


Abstract
Classic non-homologous end joining (C-NHEJ) is the predominant DNA double-strand break repair pathway in humans. Although seven genes Ku70, Ku86, DNA-PKcs, Artemis, DNA Ligase IV (LIGIV), X-ray cross-complementing group 4 and XRCC4-like factor are required for C-NHEJ, several of them also have ancillary functions. For example, Ku70:Ku86 possesses an essential telomere maintenance activity. In contrast, LIGIV is believed to function exclusively in C-NHEJ. Moreover, a viable LIGIV-null human B-cell line and LIGIV-reduced patient cell lines have been described. Together, these observations suggest that LIGIV (and hence C-NHEJ), albeit important, is nonetheless dispensable, whereas Ku70:Ku86 and telomere maintenance are essential. To confirm this hypothesis, we inactivated LIGIV in the epithelial human cell line, HCT116. The resulting LIGIV-null cell line was viable, verifying that the gene and C-NHEJ are not essential. However, functional inactivation of RAD54B, a key homologous recombination factor, in the LIGIV-null background yielded no viable clones, suggesting that the combined absence of RAD54B/homologous recombination and C-NHEJ is synthetically lethal. Finally, we demonstrate that LIGIV is differentially required for certain chromosome fusion events induced by telomere dysfunction—used for those owing to the overexpression of a dominant negative version of telomere recognition factor 2, but not used for those owing to absence of Ku70:Ku86.
INTRODUCTION
A DNA double-strand break (DSB) is one of the most deleterious lesions that can occur in cells because even a single unrepaired DNA DSB can stop the cell cycle and induce cell death (1,2). To protect themselves from DSBs, cells have developed at least two major DNA DSB repair pathways: homologous recombination (HR) and classic non-homologous end joining (C-NHEJ) (3,4). To enact repair, HR uses extensive sequence homology—generally sequences longer than 30 nucleotides (nt)—and generates repaired products that are essentially ‘error free’. In contrast, C-NHEJ requires only 0–4 nt of homology, but because of attendant insertions and deletions is consequently more ‘error-prone’. Recently, a sub- or back-up pathway of C-NHEJ, alternative-NHEJ (A-NHEJ) that has features reminiscent of both HR and C-NHEJ has been described (5–7). A-NHEJ, like HR, requires homologous sequences to mediate the repair reaction. In the case of A-NHEJ, however, only 5–25 nt of homology (often referred to as ‘microhomology’) is needed. Additionally, because its reaction mechanism always results in accompanying deletions, A-NHEJ is therefore similar to C-NHEJ in that it is ‘error prone’ (8). Depending on the organism and various parameters (e.g. position in the cell cycle, cell type, etc.), HR, C-NHEJ and A-NHEJ are differentially used (4,8–11). Bacteria (12) and lower eukaryotes like yeast (13) use HR almost exclusively for all DSB repair events. In contrast, higher eukaryotes such as humans use C-NHEJ more often than HR, and C-NHEJ is by far the predominant repair mechanism used during G0/G1 phases in human cells (7). This usage bias is, however, not exclusive. For example, during late S and G2 phases in human cells, HR is more active because a proximal homology donor becomes available in the form of a sister chromatid (14,15). In summary, higher eukaryotes have multiple options available to them in terms of the pathways that can be used to repair a DSB. This pathway choice flexibility is beneficial in certain circumstances and has evolutionarily been selected for. It is clear, however, that as each pathway makes a biologically and functionally distinct product, this choice must be tightly regulated such that the right product is generated in the correct biological context.
In addition to DSB repair, the C-NHEJ pathway is also required for variable(diversity)joining [V(D)J] recombination, class switch recombination (CSR) and telomere maintenance (16,17). V(D)J recombination is the initial step of antigen receptor maturation that occurs in early B- and T-lymphocytes, whereas CSR occurs subsequently and exclusively in more mature B-cells. Although V(D)J recombination and CSR are lymphoid-restricted processes, all nucleated cells in the human body have chromosomes, and all linear chromosomes have ends (‘telomeres’). Telomeres consist of a repetitive tract of DNA that assembles into a structure called a t-loop, which is a variation of a classical D-loop (18). The t-loop is coated by a proteinaceous cap that essentially keeps the chromosome ends invisible to all of the DNA DSB pathways. The core proteins that bind to telomeres are collectively called ‘shelterin’ (19,20). There are, additionally, a bevy of shelterin-associated proteins found at telomeres, and unexpectedly some of these correspond to C-NHEJ factors. For example, the Ku heterodimer, and possibly DNA-PKcs, is part of the t-loop-associated complex (21–23). Interestingly, telomere protection by the Ku complex is essential in human cells because Ku loss-of-function mutations provoke cell death triggered by telomere dysfunction (24,25). All of the C-NHEJ factors, however, are unlikely to be involved in telomere maintenance, and there is, for example, little evidence for a role for DNA Ligase IV (LIGIV), or its accessory factors, X-ray cross-complementing group 4 (XRCC4) and XRCC4-like factor (XLF) in normal telomere maintenance.
Mutation of any C-NHEJ component results in pathological phenotypes. For example, Ku70, Ku86 and DNA-PKcs knockout mice are viable, but they present with severe growth defects, severe-combined immune deficiency (SCID) and profound hypersensitivity to ionizing radiation (IR) (26–28). In humans, the phenotypes are actually more serious. There are no known Ku70 or Ku86 defective patients, a fact that has been correlated with Ku’s essential role in telomere maintenance (24,29,30), and only one hypomorphic DNA-PKcs-deficient patient has been reported (31), suggesting that, like Ku, complete DNA-PKcs loss-of-function mutations may not be tolerated in humans. LIGIV and XRCC4 knockout mice are embryonic lethal, and although representative MEFs derived from these animals are viable, they show severe IR sensitivity and defects in V(D)J recombination (32,33). Seven human LIGIV patients have been reported so far in the literature, and four of them presented with a ‘LIGIV syndrome’, which is characterized by severe IR sensitivity, chromosomal instability, unusual facial features and developmental and growth delay (34). The other LIGIV patients displayed only a subset of these features but were also afflicted with leukemia and radiation sensitive SCID (35). Importantly, however, all seven LIGIV patients described to date had non-null, hypomorphic mutations (34–36), suggesting that LIGIV, like Ku70, Ku86 and DNA-PKcs, is essential.
Michael Lieber’s laboratory has studied the function of LIGIV for V(D)J recombination and IR sensitivity in the human B-lymphoid precursor cell line, NALM-6 (37). These researchers generated, by classical gene-targeting technology, a LIGIV-deficient NALM-6 cell line, which was viable. The viability of the cell line was unexpected based on the phenotypes of the knockout mouse models and the reports for LIGIV patients described earlier in the text. One possible explanation for these results is that the loss-of-function of LIGIV may be tolerated specifically only in NALM-6 cells, but that this condition is not generally applicable. This hypothesis takes into account the fact that lymphoid cells tend to use more HR than C-NHEJ (38) and is consistent with the demonstration that NALM-6 cells are specifically hyper-recombinogenic (39) so that the absence of LIGIV might be less toxic to NALM-6 than most of the other cell types (40). One way to address these issues would be by inactivating LIGIV in a non-B human somatic cell line.
Accordingly, we report here the inactivation of LIGIV in the epithelial human colorectal cancer cell line, HCT116, using recombinant adeno-associated virus (rAAV) gene-targeting methodologies (41,42). Two rounds of gene targeting were used to generate a LIGIV-null HCT116 cell line, which was viable. The ability to obtain a viable LIGIV-null clone is consistent with the existence of the LIGIV-null NALM-6 cell line (40,43,44) and suggests that neither LIGIV nor C-NHEJ is essential in somatic cells. Importantly, however, a subsequent attempt to inactivate RAD54B, a key, albeit non-essential, HR factor proved unsuccessful. Thus, human somatic cells can survive with reduced HR or C-NHEJ, but they are not viable when they have deficits in both pathways. Finally, the role of LIGIV in telomere dysfunction was addressed using two disparate methodologies. In one instance, a dominant negative version of a key shelterin component, telomere recognition factor 2 (TRF2DN), was overexpressed, whereas in another instance, rapid telomere loss was induced by the functional inactivation of Ku86. Surprisingly, LIGIV was required for the chromosome fusion events mediated by TRF2DN overexpression, but not those induced by the absence of Ku. All of these observations suggest that although LIGIV (and by extension, C-NHEJ) is not essential for normal human telomere maintenance nor human somatic cell viability, it nonetheless does have important roles in maintaining cellular homeostasis including, specifically, genomic stability following telomere dysfunction.
MATERIALS AND METHODS
Cell culture
HCT116 cells (ATCC) were grown in McCoy’s 5A media supplemented with 10% fetal calf serum and 100 U/ml penicillin and 100 U/ml streptomycin and glutamine. All cells were grown at 37°C in a humidified incubator with 5% CO2.
Targeting vector construction
The LIGIV targeting vector was constructed using basic rAAV methodologies as described (41,42). The right and left homology arms were amplified by polymerase chain reaction (PCR) using genomic DNA purified from wild-type HCT116 cells as the template. The primers used to make left homology arm of LIGIV-targeting vector were P2.1 and P2.2. The right homology arm was constructed by PCR using the primers P2.3 and P2.4. As fusion PCR templates, the two arm constructs and a 4-kb PvuI-digested fragment of the plasmid Need-A-Knockout-neomycin resistance (pNeDaKO-Neo) vector (41) were used. Fusion PCR products were amplified by P2.1 and P2.4 primers and then purified by gel-extraction. The fusion construct was subsequently NotI digested and then cloned into a NotI-digested plasmid adeno-associated virus multi-cloning site (pAAV-MCS) vector. For LIGIV screening, the RArmF/KO3′R primer set and the P1F/P2R primer set were used for the first and second rounds, respectively.
Construction:
P2.1:
5′-ATACATACGCGGCCGCGCAGAAACATGCAGTATTTTCCCCTA-3′
P2.2:
5′-GCTCCAGCTTTTGTTCCCTTTAGCAAAGCGGTGATGAATCTTCTCGT-3′
P2.3:
5′-CGCCCTATAGTGAGTCGTATTACAGATGGAAAAGATGCCCTCAAACT-3′
P2.4:
5′-ATACATACGCGGCCGCTTGTGTTTTCTGCACTATTTCTATTC-3′
Screening:
RArmF:
5′-CGCCCTATAGTGAGTCGTATTAC-3′
KO3’R:
5′-AAAATGAGACATCATTCCACCCCGTGAT-3′
P1F:
5′-GGGTTGGAGCAAAACAGTTATTAAATGTAG-3′
P2R:
5′-CAATTGAGTCTAAAAGGTCGTTTACTTGC-3′
The Rad54B-targeting vector construction and screening for correctly targeted clones were performed in a fashion similar to that described earlier in the text. The 661 bp of DNA needed for the left homology arm was generated by PCR using the E3-LARM-NotI-F1 and E3-LARM-SacII-R1 primers. The 1620 bp of right homology arm DNA was constructed by PCR using the E3-RARM-KpnI-F1 and E3-RARM-NotI-R3 primers, and the vector was assembled in a three-step ligation reaction. Primer sets #1 and 2 were subsequently used for screening for correctly targeted clones. Primer set #1 consisted of Rad54B-E3-CreS-F1 and Rad54B-E3-CreS-R1. Primer set #2 consisted of Rad54B-E3-LARM-SC-F4 and NeoR2.
