath (38) with a coupling constant of 0.1 ps. The LINCS (39) algorithm was used to constrain bond lengths within the solute. The SETTLE (40) algorithm was used to constrain the bond lengths and the bond angle in water. The integration time step was 2 fs. The reaction field method (41) [reaction-field-zero as implemented in GROMACS 4.5.1 (42)] was used to account for electrostatic interactions with a twin-range cut-off of 0.9/1.2 nm. Interactions within the short-range cut-off were evaluated at every step, whereas interactions within the long cut-off were updated every five steps, together with the pair list. The equations of motion were integrated using the leapfrog algorithm. The snapshot at 1.0 ns from the NVT simulation was used as input for the anisotropic normal mode analysis (ANM) (43) calculation. The all-atom ANM calculation was implemented using the ProDy package (44), with a cut-off value of 15 Å and a uniform force constant. The non-trivial three slowest modes were used to generate a 200-conformer ensemble within a root-mean-square value of 3.0 Å. Subsequently, each conformer was energy minimized in the gas phase for 200 steps. The top 100 structures that displayed proper stereochemistry were used in a docking simulation with the structure for FPA using the virtual screening software, PyRx, in conjunction with the docking software, AutoDock Vina (45,46). Affinity scores in PyRx are reported as binding energies (ΔG), which were then converted to apparent inhibitory constants (Ki) through the equation, Ki = e(ΔG/RT), where T = 298.15 K and R = 0.001968 kcal/mol/K. The top scoring DBD-F structure was compared with the original 2B29 structure in docking both FPA and a portion of the p53TAD, 40MDDLMLSPDDI51, and graphically reported using the software program Pymol (47).
Full length p53 binding assays
Clarified bacterial lysate (100 μl) expressing GST or p53–GST was added to each well of a glutathione-coated 96 well plate (Pierce). After 30 min, the plate was washed 3× with PBST (PBS, 0.1% tween-20), then incubated for 30 min in PBS with 250 nM RPA. Plates were washed 3× with PBST and incubated with a polyclonal antibody for GST (kindly provided by Dr James Wahl, 1:100), and RPA70 (Bethyl, 1:1000) for 30 min. Wells were washed 3× with PBS and incubated with secondary fluorescent antibodies Alexa Fluor 680 (Invitrogen, 1:1000) and Dylight 800 (ThermoScientific, 1:1000). Wells were washed 3× with PBST and scanned on near infrared scanner (LI-COR Biosciences). Binding efficiency was determined by the amount of RPA signal normalized to GST signal. Western blots of bacterial lysates are shown in Supplementary Figure S4.
RESULTS
FPA, p53 and ssDNA compete for binding to RPA
We previously demonstrated that FPA prevented helix destabilization by RPA (17). A potential explanation for the loss of helix destabilization by RPA is that FPA binds to DBD-F and prevents DBD-F–ssDNA intermediates from forming before ssDNA binding by the high affinity DBDs. To test this model, we separated RPA into two ssDNA-binding entities, a heterotrimer without DBD-F (RPAΔF) and DBD-F fused to MBP (DBD-FMBP). The capacity of FPA to selectively inhibit ssDNA binding to DBD-FMBP and not RPAΔF was observed via EMSA (Figure 2A and B). MBP alone does not bind ssDNA (Supplementary Figure S1A). Combined, these data indicate that the mechanism of DNA unwinding by RPA is dependent on DBD-F, and suggest that a peptide that mimics ssDNA, such as p53TAD2, should also be an effective inhibitor of helix destabilization. Indeed, an X-rhodamine-labelled p53TAD2 peptide inhibited helix destabilization (Figure 2C). p53TAD2 was not effective in preventing the binding of RPA to ssDNA, indicating p53TAD2 does not compete with ssDNA for binding to the central high affinity DNA binding core, DBD-A and B (Figure 2D). The band observed directly above the free dsDNA probe was determined to be due to the fluorescence of the X-rhodamine label of the p53TAD2 peptide and does not interact with dsDNA or have helix destabilization activity (Supplementary Figure S1B). These data suggest that protein–protein interactions at the DBD-F would have an effect on some aspect of DNA processing by RPA, and that FPA can inhibit both types of substrates, proteins and ssDNA, from interacting with the DBD-F.
Figure 2.