Construction:
E3-LARM-NotI-F1:
5′-ACATAAGCGGCCGCTTTAAGTATTGATTTTAGTATTGAGAAATTTAAC-3′
E3-LARM-SacII-R1:
5′-GGCGGCCCGCGGCTAAAAGAAACAAATATATATTTAAATCAGAACTC-3′
E3-RARM-KpnI-F1:
5′-CCGGTACCGACTGCTTTTTATTGATAAGGTTTATGCTTGACC-3′
E3-RARM-NotI-R3:
5′-ACATAAGCGGCCGCGGTGATGGGGAAAATGACATATGTTATTTAACTGG-3′
Screening:
Rad54B-E3-CreS-F1:
5′-GAGTTCTGATTTAAATATATATTTGTTTCTTTTAG-3′
Rad54B-E3-CreS-R1:
5′-CAAGCATAAACCTTATCAATAAAAAGC-3′
Rad54B-E3-LARM-SC-F4:
5′-CCAACATAGTGAGATTACCATTATCTCACC-3′
NeoR2:
5′-AAAGCGCCTCCCCTACCCGGTAGGGCG-3′
Packaging and isolating virus
AAV-293 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) media supplemented with 10% fetal calf serum and 100 U/ml penicillin and 100 U/ml streptomycin and glutamine at 37°C in a humidified incubator with 5% CO2. The cells were subcultured into a 10 cm culture dish 1 day before transfection. pAAV-replication/capsid (RC) and pAAV-helper plasmids from the AAV Helper-Free system and the targeting vector (8 µg of each) were co-transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. Virus was harvested 2 days after transfection by collecting the cells in 1 ml DMEM media and then performing a freeze and thaw cycle three times with vigorous vortexing in between. The resulting cellular debris was clarified by centrifuging at 13 000 rpm for 2 min, after which the virus-containing supernatant was collected and used for a subsequent infection.
rAAV infections
HCT116 cells were plated on a six-well plate 1 day before infection. When the cells were ∼65% confluent, fresh media (1 ml) was added with an adequate amount of virus. After a 2-h incubation at 37°C, 4 ml of fresh media was added to the virus-containing media. Two days after infection, the cells were trypsinized and transferred at 1000 cells/well on 96-well plates and selection was started using 1 mg/ml gentimycin 418 (G418).
Isolation of genomic DNA and genomic PCR
Genomic DNA for PCR screening was isolated using a Gentra Puregene Cell Kit according to the manufacturer’s instructions. The DNA was dissolved in a final volume of 25 µl, 1 µl of which was subsequently used in each PCR reaction.
Immunoblotting
LIGIV expression was characterized by first preparing nuclear extracts using Sigma’s CelLytic NuCLEAR Extraction Kit according to the manufacturer’s instructions. Nuclear extracts (30 µg) were electrophoresed on a 7.5% SDS-polyacrylamide gel and a rabbit, anti-human LIGIV antibody (Serotec) was used at a 1:1000 dilution. To screen for complemented clones, whole cell extracts were prepared with radio immunoprecipitation assay (RIPA) buffer. LIGIV (Abcam) and HA (hemagglutinin antigen) antibodies (Covance) were used at 1:1000 and 1:5000 dilutions, respectively. Actin expression was used as a loading control using an actin antibody (SantaCruz) at a 1:250 dilution.
Etoposide sensitivity
Etoposide sensitivity assays were performed as described (45) with a slight modification. Three hundred to fifty thousand (depending on the drug concentration and the cell line involved) cells were plated ∼17–19 h before drug treatment. Etoposide was dissolved in dimethyl sulfoxide to generate a 10 mM stock solution. The cells were treated with etoposide at varying concentrations and then incubated for an additional 7–10 days. The cells were subsequently fixed with a solution of 10% methanol and 10% acetic acid and stained with crystal violet. A colony was scored as viable when it consisted of ≥50 cells.
Microhomology assay
The microhomology assay was performed as described (46). The pDVG94 plasmid (2.5 μg) was restriction digested with EcoRV and AfeI, purified and then transfected using Lipofectamine 2000. Plasmid DNA was recovered 48 h after transfection using a modified Qiagen mini-preparation protocol (10). The repaired junctions were PCR amplified using FM30 and 5′-radiolabeled DAR5 primers. The resultant radioactive PCR products were treated with BstXI, and the restriction digested PCR products were separated on a 6% Tris/Borate/ethylenediaminetetraacetic acid polyacrylamide gel. The gel was subsequently dried and exposed to X-ray film for ∼10–15 min.
DAR5:
5′-TGCTTCCGGCTCGTATGTTGGTTGGAAT-3′
FM30:
5′-CTCCATTTTAGCTTCCTTAGCTCCTG-3′
Complementation
A wild-type LIGIV cDNA and a wild-type LIGIV cDNA with a hemagglutinin (HA)-epitope tag fused in-frame at the C-terminal end were constructed. These cDNAs were cloned into pcDNA3.1(+) using BamHI and EcoRI. The constructs were subsequently linearized in the vector backbone with PvuI and transfected into LIGIV-null cells. Forty-eight hours after transfection, the cells were subcultured under limiting dilution into 96-well plates with 1 mg/ml G418. Colonies were expanded for 4–5 weeks and then complemented clones were identified by immunoblotting for LIGIV expression as described earlier in the text.
Gross chromosomal rearrangements test
G-banding cytogenetic analyses were performed in the Cytogenetics Core Laboratory at the University of Minnesota.
Telomeric fluorescence in situ hybridization and sub-telomeric fluorescence in situ hybridization
For telomeric fluorescence in situ hybridization (tFISH), cells were treated with AdCre and left to grow for 3 days. On day 3, cells were treated with 10 µg/mL colcemid for 2 h, harvested and prepared using the manufacturer’s (Dako; tFISH kit) protocol. FISH was performed using a protein:nucleic acid telomere-specific probe [Cy3 conjugated to (T2AG3)3] in accordance with manufacturer’s protocol (Dako). Coverslips were mounted using Prolong Gold Antifade reagent, and metaphases were viewed using an Olympus FluoView FV1000 BX2 upright confocal microscope at the University of Minnesota Imaging Center. tFISH signals were analysed by hand, and the signal-free ends were scored blindly.
For sub-tFISH, cells were processed precisely as described earlier in the text with only the hybridization probes being different. Sub-telomeric probes against 1q, 4p, or, as a control, to the centromeric region of chromosome 6 were diluted to 1× in hybridization buffer (HB500L) and applied to slides according to the manufacturer (Cytocell). Hybridization, viewing and scoring were carried out as described earlier in the text.
TRF2ΔBΔM expression
HEK 293 cells stably expressing retroviral envelope protein were plated into six-well plates 30 h before transfection such that at the time of transfection they were ∼50% confluent. The HEK 293 cells were then transfected with pLPC-TRF2ΔBΔM vector (47) using Lipofectamine 2000 (Invitrogen). Twelve hours after transfection, the DMEM media was exchanged. The virus-containing supernatant was collected 72 h after transfection and filtered through a 0.45 μm filter. This virus-containing media, supplemented with 15% fetal calf serum and polybrene (4 μg/ml) was added to the target cells, which had been subcultured a day before infection. The cells were maintained with virus for 120 h and then harvested for analysis.
Cre recombination of the Ku86 conditionally null cell lines
Ku86-null HCT116 cells were generated when needed as described (24). Briefly, Ku86flox/− cells were first subcultured in six-well plates at 5 × 104 cells/well, and 16–18 h later, 5 × 108 adenoviral particles (AdCre or AdCMV; Vector Development Laboratories) were added in 2 ml media. After 3 days of incubation, cells were harvested and used for sub-tFISH analysis as described earlier in the text.
RESULTS
Inactivation of LIGIV in the HCT116 cell line
rAAV gene-targeting methodology was used to inactivate the LIGIV gene in the HCT116 colorectal carcinoma cell line (41,42). This cell line has been used extensively for similar gene-targeting studies (48). The cell line is immortalized, transformed and mismatch repair defective (49–51). However, the cell line is also mostly diploid, has a stable karyotype and is wild-type for almost all other DNA repair and checkpoint genes (48). The LIGIV gene is located on chromosome 13 and encompasses three exons. However, as the LIGIV coding sequence resides only in exon 3, we disrupted the first 302 bp of coding sequence within that exon by replacing it with the neomycin phosphotransferase (NEO) gene. The relevant rAAV-targeting vector (rAAVLIGIV) consisted of two ∼900-bp long homology arms (which, on the chromosome, flanked the 302-bp region to be deleted) and a NEO selection cassette, which was itself flanked by locus-of-crossover for bacteriophage P1 (LoxP) sites (Figure 1A).
Figure 1.
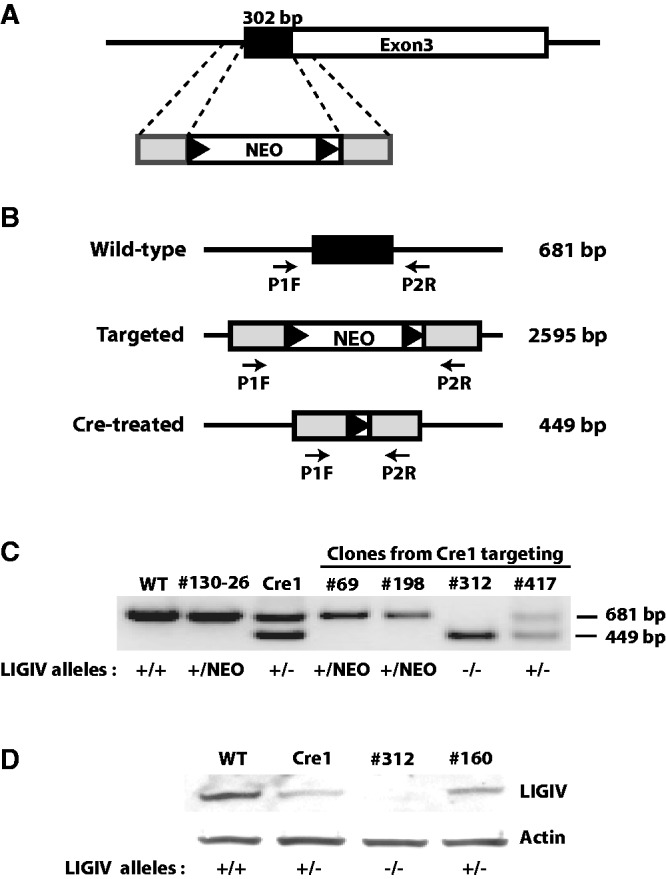
The targeting strategy and the primers used for the LIGIV knockout. (A) The black rectangle represents the first 302 bp of LIGIV coding sequence, which resides in the third exon and which was replaced by the NEO gene using rAAV-mediated gene targeting. The gray rectangles represent the ∼900 bp of homology arms flanking the targeting region. The black triangles represent LoxP sites. (B) Primers used for the second round of screening. The P1F/P2R primer set gives different-sized PCR products depending on the allelic states of the LIGIV gene; 681 bp for the wild-type allele, 2595 bp for the NEO-targeted allele and 449 bp for a Cre-treated allele. Under the PCR conditions used here with a short extension time, the 2595-bp product was not generated. (C) PCR screening. #130-26 is one of the LIGIV+/NEO (+/NEO) subclones. Cre1 is a G418-sensitive LIGIV heterozygous cell (+/−) generated by Cre-recombination of #130-26. Cre1 was used for the second round of targeting. Cell lines #69 and #198 are retargeted clones (+/NEO) from the second round of gene targeting. Clone #312 is LIGIV−/− (−/−), and #417 is an example of a LIGIV+/− (+/−) clone that suffered a random integration during the second round of gene targeting. (D) Western blot analysis confirmed that clone #312 is a LIGIV−/− (−/−) cell. Cre1 is described in (C) and is a LIGIV heterozygous cell (+/−). Clone #160 is analogous to clone #417 in (C) and is a LIGIV+/− clone (+/−) that suffered a random integration during the second round of gene targeting. The parental HCT116 (WT) cell line was used as a positive control. Actin was used as a loading control.
HCT116 cells were infected with the rAAVLIGIV vector, and in the first round of targeting, 177 G418-resistant clones were subsequently screened by PCR using primers that were specific to the targeting vector and a unique sequence in the flanking DNA. Two correctly targeted clones (#130 and #163) were identified for a relative gene targeting efficiency of 1.1% (2/177), which is similar to the frequencies reported for other rAAV-mediated gene targeting studies using basic vectors (42,48). The isolated clones were subsequently subcloned to ensure that they originated from a single cell. One of the subclones (#130-26) was designated as LIGIV+/NEO and then treated transiently with a Cre-recombinase expression vector to remove the NEO selection cassette (which is flanked by LoxP sites; Figure 1A). A G418-sensitive derivative subclone was obtained from this protocol and renamed as LIGIV+/− or Cre1. Cre1 cells were then subjected to a second round of gene targeting—using the same rAAVLIGIV vector that had been used in the first round—to inactivate the remaining wild-type allele. In the second round of gene targeting, a total of 673 clones were screened, and 16 correctly targeted clones were obtained (relative gene targeting frequency: 2.4%). Fifteen of these clones were retargeted to the already inactivated allele and were therefore still heterozygous. Only one clone (#312, see later in the text) was targeted at the remaining wild-type allele and was designated as LIGIVNEO/−. This clone was subsequently converted into a LIGIV−/− cell line by transient treatment with the Cre recombinase to remove the NEO selection cassette as described earlier in the text.
For the second round of screening, a primer set was used that could distinguish the three possible different LIGIV allelic states: a wild-type allele, a NEO-targeted allele and the Cre-treated allele with a residual single LoxP site. These three configurations generated 681, 2595 and 449-bp PCR products, respectively (Figure 1B). Retargeted clones, where the targeting construct integrated into the previously inactivated allele, had a diagnostic pattern where the 449-bp PCR product derived from the Cre-treated allele disappeared but where the 681-bp PCR product representative of the wild-type allele was still present (e.g. clones #69 and #198, Figure 1C). In contrast, randomly targeted second round clones still retained both the 449 and 681-bp PCR products in a manner unchanged from the parental Cre1 clone (e.g. clones #417 and Cre1, Figure 1C). Apart from all these clones was the pattern generated by clone #312, which did not produce the 681-bp PCR fragment from the wild-type allele but did generate the 449-bp PCR product originating from the Cre-treated allele, suggesting that it was a true null clone (clone #312, Figure 1C). In this PCR analysis, only a 2 min extension time was used; therefore, the 2595-bp product resulting from amplification of the NEO-containing targeted allele was not detected (Figure 1C). Subsequently, we confirmed that the founding LIGIV+/− cell lines, Cre1 and #160, have reduced amounts of LIGIV protein in comparison with the parental strain and that the LIGIV−/− cell line, #312, expressed no LIGIV protein detectable by western blot analysis (Figure 1D). From these molecular and biochemical analyses, we concluded that clone #312 corresponded to a viable LIGIV-null (LIG4−/−) cell line, and this demonstrated that LIGIV is not essential in the HCT116 genetic background.
A LIGIV deficiency causes a mild growth defect
Three thousand cells corresponding to each of the parental (i.e. LIGIV+/+), LIGIV+/− (Cre1 or #160) or LIG4−/− cell lines were seeded on a six-well plate on day 0, and the growth of cells from day 6 to 10 was determined. LIGIV+/− cells grew at a rate that was slightly, but significantly, reduced from the parental cell line (except on day 10) demonstrating that there is a mild haploinsufficiency associated with LIGIV (Figure 2A). Moreover, LIGIV−/− cells grew slower than either of the two LIGIV+/− cell lines, especially at earlier time points (Figure 2A). The growth defect associated with the LIGIV-null cell line, however, in contrast to the growth defect reported for DNA-PKcs (52), another key C-NHEJ gene, was in comparison mild.
Figure 2.
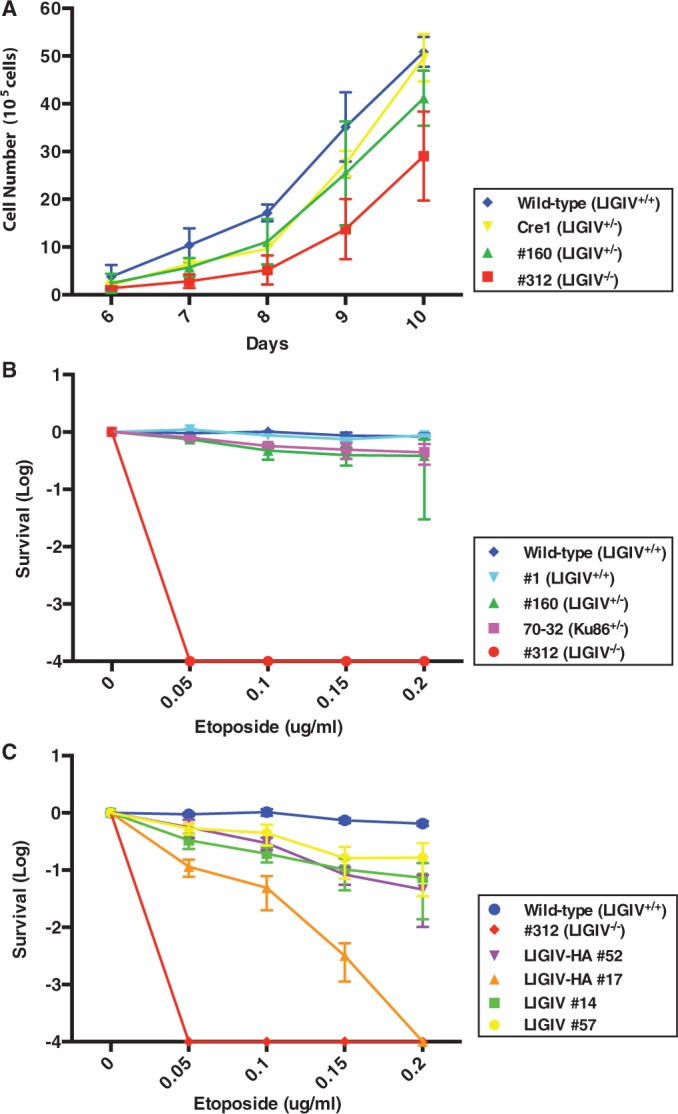
The growth properties and etoposide sensitivity of LIGIV-null cells. (A) The LIGIV−/− cell line has a mild growth defect. On day 0, 3000 cells were seeded into six-well plates, and the cells were counted from day 6 to 10. Wild-type (LIGIV+/+), #312 (LIGIV−/−) and Cre1 and #160 (both LIGIV+/−) were assessed. The data shown here represent the average of six counts derived from four independent sets of duplicates excluding the maximum and minimum values. (B) LIGIV−/− cells are extremely sensitive to etoposide. On day 0, 300–50 000 cells (depending on the cell line and drug concentration) were subcultured with different concentrations of etoposide. Ten to fourteen days later, the cells were fixed with methanol:acetic acid and stained with crystal violet. The number of colonies that survived at different concentrations of etoposide was normalized by the survivability of untreated cells, which was set as 1. Cell line designations are defined in (A). In addition, clone #1 (LIGIV+/+) also has a wild-type genotype and clone 70-32 is an etoposide-sensitive Ku86+/− HCT116 cell line that were used as additional positive controls. Each value represents the average of two independent experiments. (C) Complementation partially rescues the etoposide sensitivity of LIGIV−/− cells. Etoposide sensitivity was preformed as described in (B). The complemented cell lines and their level of expression of LIGIV are described in the text.
LIGIV−/− cells are extremely sensitive to DNA damaging agents
The first LIGIV patient described in the literature developed leukemia, but she actually died from an extreme adverse response to the radiation treatment for this cancer and not from the leukemia itself (35). To experimentally determine whether this effect could be recapitulated in our somatic cell model, we carried out colony forming assays in the presence or absence of etoposide, a topoisomerase II inhibitor and a strong radiomimetic drug (53) (Figure 2B). Positive control cell lines included the parental, wild-type cell line and clone #1, which was a randomly targeted clone from the first round of gene targeting that therefore retained the LIGIV+/+ genotype. As negative controls, a clone, #70-32, which has a Ku86+/− genotype and is known to be slightly etoposide-sensitive (30), was used. Compared with the wild-type and randomly targeted clone #1, the LIGIV+/− clone showed a mild hypersensitivity comparable with that of the Ku86+/− cell line (Figure 2B). In stark contrast, the LIGIV−/− cell line was extraordinarily sensitive to even low concentrations of etoposide (Figure 2B). Thus, as expected, LIGIV is required for the repair of etoposide-induced DNA DSBs.