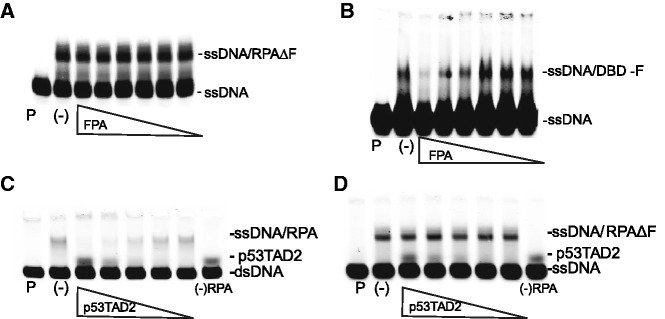
p53TAD2 and FPA bind to DBD-FMBP and RPA and affect DNA binding. For (A–D): Lane with DNA probe in the absence of RPA is denoted by ‘P’. ssDNA–RPA binding without FPA is marked by a (-). Concentration of labelled DNA probe is 10 nM. For (A) and (B), the concentration of FPA is 300, 100, 30, 10, 3, 1 µM, respectively. (A) Lack of inhibition of ssDNA–RPAΔF formation in the presence of FPA as determined by EMSA. The concentration of RPAΔF is 20 nM. (B) Inhibition of ssDNA–DBD-FMBP binding by FPA. The concentration of the DBD-FMBP is 2 µM. For (C) and (D), the p53TAD2 concentration is 24, 12, 6, 3, 1.5 µM, respectively. (C) Inhibition of helix destabilization by p53TAD2. The amount of helix destabilization was determined by the appearance of an upward shifted band consisting of RPA bound to unwound ssDNA derived from dsDNA. Lane with labelled DNA and p53TAD2 in the absence of RPA is denoted by a ‘*’. (D) Lack of inhibition of ssDNA–RPA binding by p53TAD2. The p53TAD2 peptide can be detected at higher concentrations due to the fluorescence of the x-rhodamine label, and does not bind DNA, as determined in figure S3C.
Effect of FPA analogues on RPA and DBD-FMBP binding
With the evidence of FPA directly binding and inhibiting the DBD-F, we attempted to determine the structural components of this compound that are essential for activity. A survey of the Open Chemical Repository of the Developmental Therapeutics Program at the National Cancer Institute (NCI) identified seven structural analogues of FPA, referred to in the NCI databank as NSC15520 (Figure 3A). All of the compounds have at least three fused six-membered rings, and three compounds have nearly exact structural backbones as that of FPA, but have substituted the 1,4-dicarboxylic acid with an anhydride (NSC204220), a 1,4-diol (NSC49890), or a 1,4-dione (NSC2961) (Figure 3A, #s 2, 4 and 5). The anhydride-containing NSC98616 has a slightly altered backbone structure as well as a hydroxyl group substituted for the singular carboxyl group (Figure 3A, # 3). Also included in the study were three abietic acid analogues (NSC4827, NSC5007, NSC18746) (Figure 3A, #s 6, 7 and 8), as abietic acid provides the backbone structure in the reaction with fumaric acid to create FPA (48).
Figure 3.

Effect of structural analogues of FPA on DBD-FMBP inhibition. (A) Structures of lead compound FPA and structural analogues used in this study. (B) Effect of FPA-related compounds on DNA binding as measured by EMSAs. 200 µM of each compound was used. Top panel: addition of dsDNA to compounds preincubated with RPA. Bottom panel: addition of ssDNA to compounds preincubated with RPA. Lane with DNA in the absence of RPA is demarked by ‘P’. RPA–DNA binding without inhibitor is marked by a (-). Numbers correspond to structures in ‘A’. (C) Structure of FPA sub-structures succinic acid, 1,4-cyclohexanedicarboxylic acid and FPA (1–3, respectively). (D) Effect of compounds in ‘C’ (200 µM) on helix destabilization via EMSA. Lane with ssDNA probe in the absence of RPA is demarked by ‘P’. RPA–DNA binding without inhibitor is denoted by a (-).
When assayed for helix destabilization activity, four compounds (FPA, the diol-containing NSC49890, and the anhydride compounds NSC204220 and NSC98616) inhibited helix destabilization by RPA (Figure 3B, top panel, lanes 1–4), but only FPA showed no evidence of inhibiting RPA binding to ssDNA (Figure 3B, bottom panel, lane 1). These data indicate that only FPA, with the 1,4-dicarboxylic moiety, is capable of selectively inhibiting DBD-F of RPA, whereas the other compounds (NSC204220, NSC98616, NSC49890) likely bind to residues within the other DBDs, A–E. Abietic acid analogues NSC4827, NSC5007 and NSC18746 showed no effect on RPA helix destabilization and ssDNA binding, suggesting that either the transition of abietic acid to a nonplanar molecule, the addition of negatively charged moieties, or both, are required for RPA inhibition. NSC2961, a 1,4-dione, also did not interact with RPA, accentuating the importance of the 1,4-dicarboxylic acid for inhibition of DBD-F.
The binding of FPA and structural analogues of FPA to DBD-FMBP was also investigated through fluorescent anisotropy. Here, compounds were tested for the ability to inhibit binding of p53TAD2 to DBD-FMBP. Of the compounds tested, FPA showed the most potent inhibition compared with the other seven compounds, further supporting FPA as a DBD-F-specific inhibitor (Table 1).
Table 1.
Kd values for FPA and derivatives binding to DBD-FMBP prebound to p53TAD2 as determined by fluorescence anisotropy
| # | Inhibitor | Kd (µM) | SD (µM) |
|---|---|---|---|
| 1 | FPA | 9.0 | 1.2 |
| 2 | NSC204220 | 103 | 9 |
| 3 | NSC98616 | ND | ND |
| 4 | NSC49890 | ND | ND |
| 5 | NSC2961 | ND | ND |
| 6 | NSC4827 | ND | ND |
| 7 | NSC5007 | ND | ND |
| 8 | NSC18746 | 660 | 250 |
ND, not determined.
Because the 1,4-dicarboxylic acid moiety appears to be essential for the binding activity of FPA with DBD-FMBP, we compared the specificity of FPA with other 1,4-dicarboxylic acids (Figure 3C). In these experiments, both 1,4-cyclohexanedicarboxylic and succinic acids (Figure 3C, compounds 1 and 2, respectively) are FPA sub-structures. These three compounds were analysed for their ability to inhibit the helix destabilization activity of RPA (Figure 3D). As anticipated, FPA inhibited helix destabilization of a dsDNA template by RPA, whereas 1,4-cyclohexanedicarboxylic and succinic acids had no effect (Figure 3D, lane 3). To further confirm the selective inhibition of FPA, we performed competitive titration assays using fluorescence anisotropy. Only FPA competed with X-rhodamine-labelled p53TAD2 for binding to DBD-FMBP, as reported in Table 1, with 1,4-cyclohexanedicarboxylic and succinic acids showing no evidence of binding (data not shown). Taken together, these results show that both the electrostatic charge of the dicarboxylic acid moiety and the hydrophobic backbone of FPA are necessary for binding to DBD-FMBP.
FPA and p53TAD2 directly bind to RPA independently and competitively
The ability of FPA to inhibit DBD-F binding to ssDNA suggested, but did not prove, a direct interaction of FPA with full length RPA. To assess direct binding to full length RPA, we determined the ability of FPA to quench tryptophan residues within RPA (Table 2, Supplementary Figure S2C). The addition of FPA to RPA resulted in the quenching of the fluorescence of one or more tryptophan residues within RPA, allowing the binding constant of FPA to RPA to be determined (Kd = 29.0 ± 3.5μM). DBD-F itself does not contain tryptophan residues. Therefore, these data do not confirm direct binding to DBD-F itself; however, based on our previous data (17) and the other data presented here, it suggests that the quenching of tryptophan by FPA in RPA is likely due to a conformational change induced by direct binding, and not by a direct interaction of tryptophan with the inhibitor.
Table 2.
Kd values of DBD-FMBP and various RPAs for RPA and p53TAD2 substrates as determined by either fluorescence quenching or fluorescence anisotropy
To assert that p53TAD2 and FPA compete for the same binding site, we conducted fluorescence anisotropy titrations. We observed X-rhodamine-labelled p53TAD2 bound to RPA (Supplementary Figure S2A). In a competition assay, addition of FPA to a preformed p53TAD2–RPA complex resulted in a decrease in anisotropy, indicating that FPA bound to RPA, replacing p53TAD2 (Supplementary Figure S2B). A Newton–Raphson matrix routine for two nonlinear binding equations was embedded in a two-dimensional least squares grid search in order to determine the two optimal binding constants and determine the joint 68% confidence region for FPA (Kd = 29.0 ± 3.5 μM) and p53TAD2 (Kd = 6.06 ± 0.08 μM). Increasing concentrations of FPA up to 275 μM showed no evidence of binding to p53TAD2 (Supplementary Figure S3A). The competition experiment showed no evidence for joint occupancy of p53TAD2 and FPA in the DBD-F of the full length RPA, and was determined to be a valid method for determining the affinity of a ligand by replacement of a competing fluorescent ligand. These results demonstrate that FPA binds directly to RPA, and that both FPA and p53TAD2 bind RPA independently and competitively.
p53TAD2 and FPA bind specifically to the DBD-F of RPA
To ensure that the competition between FPA and p53TAD2 for RPA is specific for DBD-F, their respective binding to DBD-FMBP was explored. The p53TAD2 bound to DBD-FMBP with high affinity (Kd of 1.03 ± 0.25μM) as determined by fluorescence quenching of the X-rhodamine label (Table 2 and Supplementary Figure S2D, filled circles). Addition of MBP to p53TAD2 gave no change in fluorescence under similar concentrations (Supplementary Figure S2D, hollow circles). These results showed that the isolated DBD-FMBP is competently folded for binding protein.
FPA binding to DBD-F could not be detected directly, as there were no tryptophans in DBD-F. Therefore, a fluorescence anisotropy competition assay was used to determine the affinity of FPA for DBD-FMBP. The fraction of p53TAD2-bound DBD-FMBP decreased upon addition of FPA (Supplementary Figure S2E). From these findings, the two binding constants for both ligands were determined to be Kd values at 1.2 and 9.0 μM, respectively (Table 2 and Supplementary Figure S2F).
In comparison with full length RPA, DBD-FMBP had substantially higher affinity for both p53TAD2 and FPA than the full length RPA (6-fold and 3-fold, respectively). A similar competition experiment involving X-rhodamine-labelled p53TAD2 and p53TAD2-HA showed that the Kd values for binding DBD-FMBP were not significantly different at the 95% confidence level. Therefore, the presence of the X-rhodamine dye on the N-terminus of p53TAD2 did not significantly alter binding (Kd = 0.67 µM, data not shown). Furthermore, the critical amino acids for contacting the DBD-F are indeed within the p53TAD2, as addition of the 10 amino acids of the HA tag did not alter binding.