A LIGIV deficiency induces genomic instability
One of the deleterious consequences of unrepaired DNA DSBs is genomic instability, which predisposes cells to cancer (43,54). Because LIGIV is essential for C-NHEJ activity, we expected that LIGIV−/− cells might be genomically unstable, and tested this by measuring karyotypic abnormalities using standard G-banding of metaphase chromosomes. The parental HCT116 wild-type cell line shows four pre-existing karyotypic aberrations: (i) a duplication on one of the chromosome 10s; (ii) a translocation involving chromosome 16; (iii) a translocation involving chromosome 18; and (iv) the variable retention of the Y chromosome [Supplementary Table S1 and Supplementary Figure S1A; (51,55)]. Twenty metaphases from three independent LIGIV heterozygous clones were analysed: #163-7 and #130-26 (Supplementary Figure S1B) are two LIGIV+/− subclones obtained from the first round of targeting, whereas #69 is one of the second-round retargeted LIGIV+/NEO clones (Figure 1C). Collectively, the LIGIV+/− cells showed an average of 10% (range of 0–20%) gross chromosomal rearrangements/abnormalities, which was almost the same as that of the parental cell and similar to what has been reported previously for the HCT116 cell line [Supplementary Table S1; (29,52)], suggesting that there is no haploinsufficiency for genomic instability, despite the slower cell growth. In contrast, the LIGIV−/− cell line had a 4-fold higher gross chromosomal rearrangement rate of 42.5%, suggesting that LIGIV is a suppressor of genomic instability (Supplementary Table S1 and Supplementary Figure S1C). In addition, 55% of all LIGIV−/− metaphases contained cytologically detectable, non-clonal chromosome or chromatid breaks (Supplementary Figure S1D), which was 5-fold elevated over the parental cell line. From these experiments, we concluded that although LIGIV−/− cells are viable, they are also genetically unstable.
LIGIV−/− cells show an increased use of microhomology during DNA end joining
The A-NHEJ DNA DSB repair pathway is generally only detectable when C-NHEJ is deficient. Indeed, microhomology-mediated end joining (the hallmark repair signature of A-NHEJ) is increased in the 180BR cell line, which was derived from a LIGIV patient (56). To see whether these attributes could be extended to our cell line, we tested the DNA end-joining activity of LIGIV−/− cells using a reporter substrate, pDVG94, that can differentiate C-NHEJ and A-NHEJ products (10,46,57). Digestion of pDVG94 with EcoRV and AfeI restriction enzymes generates a blunt-ended linear substrate that has 6-bp repeats of homologous sequence (ATCAGC) at both ends (Figure 3A, left panel). When this substrate is transfected into mammalian cells, it can be joined by either C-NHEJ or A-NHEJ, but only when it is repaired by A-NHEJ will a novel BstXI restriction recognition site be generated (Figure 3A, left panel). Consequently, this linear substrate was transfected into cells and recovered 48 h later. Repaired junctions were amplified by PCR with radiolabeled primers, and the resulting 180-bp radiolabeled PCR products were digested with BstXI. The amount of 180-bp uncut product represents the repair carried out by C-NHEJ, whereas the 120-bp BstXI-digested product corresponds to repair by A-NHEJ (Figure 3A, right panel). Wild-type cells had only 0.2% A-NHEJ activity (Figure 3B, lane 2), consistent with previous analyses (10). In contrast, >99% of the repair in LIGIV−/− cells was A-NHEJ mediated (Figure 3B, lane 4). Based on these results, we concluded that LIGIV is the major, if not exclusive, C-NHEJ pathway ligase and that in the absence of LIGIV cells use the A-NHEJ pathway of DSB repair.
Figure 3.

LIGIV−/− cells use microhomology for DSB repair. (A) Left Panel. After EcoRV and AfeI restriction enzyme digestion, the reporter substrate, pDVG94, becomes a blunt-ended linear plasmid with 6-bp direct repeats at both ends. Repair of the plasmid via C-NHEJ retains part of both repeats, whereas A-NHEJ generates only a single repeat, which can be subsequently cleaved by BstXI. Right Panel. Repaired plasmids can be isolated and the resulting junctions can then be amplified by PCR using radiolabeled primers to generate a 180-bp PCR product, which can be subjected to BstXI digestion. The ∼180-bp uncut product represents repair via C-NHEJ, whereas the 120-bp digested product represents A-NHEJ-mediated repair. (B) LIGIV−/− cells are virtually devoid of C-NHEJ end-joining activity. The assay described in (A) was carried out, and the products were separated by gel electrophoresis and analysed by autoradiography. The indicated samples are arranged in pairs without (−) and with (+) BstXI digestion. Below each BstXI digested lane is the percentage of A-NHEJ used during repair based on densitometric quantitation of the autoradiogram. In LIGIV−/− cells, virtually the entire repair is mediated by A-NHEJ (lane 4). After complementation (lanes 6, 8, 10, and 12), the ratio between the 180- versus 120-bp products becomes more similar to that of wild-type (lane 2), indicating the restoration of functional C-NHEJ activity in the complemented cells.
Complementation experiments using the re-expression of a wild-type LIGIV cDNA
To confirm that the phenotypes we observed in LIGIV−/− cells were owing to the absence of LIGIV, we attempted to rescue the LIGIV−/− cells using the expression of a LIGIV cDNA. A wild-type LIGIV cDNA was cloned into the pcDNA3.1(+) mammalian expression vector with or without an HA epitope tag. These constructs were transfected into the LIGIV−/− cells, and G418-resistant colonies were screened by immunoblot analysis for the expression of LIGIV. Four clones, two each for each of the cDNAs, were used for further characterization. Clones #14 and #57 expressed untagged wild-type LIGIV, whereas clones #17 and #52 were complemented with the HA epitope-tagged wild-type LIGIV. Clones #14, #52 and #57 showed higher levels of LIGIV expression than clone #17, whose expression was detectable only with an HA antibody (Supplementary Figure S2). Correspondingly, the highest LIGIV-expressing clones (#14, #52 and #57) showed better (albeit not wild-type levels) of complementation for etoposide sensitivity than clone #17; however, all of the clones were significantly more resistance than the null cell line (Figure 2C). In addition, we tested the DNA end-joining activity of the complemented clones. All four clones showed a significant rescue towards the wild-type profile (i.e. reduced A-NHEJ activity; Figure 3B, lanes 6, 8, 10 and 12) in comparison with the null clone (Figure 3B, lane 4). Again, the three highest LIGIV-expressing clones (clones #14, #52 and #57) showed much better levels of complementation (Figure 3B, lanes 6, 8 and 12) than clone #17 (Figure 3B, lane 10). The ability to complement the etoposide sensitivity and repair defects of the LIGIV-null cell line by the re-expression of a LIGIV cDNA strongly suggests that the phenotypes of this cell line are due specifically to the loss-of-function of LIGIV.
The absence of LIGIV has a modest stimulatory effect on the frequency of correct gene targeting
In the course of generating the LIGIV-null cell line, we noted that the recovery of correctly targeted clones during the second round of targeting (when the LIGIV levels had already been reduced to 50%) was 2.0-fold greater than during the first round of targeting (when LIGIV levels were still at 100%): 2.2% targeting in the second round versus 1.1% in the first round. This subtle enhancement implied that the absence of LIGIV (and consequently C-NHEJ) might improve gene-targeting frequency. To extend this observation, a rAAV-mediated gene-targeting experiment was carried out at an independent locus (Ku70) in LIGIV heterozygous cells, but no statistically significant improvement in the relative gene-targeting frequency was observed (Table 1). Nonetheless, we then extended these studies in the LIGIV-null background to three disparate loci: hypoxanthine-guanine phosphoribosyl transferase (HPRT), RAD54B (see also later in the text) and Ku70. Reproducibly, a 2- to 3-fold increase in the relative gene-targeting frequency was observed (Table 1). Thus, the absence of LIGIV does appear to enhance the relative frequency of rAAV-mediated gene targeting, although the effect is modest, and it is substantially weaker than that observed in cells with Ku loss-of-function mutations (30).
Table 1.
Relative gene targeting frequency in LIGIV-deficient cell lines
| Target | HPRT | RAD54B | Ku70 |
|---|---|---|---|
| Cell line | |||
| LIGIV+/+ | 1.0 | 1.0 | 1.0 |
| LIGIV+/− | n.a.* | n.a.* | 1.1 |
| LIGIV−/− | 1.9 | 2.4 | 3.2 |
*n.a. = not available.
Synthetic lethality with RAD54B loss-of-function mutations
RAD54 is an important HR factor with ATP-dependent chromatin remodeling activity (58). Mammals contain two RAD54 genes (RAD54 and RAD54B), neither of which is essential (59). Although loss-of-function mutations in either gene produce relatively mild deficits in the mouse, the combined inactivation of both genes greatly impairs HR (59). In human HCT116 cells, the RAD54B gene has been inactivated by gene targeting, and although the gene is non-essential, its absence alone impairs HR (60). Given that neither LIGIV nor RAD54B are essential genes, we decided that it might be possible to inactivate both genes (and, perforce, C-NHEJ and HR) and construct an ‘A-NHEJ-only’ human cell line. To this end, we disrupted the RAD54B gene in the LIGIV-null cell line. This experiment was complicated by the fact that HCT116 cell line has three copies of the Rad54B gene owing to a duplication on chromosome 10 (60), which necessitated three rounds of gene targeting. Undeterred, we sequentially generated LIGIV−/−:RAD54B+/+/+ > LIGIV−/−:RAD54B+/+/− > LIGIV−/−:RAD54B+/−/− > and finally LIGIV−/−:RAD54B−/−/− cells via rAAV-mediated gene targeting. In the final round of targeting, 11 correct targeting events were recovered (Table 2A). Ten of those events were re-targeting events, but one clone corresponded to the desired LIGIV−/−:RAD54B−/−/− genotype. Unfortunately, however, that clone grew extremely slowly, and within a month, the clone died without ever expanding beyond a single well of a 96-well plate (Table 2). To confirm that the absence of RAD54B by itself was not lethal, we carried out a parallel RAD54B knockout experiment in a Ku86 conditionally null [Ku86flox/−; (24)] background. After the third round of targeting, seven clones were recovered. Two of these corresponded to re-targeting events, whereas the other five clones, all of which grew normally, were the desired Ku86flox/−:RAD54B−/−/− genotype (Table 2B). The Rad54B retargeting frequencies in Ku86flox/−:RAD54B+/−/− and LIGIV−/−:RAD54B+/−/− cells were significantly different (P = 0.013), and confirmed (60) that the absence of RAD54B by itself is not lethal. Moreover, the combined gene-targeting experiments (Table 2) suggested that the dual absence of LIGIV and RAD54B is synthetically lethal. Thus, although HCT116 cells lacking C-NHEJ (LIGIV−/−) or HR (RAD54B−/−/−) are viable, an ‘A-NHEJ only’ cell line is not.
Table 2.
Summary of the Rad54B gene targeting frequency in LIGIV-null and Ku86-conditional null cell lines
| Targeted cell type | G418 positive | Correct targeting | Targeting frequency (%) | Retargeted clone | ||||
|---|---|---|---|---|---|---|---|---|
| LIGIV-null HCT116 | ||||||||
| Rad54B+/+/+ | 34 | 3 | 8.8 | – | ||||
| Rad54B+/+/− | 91 | 12 | 13.2 | 8 | ||||
| Rad54B+/−/− | 232 | 11 | 4.7 | 10* | ||||
| Ku86 conditional-null HCT116 | ||||||||
| Rad54B+/+/+ | 70 | 3 | 4.3 | – | ||||
| Rad54B+/+/− | 229 | 12 | 5.2 | 5 | ||||
| Rad54B+/−/− | 121 | 7 | 5.8 | 2 | ||||
*One LIGIV−/−: RAD54B−/−/− clone died within one month.
Loss of Ku86 induces LIGIV-independent sister chromatid fusions
In a previous study using the Ku86flox/− cell line, we observed that the removal of all Ku86 expression from the HCT116 cell line resulted in rapid telomere loss and the apparent high frequency fusion of sister chromatids (24,25). Where it has been examined, telomere fusions in mammalian cells generally [see, however, (61,62) for reports to the contrary] are mediated by LIGIV. This includes instances where telomeres from one chromosome are fusing to another chromosome (63–65), as well as instances where the sister chromatids are fused (66). Thus, it seemed plausible that the sister chromatid fusions in Ku86-null cells would also be mediated by LIGIV. Therefore, a Ku86flox/−:LIGIV−/− cell line was constructed by rAAV-mediated gene-targeting using the same rAAVLIGIV vector used to generate the single mutant LIGIV−/− cell line. In the first round of targeting, 156 G418-resistant clones were screened by PCR, and one correctly targeted clone was identified for a relative gene-targeting efficiency of 0.6% (1/156). This clone was treated with Cre recombinase to remove the G418-resistance gene and subsequently re-infected to inactivate the second LIGIV allele. In the second round of targeting, 5 of 164 G418-resistant clones were correctly targeted (for a relative gene-targeting efficiency of 3.1%; 5/164), among which three were re-targeted and two were null. One of the Ku86flox/−:LIGIVNEO/− clones was again treated with the Cre recombinase to generate a Ku86flox/−:LIGIV−/− clone.
The Ku86flox/−:LIGIV−/− and, as a control, Ku86flox/− cells were subsequently re-infected with AdCre virus to convert the cells to a Ku86-null state, and 72 h after the infection, the cells were arrested at metaphase and the chromosome spreads were then analysed by FISH using a sub-telomeric probe to chromosome 1q and 4p, which are homologous to sequences located 80 and 73 kb, respectively, centromeric to the telomere. We predicted that if the sister chromatids of Ku86-null cells do fuse, that the discrete, side-by-side two-spots of hybridization (i.e. ‘doublets’; Figure 4C, D and G) expected in a normal metaphase might be replaced by a single spot of hybridization (i.e. ‘singlets’; Figure 4B, E, H and I), owing to the physical proximity of the fused sub-telomeric regions. This is a prediction that has been validated by experimentation in other laboratories (66,67). An increased incidence of singlets was observed in Ku86flox/− cells treated with AdCre (i.e. Ku86−/−; Figure 4E and F). Eighteen metaphases of Ku86−/−:LIGIV−/− cells were then analysed for telomeric fusions at chromosome 1q. Surprisingly, the absence of LIGIV had no difference on the frequency of singlet 1q signals (Figure 4F through I). Overall, in wild-type (i.e. Ku86+/+:LIGIV+/+) cells, 9.2% singlets—denoting a sister chromatid fusion—were detected, and this increased to 17.4% in untreated Ku86flox/− cells (Figure 4F). In contrast, in Ku86−/− cells, sister chromatid fusions were observed 52.2% of the time, which was similar to the 47.2% of singlets observed in Ku86−/−:LIGIV−/− cells (Figure 4F). Thus, sister chromatid fusions in Ku86−/− cells appeared to occur independently of LIGIV (and, consequently, C-NHEJ).
Figure 4.
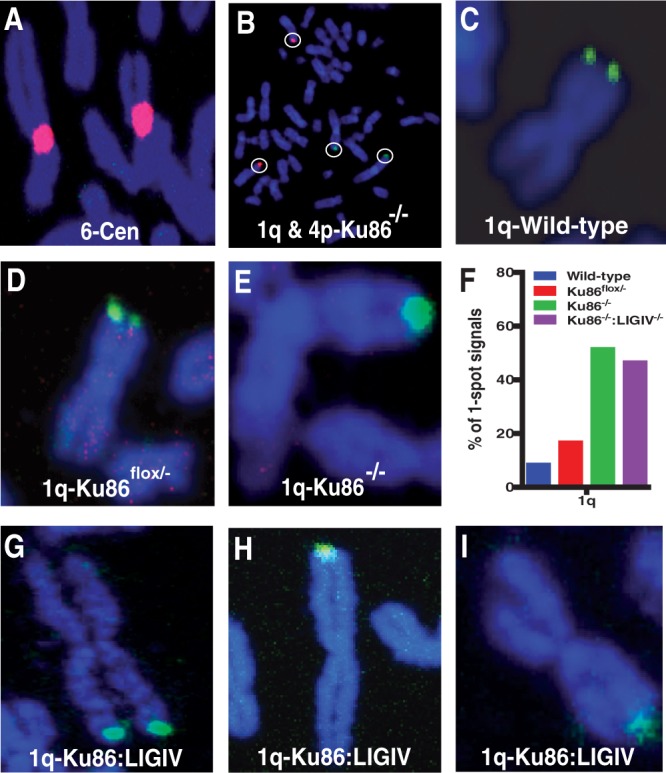
Cytological evidence for LIGIV-independent sister chromatid telomere fusions in human Ku86-null cells. (A) A probe homologous to the centromeric portion of chromosome 6, which served as a hybridization control. (B) Human subtelomeric probes to 1q (green) and 4p (red) in a Ku86-null metaphase cell, where only four signals are seen instead of the expected eight. (E, H and I) Close-ups of metaphase chromosomes stained with the 1q subtelomeric probe, which are suggestive of the sisters being fused. (C, D and G) Close-ups of metaphase chromosomes stained with the 1q sub-telomeric probe, which are suggestive of the sisters not being fused. Images A and D are derived from Ku86flox/− cells. Images B and E are derived from Ku86-null cells. Image C comes from a wild-type (Ku86+/+:LIGIV+/+) cell line, whereas images G through I are derived from Ku86:LIGIV doubly null cells. (F) Quantitation of the frequency of single-spot versus two-spot subtelomeric signals using the 1q probe. The blue bar corresponds to wild type cells; the red bar to Ku86flox/− cells not treated with AdCre; the green bar to Ku86-null cells and the purple bar to Ku86−/−:LIGIV−/−cells. Eighteen to forty-five metaphase images were scored for each data set.
The expression of TRF2ΔBΔM induces LIGIV-dependent chromosome fusions
The lack of an impact owing to the absence of LIGIV on telomeric fusions induced by the loss-of-function of Ku86 was unexpected. Thus, as an additional control, we investigated an aspect of telomere fusions where LIGIV was anticipated to have a larger role. Shelterin, a complex of six core proteins, binds to and protects mammalian telomeres (19,20). TRF2 is a key Shelterin component, and the de Lange laboratory has demonstrated that the expression of TRF2ΔBΔM, a dominant-negative form of TRF2, interferes with telomere binding of endogenous TRF2, resulting in LIGIV-dependent chromosome (telomere) fusions in murine cells (64). To see whether this effect could be observed in human cells, wild-type and LIGIV−/− HCT116 cells were infected with either a control retrovirus (pLPC) or one expressing TRF2ΔBΔM, and 120 h after infection, the cells were arrested at metaphase, hybridized with a telomere-specific probe and analysed by FISH. In either the wild-type (green bars) or wild-type cells infected with the control pLPC retrovirus (red bars), few sister chromatid (Figure 5C) or chromosome fusions (Figure 5D) were observed. In striking contrast, when TRF2ΔBΔM was expressed in wild-type cells, 10% of cells contained sister:sister chromatid fusions and 66.7% of cells had chromosome:chromosome fusions (Figure 5A panels i and ii, 5B, 5C–D, blue bars). Impressively, in TRF2ΔBΔM-expressing LIGIV−/− cells, the frequency of cells carrying chromosome fusions was significantly reduced from 66.7 to 22.5% (Figure 5A panels iii and iv, 5F), and the number of fusions per cell was also greatly reduced (compare Figure 5F with D). In addition, the number of sister:sister chromatid fusions was also reduced in the TRF2ΔBΔM-expressing LIGIV−/− cells. Overall, these results confirmed that the loss of functional TRF2 in human cells stimulates chromosome fusions (64,68) and demonstrates that they are LIGIV dependent.
Figure 5.