R41E and R43E mutations in RPA70 reveal the specific nature of DBD-F for protein, DNA and FPA
Evidence for the requirement of DBD-F in recombination was first derived from the yeast strain rfa-t11, containing a K45E mutation in Rfa1, the yeast homolog of RPA70 (49). Although it has been inferred that the mutation causes the interruption of RPA with several RPA substrates such as p53, only Ddc2, the yeast homolog of ATRIP, has been shown to be directly affected by the mutation using full length proteins (50), and protein–protein interaction studies involving full length human RPA with analogous mutations to rfa-t11 have not been demonstrated. To confirm that FPA interactions with human RPA are specific to the analogous K45 mutation of rfa-t11, we generated a glutamic acid-substituted mutant (R41E, R43E), as NMR studies have indicated that both residues are essential for p53TAD2 binding (51).
The double mutant retained the ability to bind ssDNA, as no amino acid substitutions were made on the high-affinity DBDs (Figure 4A, left panel). When tested for helix destabilization activity, the mutant was limited in unwinding DNA, similar to RPAΔF (Figure 4A, right panel). The small amount of unwinding observed was not further inhibited by FPA at concentrations up to 1 mM, suggesting that the nominal unwinding that does occur by RPA (R41E, R43E) is DBD-F independent. The R41E and R43E mutations highlight their importance in helix destabilization, and strongly suggest that the recombination deficient rfa-t11 yeast strain likewise contains an RPA that is deficient in this activity.
Figure 4.

The K41E,K43E mutation affects RPA binding to protein, DNA and FPA. (A) Left panel: wild-type RPA and two mutant RPAs are all capable of competently binding ssDNA via EMSA. Right panel: only wild-type RPA is capable of effectively unwinding dsDNA. (B) The small amount of dsDNA that is unwound by RPA (R41E, R43E) is not effected by moderate concentrations of FPA. Concentrations of FPA are 31, 62, 125, 250, 500, 1000 µM, respectively. (C) ELISA-based assay showing the ability of FPA to prevent p53 binding to full length RPA. Bacterial lysates expressing p53–GST were bound to a glutathione-coated plate, and then incubated with 250 nM RPA in the presence of the indicated concentration of FPA. 100% RPA signal is normalized to wells with 0 µM inhibitor. 0% RPA signal is normalized to the absence of RPA (0*). Error bars represent the standard error of the mean. (D) RPA(R41E, R43E) and RPAΔF are unable to bind p53 as determined by assay described in ‘A’. (E) Tryptophan quenching of RPA(K41E,K43E) by FPA. Kd = 230 µM ± 18 µM.
To determine whether the RPA (R41E, R43E) mutation has an effect on p53 binding, we devised an ELISA-based binding assay (Figure 4C and D). Wild-type RPA bound readily to the immobilized p53–GST fusion protein, and was effectively inhibited from binding to p53 by FPA at 100 µM. GST alone does not bind RPA (Supplementary Figure S3B). In a comparison of RPA, RPAΔF and RPA (R41E, R43E), only RPA was capable of effectively binding p53–GST. These data support R41 and R43 as essential residues in DBD-F for p53 binding.
We then tested whether RPA (R41E, R43E) would undergo tryptophan quenching in the presence of FPA (Table 2 and Figure 4E). Half-maximal quenching was seen at a concentration of 230 μM, approximately 8-fold higher concentration than required for RPA. This suggests that the double mutation affects the ability of RPA to change conformation in the presence of FPA, providing further evidence of FPA specificity for DBD-F.
Modelling of the DBD-F reveals large scale changes within the basic cleft
The ability of DBD-F to bind several disparate ligands (ssDNA, FPA, proteins) suggests that the domain possesses structural and dynamic features. Recent work suggests that conformational changes at the binding site are often responsible for the ability of proteins to bind different ligands (52,53). Using MD simulations, we determined the extent by which DBD-F is susceptible to conformational change (Figure 5). The original crystal structure of DBD-F shows a wide gap within the basic cleft, with the walls of the cleft opening outward, creating a large area for binding (24) as represented in Figure 5A. However, when analysed by docking software, FPA was predicted to bind with low affinity (Ki = 204.7 µM) at a position adjacent to the cleft. A portion of the p53TAD2 predictively bound at higher affinity (Ki = 24.5 µM), but does not lie in line with the basic cleft of DBD-F (Figure 5A).
Figure 5.

Docking simulations of a crystal structure of DBD-F and conformer optimized for FPA binding. For both structures, FPA is shown in stick form, whereas the p53TAD2 sequence ‘MDDLMLSPDDI’ is represented by a ribbon structure. (A) The original crystal structure of DBD-F docked to FPA and p53TAD2 [−5.5 and −4.4 kcal/mol (24.5 µM and 204.7 µM), respectively]. (B) A DBD-F model optimized for FPA binding docked FPA and p53TAD2 [−6.6 and −5.9 kcal/mol (2.9 µM and 11.3 µM), respectively].
When MD simulations were used to find alternative conformations of the DBD-F, a DBD-F representation predicted to bind FPA at 18-fold greater affinity was found (Ki = 11.3 µM) (Figure 5B). This representation possessed a basic cleft in a more restricted conformation, with the walls of the cleft almost wrapping around the inhibitor. The p53TAD2 peptide also was predicted to bind at 8-fold higher affinity (Ki = 2.9 µM) than to the original crystal structure of DBD-F, enveloping the predicted binding site of FPA. Though the docking scores should be considered as a relative ranking parameter, they do suggest that the newly ascribed DBD-F representation may be more accurate than the original crystal structure. This new representation of DBD-F may help to explain the exclusive binding of dissimilar ligands and provide a more accurate model for DBD-F in vivo.