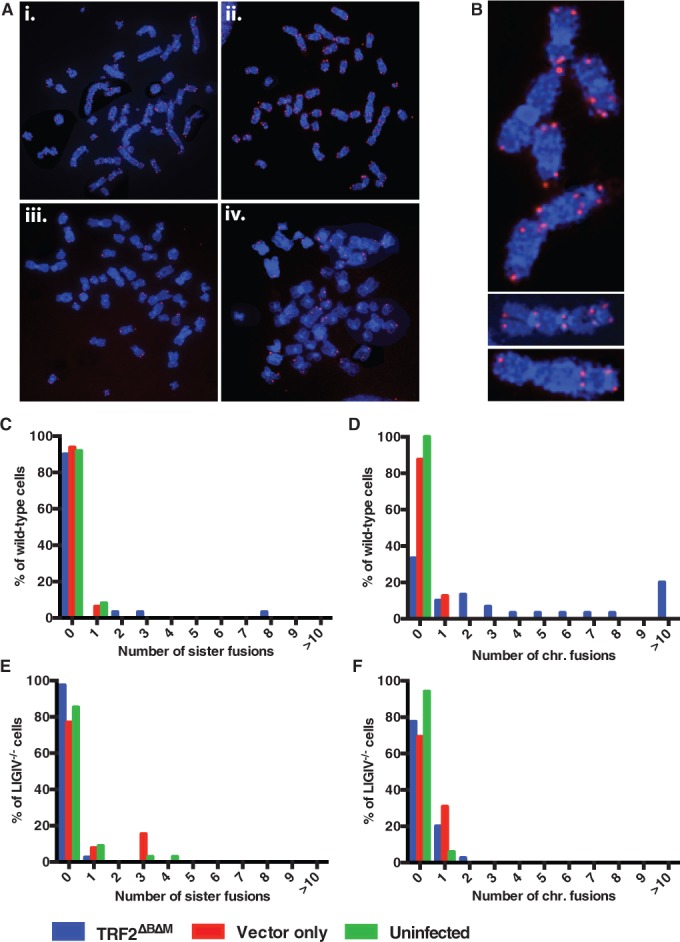
LIGIV-dependent chromosome fusions induced by TRF2ΔBΔM were monitored by FISH analysis with a telomere-specific Cy3-(C3TA2)3 protein–nucleic acid probe. Telomeres are seen as red dots, and metaphase chromosomes are stained blue. (A) i and ii are metaphase chromosome spread examples of wild-type HCT116 cells expressing TRF2ΔBΔM, whereas iii and iv are two independent examples of LIGIV−/− cells expressing TRF2ΔBΔM. (B) From the metaphase spreads (i and ii) shown in (A), a few chromosomes are enlarged to highlight the frequent telomere fusions. (C–F) The number of sister chromatid and chromosome fusions were plotted as a percentage scale. The blue bars represent cells expressing a dominant-negative TRF2 (TRF2ΔBΔM). Red and green bars stand for retroviral vector-only expressing (Vector only) and uninfected (Uninfected) cells, respectively. In (C) and (D), 30, 16 and 27 wild-type metaphase cells were scored for TRF2ΔBΔM, Vector only and Uninfected, respectively. In (E) and (F), 40, 13 and 34 LIGIV−/− metaphase cells were analysed for TRF2ΔBΔM, Vector only and Uninfected, respectively.
DISCUSSION
In this study, we generated a viable LIGIV−/− HCT116 cell line using rAAV-mediated gene-targeting methodology. The successful inactivation of LIGIV in the HCT116 cell line, which is less reliant on HR than the NALM-6 cell line (39), suggests that LIGIV is not essential for the viability of human somatic cells, regardless of the cell type (40,43). With that said, we were initially surprised at the large bias toward retargeting at the already-inactivated locus (15 events) compared with targeting of the wild-type locus (one event), during the construction of the LIGIV-null cell line. Such a disequilibrium in the gene-targeting frequency is usually indicative of genes that provide a significant growth disadvantage when absent (30,52,69), as multiple independent studies using rAAV-mediated gene knockout strategies have demonstrated that there is no allelic preference in gene targeting (70–73). Inconsistent with this view, however, is the fact that the LIGIV-null cell line displays only mild growth defects (Figure 2A). In contrast, when DNA-PKcs was inactivated in the HCT116 cell line not only was a highly skewed second round gene-targeting frequency (1 of 17) obtained but the resulting DNA-PKcs-null cell line showed a severe proliferation defect with a cell doubling time on the order of ∼40 h (52). Lastly, when LIGIV was inactivated in a Ku86flox/− genetic background, no bias in gene targeting was observed (two null clones from five second round events), and these two clones, like the LIGIV-null single mutant cell line, showed near wild-type growth characteristics (data not shown). In summary, we conclude that although cells lacking LIGIV are likely to be at a disadvantage for growth and survival in comparison with LIGIV-proficient cells, LIGIV is non-essential.
As LIGIV is absolutely required for C-NHEJ, it would be logical to extrapolate our results to conclude that C-NHEJ is also non-essential in human somatic cells. Confusing this interpretation, however, is the observation that two other major C-NHEJ components, Ku70 and Ku86, are essential in human somatic cells (30,74). We believe this discrepancy is owing to the fact that LIGIV likely functions exclusively in C-NHEJ, whereas Ku has additional (essential) roles in homeostatic telomere maintenance (24,25). Unlike LIGIV, murine Ku70 and Ku86 are physically associated with telomeres (17,21), and once either Ku subunit is depleted, the telomeres shorten and then fuse (17). In human cells, a Ku86 deficiency causes telomere loss, sister chromatid fusion and t-circle formation, which together eventually induce cell death within a week (24). The lack of these phenotypes in human LIGIV-null cells implies that only Ku, but not LIGIV nor C-NHEJ, is involved in normal telomere maintenance in human cell. Consistent with these observations are the recent results demonstrating that the functional inactivation of the two LIGIV accessory factors, XLF (F. Fattah and E.A. Hendrickson, manuscript in preparation) and XRCC4 (B. Ruis and E. A. Hendrickson, manuscript in preparation) result in viable human cell lines. Thus, the preponderance of data suggests that not only is LIGIV not essential in human somatic cells but that C-NHEJ is non-essential as well.
To date, only seven LIGIV-defective patients have been reported in a world-wide population of 7 billion (36,75,76). This dearth of patients suggests that there is a profound selective disadvantage at the organismal level to having reduced levels of, or being totally without, LIGIV. Four of the LIGIV-defective patients presented with a ‘LIGIV syndrome’, which is associated with chromosomal instability, pancytopenia, developmental and growth delay and dysmorphic facial features (34). Two other patients had leukemia (i.e. cancer predisposition) (35,77), and the last patient presented as a T−B−NK+ radiation sensitive SCID (76). The LIGIV mutations in all of these patients have been characterized at the DNA level. One patient had a mutation in the putative nuclear localization signal so that the protein, which was otherwise presumably functional, was mislocalized in the cytoplasm. Other patients had hypomorphic mutations, which did not completely abolish, but significantly reduced, enzyme function (78), and the severity of the clinical features was correlated with the level of residual LIGIV activity (77). In summary, even though LIGIV is unequivocally dispensable for human somatic cell survival, the absence of even a single true LIGIV-null patient suggests that there is an essential role for LIGIV at the organismal level—presumably somewhere during early development. This stage may correspond to neurogenesis because two of the main clinical features of LIGIV patients are microcephaly and neurological abnormalities (34). This view is consistent with work carried out in mice. LIGIV is essential in the mouse, and the mice succumb early in development not to any obvious defect in C-NHEJ but owing to massive neuronal cell death (79,80). The extraordinary requirement for LIGIV during neural development is not obvious. The brain does, however, occupy only 2% of the total body weight, but consumes 20% of the cellular oxygen (81,82). Perhaps this high oxidative stress causes more DNA DSBs in brain tissue compared with other tissues/organs, which would clearly be detrimental to cells without functional LIGIV. This speculation is at least consistent with the extreme sensitivity to DNA damaging agents (Figure 2B) and the high frequency of spontaneous chromosomal breaks (Supplementary Figure S1) observed in LIGIV-null cells, which may resemble the cells required for early neuronal development subjected to high oxidative stress. In summary, the data are compelling that LIGIV is likely essential for organismal development but dispensable for the survival of single somatic cells.
LIGIV is not required for rAAV random integrations
The process of gene targeting in human somatic cells has been vastly improved to the extent that virtually, any gene modification in any human cell line can be easily carried out in a standard laboratory (42,48). Nonetheless, the frequency of correct gene targeting, where the donor DNA actually finds its cognate homologous chromosomal sequence and replaces it, still hovers around the 1–2% range, relative to all integration events. The corollary of this is that 98–99% of the targeting events correspond to the gene-targeting vector just being randomly inserted into the cell’s genome. The presumption has always been that the correct gene-targeting events are mediated by HR, whereas the random events are owing to some form of end joining (44,48,83). Despite the fact that this presumption is almost certainly true, we demonstrate here that C-NHEJ is not the end-joining pathway used for random rAAV integration events, although it may be more important for the random integration of standard double-stranded targeting vectors (44). Instead, the frequency of correct gene targeting was only modestly improved ∼1.9- to 3.2-fold (Table 1) in the LIGIV-null cell line demonstrating that the absence of LIGIV (and perforce C-NHEJ) had little impact on random rAAV integrations. This observation, by extrapolation, implies that some other form of end joining, most likely A-NHEJ, is responsible for rAAV random integrations. Indeed, in previous work characterizing rAAV-mediated gene targeting in Ku-reduced human cell lines we hypothesized that random rAAV integration events were likely mediated by A-NHEJ rather than C-NHEJ (30); a speculation that is supported strongly by the lack of impact that LIGIV loss-of-function mutations have on gene targeting reported here. Together, these studies suggest that abrogation of A-NHEJ, perhaps via LIGIII (5–7), might be an attractive strategy—and one, it should be added, that has significant clinical applications—to augment correct gene targeting.
Ku, and not LIGIV, is the main regulator of repair pathway choice
The viability of LIGIV-null cells suggests that other DNA DSB pathways (i.e. HR and A-NHEJ) are sufficient to deal with the endogenous DNA DSB damage that spontaneously arises in normal cells. By extension, the synthetic lethality observed with LIGIV (C-NHEJ) and RAD54B (HR) loss-of-function mutations (Table 2) implies that A-NHEJ is not sufficient. Although, this conclusion has not previously been experimentally demonstrated in humans, the result is not conceptually surprising. Thus, A-NHEJ is inherently error prone (yielding deletions) and is thought to be the mechanistic source of chromosomal translocations (84). It seems likely that a human cell trying to survive solely via A-NHEJ would ultimately (and our data would imply much sooner rather than later) mutate itself to death. To unequivocally demonstrate this, a doubly mutant cell line containing a conditionally null allele of either LIGIV or RAD54B would be required, and we are in the process of trying to construct such a cell line so that this hypothesis can be directly addressed.
These observations, however, beg a larger question of how a normal cell that suffers a DNA DSB decides which DSB repair pathway to use to enact repair. This issue is important, as the repaired products that these pathways generate are distinctly different. Numerous recent studies have suggested that in human cells this regulatory activity resides with Ku (10,85,86). C-NHEJ, the major DNA DSB pathway, is usually depicted as a sequential event with the first protein in the reaction mechanism, Ku, essentially acting to commit the DSB to C-NHEJ while simultaneously suppressing the access of HR and A-NHEJ factors to the DSB (10,86). Our work is consistent with that view. Thus, in a recent study from our laboratory, the LIGIV-null cell line was shown to have lost virtually all of its end-joining activity (10). That study and the work presented here (Figure 3) demonstrate that the miniscule residual end-joining activity that remains in LIGIV-null cells corresponds exclusively to A-NHEJ. Thus, in the presence of functional Ku, virtually, all DSB repair is apparently forced down the C-NHEJ pathway where, in LIGIV-null cells, the repair events cannot be consummated. This phenotype of LIGIV-null cells stands in stark contrast to that of Ku-reduced cells, which—while having no detectable C-NHEJ activity—actually have elevated levels of HR (87–89) and A-NHEJ (10,56,90). Nonetheless, it may be possible that LIGIV retains a subtle influence on pathway choice regulation. Mechanistically, this could be owing to a role for LIGIV in some of the upstream steps of C-NHEJ. Thus, LIGIV:XRCC4 has been reported to be required for C-NHEJ initiation by facilitating polymerase and nuclease activity in HeLa cell extract (89,91), and Ku:LIGIV complexes have been implicated in DNA end-bridging reactions (92). Moreover, in yeast, Dnl4 and Lif1, yeast orthologs of LIGIV and XRCC1, are required to stabilize NHEJ complexes and suppress HR by inhibiting resection (89). This later activity may also be the explanation for the observed 1.9- to 3.2-fold increase in rAAV-mediated gene targeting (a HR-dependent reaction) in LIGIV-null cells discussed earlier in the text. A similar ∼2-fold increase was also observed in linearized plasmid-mediated gene targeting in LIGIV-null cells, although in this case, the authors attempted to explain the data by a reduction in random integrations (44). Nonetheless, these enhancements are nowhere near the improved gene-targeting frequencies reported for Ku-reduced human cell lines (30). In summary, our data are consistent with a mechanism where the bulk of the pathway choice regulation resides within the purview of Ku, but that the presence or absence of LIGIV may influence this process.