DISCUSSION
The ability of multiple ligands to bind DBD-F is an example of DNA and protein mimicry common to DNA repair pathways, allowing RPA to ‘hand-off’ multiple DNA and protein substrates (54). RPA–p53 interactions are required for maintaining a balance between pathways that repair double strand breaks, as mutations in the p53TAD2 that prevent RPA binding result in excessive homologous recombination (55). This hyper-recombination phenotype can lead to genomic instability and cell death (56,57). An inhibitor to the DBD-F may have a similar destabilizing effect on cancer cells to that of the p53TAD2 mutant. As other proteins involved in homologous recombination also bind the DBD-F such as Mre11 and Nbs1 (10,14), it is likely that a DBD-F inhibitor, such as FPA, could have the ability to deregulate homologous recombination, allowing the selection of normal cells that have competent and redundant DNA repair pathways over cancer cells that typically have reduced DNA repair capacity.
Two components of FPA appear to be essential for the inhibition of DBD-F. The dicarboxylic moiety of FPA likely interacts with the positively charged residues of the DBD-F, R41 and R43, as removal or alteration of the moiety to hydroxyl, aldehyde or anhydride eliminates either the action or specificity of the compound to DBD-F. The backbone of FPA, derived from abietic acid, provides a complex hydrophobic helical structure that likely fits within the hydrophobic residues of DBD-F. The negatively charged and hydrophobic components of FPA combine to resemble both the ssDNA and peptide substrates of DBD-F. Further studies will entail modifications in both charged and uncharged components of FPA in newly synthesized compounds that allow for higher specificity and affinity to the DBD-F.
Tryptophan fluorescence quenching has been used previously to determine changes in RPA conformation upon phosphorylation of the N-terminal domain of RPA32 (58–61). By assessing tryptophan quenching of RPA by FPA and subsequent fluorescence anisotropy competition assays, we demonstrate that FPA binds the DBD-F of RPA. Additionally, the comparison of tryptophan fluorescence quenching of full-length RPA, RPAΔF and the RPA (R41E, R43E) mutant in the presence of FPA provide evidence that FPA induces a structural rearrangement involving DBD-F and DBDs A–E. We currently do not know which of the eight tryptophan residues in DBDs A–E that are quenched upon FPA binding. However, ssDNA is capable of quenching four residues that reside in the DBDs A–D, W212, W361, W528 and W107(RPA32), respectively, (58) and our data shows that FPA does not affect ssDNA binding of RPA, making those residues less viable candidates for quenching by FPA-bound DBD-F. This suggests that W197, W414, W442 or W2 (RPA32) are quenched by FPA binding. FPA-induced conformational change is further supported by our data that revealed a difference in ligand binding to DBD-FMBP compared with full length RPA. This was initially an unexpected result, as the flexibly linked DBD-F on RPA and the DBD-FMBP should have the same affinity to ligand if DBD-F is a truly independent domain. Combined, our data imply an interaction between DBD-F and other RPA domains without preventing those same domains from binding to ssDNA (Figure 6A). We have previously shown FPA does not affect ssDNA binding to RPA (17), nor does p53TAD2 affect RPAΔF binding to ssDNA (Figure 3D), consistent with previous findings that full-length p53 did not inhibit RPA binding to ssDNA (62). These data, and the observed binding affinity differences of ligand to DBD-FMBP and RPA, suggest that conformational changes occur in DBD-F in proximity to other RPA domains without affecting ssDNA binding of core domains DBD-A and DBD-B.
Figure 6.

(A) Comparison of Kd values for p53 peptides and FPA binding to DBD-F and RPA. Solid and hollow circles are p53TAD2 and FPA Kd values, respectively, determined in this study. Solid and hollow triangles are p53TAD2b [15 aa peptide (aa 43–57 of p53)] and FPA Kd values, respectively, as reported in previous literature and therein denoted by (†)(63). (B) Depiction of competition between p53TAD2 and an inhibitor for the DBD-F accompanied by a change in conformation.
The RPA (R41E, R43E) mutation not only links the loss of RPA–p53 interaction to the loss of recombination in the rfa-t11 yeast strain, but also raises the possibility that the helix destabilization activity of RPA may have relevance in vivo. RPA is commonly used in in vitro based recombination studies, initially binding ssDNA, and is eventually replaced by Rad51 before recombination with homologous dsDNA. Although RPA is thought to serve merely to deconvolute the ssDNA substrate before Rad51 binding, there could be a DBD-F–ssDNA intermediate step that is essential for efficient recombination to occur. The K45E mutation in rfa-t11 has a deficit in catalysing recombination in these assays, which has been attributed to a loss of protein–protein binding (7). We suggest that both protein–protein and protein–DNA interactions with DBD-F may be important in the recombination process.
The crystal structure of the DBD-F, valuable as it has been for determining the spatial organization of the domain, offers only one conformation within a wide range of possible conformations. We previously used this crystal structure to model FPA and Rad9 binding to DBD-F (17); however, the molecular docking of FPA predicted an affinity that was much lower than the actual in vitro binding affinity. By determining an optimal binding conformation towards FPA, we demonstrate how the basic cleft of the DBD-F changes shape dramatically to confine the ligand. Multiple conformational changes are possible that are initiated by different ligands, as well as DBD-F contact with the remainder of RPA. Based on the relative binding parameters of the new conformation of RPA towards FPA, we are more confident in using this model to identify new inhibitors of DBD-F in silico.
Competition displacement studies of a similar p53 fragment and FPA have been reported (63). The reported binding constant reflected markedly weaker binding, 100 μM vs. 1 μM for p53TAD2 (Figure 6B). For FPA, the binding constant was also reported as weaker (18 μM vs. 9 μM). Although we can appreciate differences in apparent binding due to small changes in peptide sequence and assay conditions, we cannot account for the 50-fold difference in the ratios of binding constants (FPA:p53TAD2 peptide) between these two studies.
The involvement of RPA in several DNA modulatory activities hinges upon its ability to bind multiple disparate substrates. Many of these activities are dependent on the versatility of DBD-F, which offers a binding site for both protein and ssDNA, and harbours a site necessary for DNA unwinding. Further study with FPA and DBD-F may resolve more in-depth aspects of RPA in DNA metabolic activities.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–4.
FUNDING
American Cancer Society [ACS RSG-10-031-01-CCG to G.G.O.]; Nebraska Department of Health and Human Services [2011–25 to G.G.O.]; National Institutes of Health (NIH) [P20RR018759 to G.G.O., P20-RR01875, GM59346, RR015468 to L.J.P.]. Funding for open access charge: NIH [P20RR018759]; American Cancer Society [RSG1003101CCG].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Marc Wold and James Wahl for kindly providing reagents and Adam Mosel for graphical assistance.
REFERENCES
- 1.Oakley GG, Patrick SM. Replication protein A: directing traffic at the intersection of replication and repair. Front. Biosci. 2010;15:883–900. doi: 10.2741/3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 3.Patrick SM, Turchi JJ. Replication protein A (RPA) binding to duplex cisplatin-damaged DNA is mediated through the generation of single-stranded DNA. J. Biol. Chem. 1999;274:14972–14978. doi: 10.1074/jbc.274.21.14972. [DOI] [PubMed] [Google Scholar]
- 4.Lao Y, Lee CG, Wold MS. Replication protein A interactions with DNA. 2. Characterization of double-stranded DNA-binding/helix-destabilization activities and the role of the zinc-finger domain in DNA interactions. Biochemistry. 1999;38:3974–3984. doi: 10.1021/bi982371m. [DOI] [PubMed] [Google Scholar]
- 5.Treuner K, Ramsperger U, Knippers R. Replication protein A induces the unwinding of long double-stranded DNA regions. J. Mol. Biol. 1996;259:104–112. doi: 10.1006/jmbi.1996.0305. [DOI] [PubMed] [Google Scholar]
- 6.Daughdrill GW, Ackerman J, Isern NG, Botuyan MV, Arrowsmith C, Wold MS, Lowry DF. The weak interdomain coupling observed in the 70 kDa subunit of human replication protein A is unaffected by ssDNA binding. Nucleic Acids Res. 2001;29:3270–3276. doi: 10.1093/nar/29.15.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kantake N, Sugiyama T, Kolodner RD, Kowalczykowski SC. The recombination-deficient mutant RPA (rfa1-t11) is displaced slowly from single-stranded DNA by Rad51 protein. J. Biol. Chem. 2003;278:23410–23417. doi: 10.1074/jbc.M302995200. [DOI] [PubMed] [Google Scholar]
- 8.Kim HS, Brill SJ. Rfc4 interacts with Rpa1 and is required for both DNA replication and DNA damage checkpoints in Saccharomyces cerevisiae. Mol. Cell. Biol. 2001;21:3725–3737. doi: 10.1128/MCB.21.11.3725-3737.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haring SJ, Mason AC, Binz SK, Wold MS. Cellular functions of human RPA1. Multiple roles of domains in replication, repair, and checkpoints. J. Biol. Chem. 2008;283:19095–19111. doi: 10.1074/jbc.M800881200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oakley GG, Tillison K, Opiyo SA, Glanzer JG, Horn JM, Patrick SM. Physical interaction between replication protein A (RPA) and MRN: involvement of RPA2 phosphorylation and the N-terminus of RPA1. Biochemistry. 2009;48:7473–7481. doi: 10.1021/bi900694p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li R, Botchan MR. The acidic transcriptional activation domains of VP16 and p53 bind the cellular replication protein A and stimulate in vitro BPV-1 DNA replication. Cell. 1993;73:1207–1221. doi: 10.1016/0092-8674(93)90649-b. [DOI] [PubMed] [Google Scholar]
- 12.He Z, Brinton BT, Greenblatt J, Hassell JA, Ingles CJ. The transactivator proteins VP16 and GAL4 bind replication factor A. Cell. 1993;73:1223–1232. doi: 10.1016/0092-8674(93)90650-f. [DOI] [PubMed] [Google Scholar]