LIGIV is required for some, but not all, types of chromosome fusions following telomere dysfunction
One of the more interesting features of human LIGIV-deficient cells is that they are proficient for certain types of chromosome fusions following telomere dysfunction, but deficient for others. Thus, loss of functional endogenous TRF2 protein by overexpression of a TRF2ΔBΔM dominant negative construct results in a high frequency of chromosome fusion events that are LIGIV dependent (Figure 5). In contrast, the sister:sister chromatid fusion events that are triggered by the removal of Ku86 are LIGIV independent (Figure 4). Our current model to explain these results proposes that TRF2 and Ku act as non-redundant impediments to chromosome fusion events (and, hence, subsequent genomic instability; Figure 6). Thus, in wild-type cells, TRF2 suppresses the LIGIV-dependent C-NHEJ pathway that often leads to chromosome:chromosome fusions (Figure 6A); an activity of TRF2’s that has been well-documented by numerous laboratories (20,64,93–95). In addition, telomeric Ku suppresses the A-NHEJ (and HR) pathways that we postulate lead to sister:sister chromatid fusions [(10,86); Figure 6A]. Thus, in wild-type cells, all pathways of DNA DSB repair are suppressed at telomeric ends, and any type of chromosome fusion event is unlikely. When endogenous TRF2 is removed from the telomere via TRF2ΔBΔM expression, the suppression on C-NHEJ that TRF2 normally supplies is also removed, and in the presence of functional Ku and LIGIV, a high frequency of chromosome:chromosome fusions ensues (Figure 6B). Interestingly, in the presence of TRF2ΔBΔM, the frequency of sister:sister chromatid fusions was also slightly elevated (Figure 5C). Although, Ku has been shown to be localized to telomeres via its interaction with TRF1 (96) or via direct binding to the telomeric DNA (23), it is also known to interact with TRF2 (97,98), and it may be that in the absence of functional TRF2, that there is less Ku localized at the telomeres to keep A-NHEJ completely inhibited. In contrast, when Ku86 is removed from cells, the C-NHEJ pathway is functionally inactive owing to the lack of Ku, and thus whether it is suppressed by TRF2 (98), is irrelevant (Figure 6C). Hence, most of the telomeric events are mediated by A-NHEJ, which results in a preponderance of sister:sister chromatid fusions. That chromosome:chromosome and chromatid:chromatid fusions are mediated predominately by C-NHEJ (i.e. LIGIV) and A-NHEJ [presumably LIGIII; (6,99)], respectively, is consistent with the empirical observation that C-NHEJ can join ends together in a modestly conservative fashion, whereas A-NHEJ is significantly more deletion prone (6,86). Consistent with these reaction mechanisms, most of the chromosome:chromosome fusions mediated by TRF2ΔBΔM expression result in the retention of telomeric tracts at the site of fusion (100), whereas the chromatid:chromatid fusions induced by the absence of Ku86 often lack telomeric tracts (24). In summary, our data suggest that LIGIV is not required for normal telomere maintenance. However, in the face of certain types of telomere dysfunction, the LIGIV-dependent C-NHEJ pathway is engaged, which leads to chromosome:chromosome fusions and subsequent cell transformation or lethality.
Figure 6.
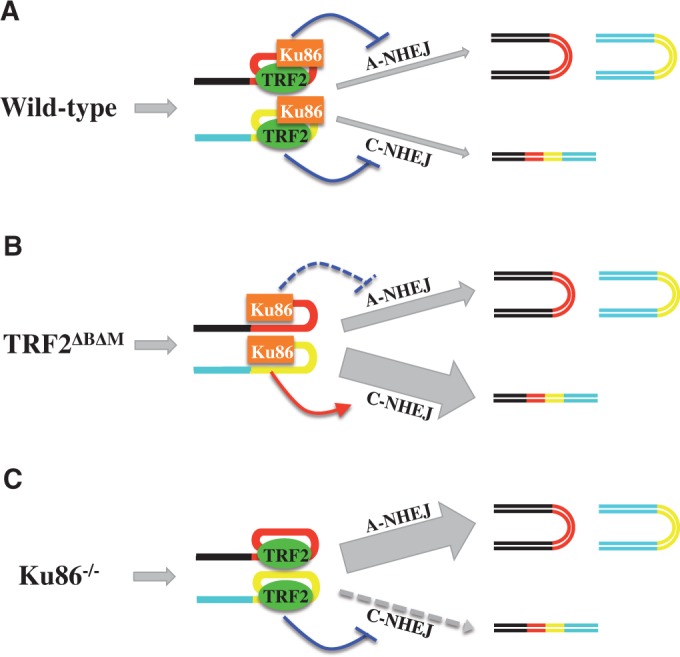
Model for the impact of LIGIV loss-of-function mutations on telomere dysfunction-induced telomere fusions. Chromosomal DNA is cartooned as blue or black lines. Telomeric sequences are shown as yellow or red lines. The impact of TRF2 (green oval) and Ku86 (orange rectangle) on the A-NHEJ and C-NHEJ pathways is indicated either by a purple line with a horizontal hatch (suppression) or red line with an arrowhead (activation). The resultant effect on the indicated pathway is shown by the size of the gray arrows. Chromosome fusions are cartooned as four-colored double horizontal lines, whereas sister:sister chromatid fusions are shown as bicolored U-shaped structures tipped onto their sides. (A) Wild-type cells. TRF2 and Ku86 are both present and suppressing C-NHEJ and A-NHEJ, respectively, and thus few fusions of any type occur. (B) In cells expressing TRF2ΔBΔM, C-NHEJ is greatly and A-NHEJ slightly, activated, respectively, resulting in a large number of chromosome:chromosome fusions and a slight increase in sister:sister chromatid fusions. (C) In cells lacking Ku86, the C-NHEJ pathway is not active, and a large number of sister:sister chromatid fusions accumulate.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1 and 2.
FUNDING
Funding for open access charge: National Institutes of Health [GM088351]; National Cancer Institute [CA154461]; Horizon Discovery, Ltd.
Conflict of interest statement. E.A.H. declares that he is a member of the scientific advisory board of Horizon Discovery, Ltd., a company that specializes in rAAV-mediated gene targeting technology and that his laboratory is funded, in part, through a research contract from the same company.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Drs. Dale Ramsden (University of North Carolina-Chapel Hill) and Titia de Lange (Rockefeller University) for the gifts of the LIGIV and TRF2DN cDNAs, respectively. The authors thank Drs. Anja-Katrin Bielinsky and Alexandra Sobeck (University of Minnesota) for their critical comments on the manuscript.