- 13.Dutta A, Ruppert JM, Aster JC, Winchester E. Inhibition of DNA replication factor RPA by p53. Nature. 1993;365:79–82. doi: 10.1038/365079a0. [DOI] [PubMed] [Google Scholar]
- 14.Olson E, Nievera CJ, Liu E, Lee AY, Chen L, Wu X. The Mre11 complex mediates the S-phase checkpoint through an interaction with replication protein A. Mol. Cell. Biol. 2007;27:6053–6067. doi: 10.1128/MCB.00532-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu X, Vaithiyalingam S, Glick GG, Mordes DA, Chazin WJ, Cortez D. The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Mol. Cell. Biol. 2008;28:7345–7353. doi: 10.1128/MCB.01079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ball HL, Ehrhardt MR, Mordes DA, Glick GG, Chazin WJ, Cortez D. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol. Cell. Biol. 2007;27:3367–3377. doi: 10.1128/MCB.02238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glanzer JG, Liu S, Oakley GG. Small molecule inhibitor of the RPA70 N-terminal protein interaction domain discovered using in silico and in vitro methods. Bioorg. Med. Chem. 2011;19:2589–2595. doi: 10.1016/j.bmc.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 19.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 20.Joerger AC, Fersht AR. The tumor suppressor p53: from structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010;2:a000919. doi: 10.1101/cshperspect.a000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Candau R, Scolnick DM, Darpino P, Ying CY, Halazonetis TD, Berger SL. Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene. 1997;15:807–816. doi: 10.1038/sj.onc.1201244. [DOI] [PubMed] [Google Scholar]
- 22.Vise PD, Baral B, Latos AJ, Daughdrill GW. NMR chemical shift and relaxation measurements provide evidence for the coupled folding and binding of the p53 transactivation domain. Nucleic Acids Res. 2005;33:2061–2077. doi: 10.1093/nar/gki336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajagopalan S, Andreeva A, Teufel DP, Freund SM, Fersht AR. Interaction between the transactivation domain of p53 and PC4 exemplifies acidic activation domains as single-stranded DNA mimics. J. Biol. Chem. 2009;284:21728–21737. doi: 10.1074/jbc.M109.006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bochkareva E, Kaustov L, Ayed A, Yi GS, Lu Y, Pineda-Lucena A, Liao JC, Okorokov AL, Milner J, Arrowsmith CH, et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl Acad. Sci. USA. 2005;102:15412–15417. doi: 10.1073/pnas.0504614102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajagopalan S, Andreeva A, Rutherford TJ, Fersht AR. Mapping the physical and functional interactions between the tumor suppressors p53 and BRCA2. Proc. Natl Acad. Sci. USA. 2010;107:8587–8592. doi: 10.1073/pnas.1003689107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaustov L, Yi GS, Ayed A, Bochkareva E, Bochkarev A, Arrowsmith CH. p53 transcriptional activation domain: a molecular chameleon? Cell Cycle. 2006;5:489–494. doi: 10.4161/cc.5.5.2489. [DOI] [PubMed] [Google Scholar]
- 27.Sirbu BM, Lachmayer SJ, Wulfing V, Marten LM, Clarkson KE, Lee LW, Gheorghiu L, Zou L, Powell SN, Dahm-Daphi J, et al. ATR-p53 restricts homologous recombination in response to replicative stress but does not limit DNA interstrand crosslink repair in lung cancer cells. PLoS One. 2011;6:e23053. doi: 10.1371/journal.pone.0023053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pretto DI, Tsutakawa S, Brosey CA, Castillo A, Chagot ME, Smith JA, Tainer JA, Chazin WJ. Structural dynamics and single-stranded DNA binding activity of the three N-terminal domains of the large subunit of replication protein A from small angle X-ray scattering. Biochemistry. 2010;49:2880–2889. doi: 10.1021/bi9019934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henricksen LA, Umbricht CB, Wold MS. Recombinant replication protein A: expression, complex formation, and functional characterization J. Biol. Chem. 1994;269:11121–11132. [PubMed] [Google Scholar]
- 30.Sheffield P, Garrard S, Derewenda Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif. 1999;15:34–39. doi: 10.1006/prep.1998.1003. [DOI] [PubMed] [Google Scholar]
- 31.Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991;10:4129–4135. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Press WH, Teukolsky SA, Vetterling WT, Flannery BP. Numerical Recipes: The Art of Scientific Computing. 3rd edn. Cambridge England: Cambridge University Press; 2007. [Google Scholar]
- 33.Draper NR, Smith H. Applied Regression Analysis. 2nd edn. John Wiley and Sons; 1981. [Google Scholar]
- 34.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 35.Lee MR, Duan Y, Kollman PA. State of the art in studying protein folding and protein structure prediction using molecular dynamics methods. J. Mol. Graph. Model. 2001;19:146–149. doi: 10.1016/s1093-3263(00)00126-1. [DOI] [PubMed] [Google Scholar]
- 36.Jorgensen WL, Chandrasekhar J, Buckner JK, Madura JD. Computer simulations of organic reactions in solution. Ann. N. Y. Acad. Sci. 1986;482:198–209. doi: 10.1111/j.1749-6632.1986.tb20951.x. [DOI] [PubMed] [Google Scholar]
- 37.Villa A, Mark AE, Saracino GA, Cosentino DP, Moro G, Salmona M. Conformational polymorphism of the PrP106-126 peptide in different environments: a molecular dynamics study. J. Phys. Chem. B. 2006;110:1423–1428. doi: 10.1021/jp052722o. [DOI] [PubMed] [Google Scholar]
- 38.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 39.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 40.Miyamoto S, Kollman PA. Settle—an analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 41.Tironi I, Sperb R, Smith P, van Gunsteren W. A generalized reaction field method for molecular-dynamics simulations. J. Phys. Chem. 1995;102:5451–5459. [Google Scholar]
- 42.van der Spoel D, Lindahl E, Hess B, van Buuren AR, Apol E, Meulenhoff PJ, Tieleman DP, Sijbers AL, Teemu M, Feenstra KA. 2010 Gromacs user manual version 4.5.4. [Google Scholar]
- 43.Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bakan A, Meireles LM, Bahar I. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics. 2011;27:1575–1577. doi: 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolf LK. Digital Briefs. Chemical & Engineering News. 2009;87:31. [Google Scholar]
- 47.Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010;24:417–422. doi: 10.1007/s10822-010-9352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wiyono B, Tachibana S, Tinambunan D. Reaction of abietic acid with maleic anhydride and fumaric acid and attempts to find the fundamental component of fortified rosin. Pak. J. Biol. Sci. 2007;10:1588–1595. doi: 10.3923/pjbs.2007.1588.1595. [DOI] [PubMed] [Google Scholar]
- 49.Umezu K, Sugawara N, Chen C, Haber JE, Kolodner RD. Genetic analysis of yeast RPA1 reveals its multiple functions in DNA metabolism. Genetics. 1998;148:989–1005. doi: 10.1093/genetics/148.3.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 51.Jacobs DM, Lipton AS, Isern NG, Daughdrill GW, Lowry DF, Gomes X, Wold MS. Human replication protein A: global fold of the N-terminal RPA-70 domain reveals a basic cleft and flexible C-terminal linker. J. Biomol. NMR. 1999;14:321–331. doi: 10.1023/a:1008373009786. [DOI] [PubMed] [Google Scholar]
- 52.Nussinov R, Ma B. Protein dynamics and conformational selection in bidirectional signal transduction. BMC Biol. 2012;10:2. doi: 10.1186/1741-7007-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boehr DD. Promiscuity in protein-RNA interactions: conformational ensembles facilitate molecular recognition in the spliceosome: conformational diversity in U2AF(6)(5) facilitates binding to diverse RNA sequences. Bioessays. 2012;34:174–180. doi: 10.1002/bies.201100152. [DOI] [PubMed] [Google Scholar]
- 54.Fanning E, Klimovich V, Nager AR. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006;34:4126–4137. doi: 10.1093/nar/gkl550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Romanova LY, Willers H, Blagosklonny MV, Powell SN. The interaction of p53 with replication protein A mediates suppression of homologous recombination. Oncogene. 2004;23:9025–9033. doi: 10.1038/sj.onc.1207982. [DOI] [PubMed] [Google Scholar]
- 56.Shammas MA, Shmookler Reis RJ, Koley H, Batchu RB, Li C, Munshi NC. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood. 2009;113:2290–2297. doi: 10.1182/blood-2007-05-089193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plo I, Nakatake M, Malivert L, de Villartay JP, Giraudier S, Villeval JL, Wiesmuller L, Vainchenker W. JAK2 stimulates homologous recombination and genetic instability: potential implication in the heterogeneity of myeloproliferative disorders. Blood. 2008;112:1402–1412. doi: 10.1182/blood-2008-01-134114. [DOI] [PubMed] [Google Scholar]
- 58.Liu Y, Kvaratskhelia M, Hess S, Qu Y, Zou Y. Modulation of replication protein A function by its hyperphosphorylation-induced conformational change involving DNA binding domain B. J. Biol. Chem. 2005;280:32775–32783. doi: 10.1074/jbc.M505705200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patrick SM, Turchi JJ. Stopped-flow kinetic analysis of replication protein A-binding DNA: damage recognition and affinity for single-stranded DNA reveal differential contributions of k(on) and k(off) rate constants. J. Biol. Chem. 2001;276:22630–22637. doi: 10.1074/jbc.M010314200. [DOI] [PubMed] [Google Scholar]
- 60.Roy R, Kozlov AG, Lohman TM, Ha T. SSB protein diffusion on single-stranded DNA stimulates RecA filament formation. Nature. 2009;461:1092–1097. doi: 10.1038/nature08442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim C, Paulus BF, Wold MS. Interactions of human replication protein A with oligonucleotides. Biochemistry. 1994;33:14197–14206. doi: 10.1021/bi00251a031. [DOI] [PubMed] [Google Scholar]
- 62.Miller SD, Moses K, Jayaraman L, Prives C. Complex formation between p53 and replication protein A inhibits the sequence-specific DNA binding of p53 and is regulated by single- stranded DNA. Mol.Cell. Biol. 1997;17:2194–2201. doi: 10.1128/mcb.17.4.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Souza-Fagundes EM, Frank AO, Feldkamp MD, Dorset DC, Chazin WJ, Rossanese OW, Olejniczak ET, Fesik SW. A high-throughput fluorescence polarization anisotropy assay for the 70N domain of replication protein A. Anal. Biochem. 2012;421:742–749. doi: 10.1016/j.ab.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.