REFERENCES
- 1.Bennett CB, Lewis AL, Baldwin KK, Resnick MA. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc. Natl Acad. Sci. USA. 1993;90:5613–5617. doi: 10.1073/pnas.90.12.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hakem R. DNA-damage repair; the good, the bad, and the ugly. Embo. J. 2008;27:589–605. doi: 10.1038/emboj.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kass EM, Jasin M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 2010;584:3703–3708. doi: 10.1016/j.febslet.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nussenzweig A, Nussenzweig MC. A backup DNA repair pathway moves to the forefront. Cell. 2007;131:223–225. doi: 10.1016/j.cell.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Iliakis G. Backup pathways of NHEJ in cells of higher eukaryotes: cell cycle dependence. Radiother. Oncol. 2009;92:310–315. doi: 10.1016/j.radonc.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 7.Mladenov E, Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat. Res. 2011;711:61–72. doi: 10.1016/j.mrfmmm.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 8.McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fattah F, Lee EH, Weisensel N, Wang Y, Lichter N, Hendrickson EA. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet. 2010;6:e1000855. doi: 10.1371/journal.pgen.1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tichy ED, Pillai R, Deng L, Liang L, Tischfield J, Schwemberger SJ, Babcock GF, Stambrook PJ. Mouse embryonic stem cells, but not somatic cells, predominantly use homologous recombination to repair double-strand DNA breaks. Stem Cells Dev. 2010;19:1699–1711. doi: 10.1089/scd.2010.0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayora S, Carrasco B, Cardenas PP, Cesar CE, Canas C, Yadav T, Marchisone C, Alonso JC. Double-strand break repair in bacteria: a view from Bacillus subtilis. FEMS Microbiol. Rev. 2011;35:1055–1081. doi: 10.1111/j.1574-6976.2011.00272.x. [DOI] [PubMed] [Google Scholar]
- 13.Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kadyk LC, Hartwell LH. Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics. 1992;132:387–402. doi: 10.1093/genetics/132.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sonoda E, Takata M, Yamashita YM, Morrison C, Takeda S. Homologous DNA recombination in vertebrate cells. Proc. Natl Acad. Sci. USA. 2001;98:8388–8394. doi: 10.1073/pnas.111006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lieber MR, Yu K, Raghavan SC. Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Repair (Amst) 2006;5:1234–1245. doi: 10.1016/j.dnarep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 17.d'Adda di Fagagna F, Hande MP, Tong WM, Roth D, Lansdorp PM, Wang ZQ, Jackson SP. Effects of DNA nonhomologous end-joining factors on telomere length and chromosomal stability in mammalian cells. Curr. Biol. 2001;11:1192–1196. doi: 10.1016/s0960-9822(01)00328-1. [DOI] [PubMed] [Google Scholar]
- 18.de Lange T. T-loops and the origin of telomeres. Nat. Rev. Mol. Cell. Biol. 2004;5:323–329. doi: 10.1038/nrm1359. [DOI] [PubMed] [Google Scholar]
- 19.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 20.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 21.Hsu HL, Gilley D, Blackburn EH, Chen DJ. Ku is associated with the telomere in mammals. Proc. Natl Acad. Sci. USA. 1999;96:12454–12458. doi: 10.1073/pnas.96.22.12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Lange T. How telomeres solve the end-protection problem. Science. 2009;326:948–952. doi: 10.1126/science.1170633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez CR, Ribes-Zamora A, Indiviglio SM, Williams CL, Haricharan S, Bertuch AA. Ku must load directly onto the chromosome end in order to mediate its telomeric functions. PLoS Genet. 2011;7:e1002233. doi: 10.1371/journal.pgen.1002233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Ghosh G, Hendrickson EA. Ku86 represses lethal telomere deletion events in human somatic cells. Proc. Natl Acad. Sci. USA. 2009;106:12430–12435. doi: 10.1073/pnas.0903362106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Indiviglio SM, Bertuch AA. Ku's essential role in keeping telomeres intact. Proc. Natl Acad. Sci. USA. 2009;106:12217–12218. doi: 10.1073/pnas.0906427106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu Y, Seidl KJ, Rathbun GA, Zhu C, Manis JP, van der Stoep N, Davidson L, Cheng HL, Sekiguchi JM, Frank K, et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity. 1997;7:653–665. doi: 10.1016/s1074-7613(00)80386-6. [DOI] [PubMed] [Google Scholar]
- 27.Nussenzweig A, Chen C, da Costa Soares V, Sanchez M, Sokol K, Nussenzweig MC, Li GC. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature. 1996;382:551–555. doi: 10.1038/382551a0. [DOI] [PubMed] [Google Scholar]
- 28.Gu Y, Jin S, Gao Y, Weaver DT, Alt FW. Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proc. Natl Acad. Sci. USA. 1997;94:8076–8081. doi: 10.1073/pnas.94.15.8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myung K, Ghosh G, Fattah FJ, Li G, Kim H, Dutia A, Pak E, Smith S, Hendrickson EA. Regulation of telomere length and suppression of genomic instability in human somatic cells by Ku86. Mol. Cell. Biol. 2004;24:5050–5059. doi: 10.1128/MCB.24.11.5050-5059.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fattah FJ, Lichter NF, Fattah KR, Oh S, Hendrickson EA. Ku70, an essential gene, modulates the frequency of rAAV-mediated gene targeting in human somatic cells. Proc. Natl Acad. Sci. USA. 2008;105:8703–8708. doi: 10.1073/pnas.0712060105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Burg M, van Dongen JJ, van Gent DC. DNA-PKcs deficiency in human: long predicted, finally found. Curr. Opin. Allergy Clin. Immunol. 2009;9:503–509. doi: 10.1097/ACI.0b013e3283327e41. [DOI] [PubMed] [Google Scholar]
- 32.Frank KM, Sekiguchi JM, Seidl KJ, Swat W, Rathbun GA, Cheng HL, Davidson L, Kangaloo L, Alt FW. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 1998;396:173–177. doi: 10.1038/24172. [DOI] [PubMed] [Google Scholar]
- 33.Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- 34.O'Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, Hirsch B, Gennery A, Palmer SE, Seidel J, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol. Cell. 2001;8:1175–1185. doi: 10.1016/s1097-2765(01)00408-7. [DOI] [PubMed] [Google Scholar]
- 35.Riballo E, Critchlow SE, Teo SH, Doherty AJ, Priestley A, Broughton B, Kysela B, Beamish H, Plowman N, Arlett CF, et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr. Biol. 1999;9:699–702. doi: 10.1016/s0960-9822(99)80311-x. [DOI] [PubMed] [Google Scholar]
- 36.O'Driscoll M, Gennery AR, Seidel J, Concannon P, Jeggo PA. An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair (Amst) 2004;3:1227–1235. doi: 10.1016/j.dnarep.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 37.Grawunder U, Zimmer D, Fugmann S, Schwarz K, Lieber MR. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol. Cell. 1998;2:477–484. doi: 10.1016/s1097-2765(00)80147-1. [DOI] [PubMed] [Google Scholar]
- 38.Puebla-Osorio N, Zhu C. DNA damage and repair during lymphoid development: antigen receptor diversity, genomic integrity and lymphomagenesis. Immunol. Res. 2008;41:103–122. doi: 10.1007/s12026-008-8015-3. [DOI] [PubMed] [Google Scholar]
- 39.Adachi N, So S, Iiizumi S, Nomura Y, Murai K, Yamakawa C, Miyagawa K, Koyama H. The human pre-B cell line Nalm-6 is highly proficient in gene targeting by homologous recombination. DNA Cell Biol. 2006;25:19–24. doi: 10.1089/dna.2006.25.19. [DOI] [PubMed] [Google Scholar]
- 40.Iiizumi S, Nomura Y, So S, Uegaki K, Aoki K, Shibahara K, Adachi N, Koyama H. Simple one-week method to construct gene-targeting vectors: application to production of human knockout cell lines. Biotechniques. 2006;41:311–316. doi: 10.2144/000112233. [DOI] [PubMed] [Google Scholar]
- 41.Kohli M, Rago C, Lengauer C, Kinzler KW, Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 2004;32:e3. doi: 10.1093/nar/gnh009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khan IF, Hirata RK, Russell DW. AAV-mediated gene targeting methods for human cells. Nat. Protoc. 2011;6:482–501. doi: 10.1038/nprot.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karanjawala ZE, Grawunder U, Hsieh CL, Lieber MR. The nonhomologous DNA end joining pathway is important for chromosome stability in primary fibroblasts. Curr. Biol. 1999;9:1501–1504. doi: 10.1016/s0960-9822(00)80123-2. [DOI] [PubMed] [Google Scholar]
- 44.Iiizumi S, Kurosawa A, So S, Ishii Y, Chikaraishi Y, Ishii A, Koyama H, Adachi N. Impact of non-homologous end-joining deficiency on random and targeted DNA integration: implications for gene targeting. Nucleic Acids Res. 2008;36:6333–6342. doi: 10.1093/nar/gkn649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He DM, Lee SE, Hendrickson EA. Restoration of X-ray and etoposide resistance, Ku-end binding activity and V(D) J recombination to the Chinese hamster sxi-3 mutant by a hamster Ku86 cDNA. Mutat. Res. 1996;363:43–56. doi: 10.1016/0921-8777(95)00060-7. [DOI] [PubMed] [Google Scholar]
- 46.Verkaik NS, Esveldt-van Lange RE, van Heemst D, Bruggenwirth HT, Hoeijmakers JH, Zdzienicka MZ, van Gent DC. Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur. J. Immunol. 2002;32:701–709. doi: 10.1002/1521-4141(200203)32:3<701::AID-IMMU701>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 47.Karlseder J, Smogorzewska A, de Lange T. Senescence induced by altered telomere state, not telomere loss. Science. 2002;295:2446–2449. doi: 10.1126/science.1069523. [DOI] [PubMed] [Google Scholar]
- 48.Hendrickson EA. Gene targeting in human somatic cells. In: Conn PM, editor. Source Book of Models for Biomedical Research. Totowa, NJ: Humana Press, Inc.; 2008. pp. 509–525. [Google Scholar]
- 49.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 50.Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 51.Masramon L, Ribas M, Cifuentes P, Arribas R, Garcia F, Egozcue J, Peinado MA, Miro R. Cytogenetic characterization of two colon cell lines by using conventional G-banding, comparative genomic hybridization, and whole chromosome painting. Cancer Genet. Cytogenet. 2000;121:17–21. doi: 10.1016/s0165-4608(00)00219-3. [DOI] [PubMed] [Google Scholar]
- 52.Ruis BL, Fattah KR, Hendrickson EA. The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Mol. Cell. Biol. 2008;28:6182–6195. doi: 10.1128/MCB.00355-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pacchierotti F, Ranaldi R. Mechanisms and risk of chemically induced aneuploidy in mammalian germ cells. Curr. Pharm. Des. 2006;12:1489–1504. doi: 10.2174/138161206776389859. [DOI] [PubMed] [Google Scholar]
- 54.Maser RS, DePinho RA. Connecting chromosomes, crisis, and cancer. Science. 2002;297:565–569. doi: 10.1126/science.297.5581.565. [DOI] [PubMed] [Google Scholar]
- 55.Abdel-Rahman WM, Katsura K, Rens W, Gorman PA, Sheer D, Bicknell D, Bodmer WF, Arends MJ, Wyllie AH, Edwards PA. Spectral karyotyping suggests additional subsets of colorectal cancers characterized by pattern of chromosome rearrangement. Proc. Natl Acad. Sci. USA. 2001;98:2538–2543. doi: 10.1073/pnas.041603298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang H, Zeng ZC, Perrault AR, Cheng X, Qin W, Iliakis G. Genetic evidence for the involvement of DNA ligase IV in the DNA-PK-dependent pathway of non-homologous end joining in mammalian cells. Nucleic Acids Res. 2001;29:1653–1660. doi: 10.1093/nar/29.8.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lou Z, Chen BP, Asaithamby A, Minter-Dykhouse K, Chen DJ, Chen J. MDC1 regulates DNA-PK autophosphorylation in response to DNA damage. J. Biol. Chem. 2004;279:46359–46362. doi: 10.1074/jbc.C400375200. [DOI] [PubMed] [Google Scholar]
- 58.Heyer WD, Li X, Rolfsmeier M, Zhang XP. Rad54: the Swiss Army knife of homologous recombination? Nucleic Acids Res. 2006;34:4115–4125. doi: 10.1093/nar/gkl481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wesoly J, Agarwal S, Sigurdsson S, Bussen W, Van Komen S, Qin J, van Steeg H, van Benthem J, Wassenaar E, Baarends WM, et al. Differential contributions of mammalian Rad54 paralogs to recombination, DNA damage repair, and meiosis. Mol. Cell. Biol. 2006;26:976–989. doi: 10.1128/MCB.26.3.976-989.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyagawa K, Tsuruga T, Kinomura A, Usui K, Katsura M, Tashiro S, Mishima H, Tanaka K. A role for RAD54B in homologous recombination in human cells. EMBO J. 2002;21:175–180. doi: 10.1093/emboj/21.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maser RS, Wong KK, Sahin E, Xia H, Naylor M, Hedberg HM, Artandi SE, DePinho RA. DNA-dependent protein kinase catalytic subunit is not required for dysfunctional telomere fusion and checkpoint response in the telomerase-deficient mouse. Mol. Cell. Biol. 2007;27:2253–2265. doi: 10.1128/MCB.01354-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rai R, Zheng H, He H, Luo Y, Multani A, Carpenter PB, Chang S. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. Embo. J. 2010;29:2598–2610. doi: 10.1038/emboj.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Celli GB, Denchi EL, de Lange T. Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat. Cell Biol. 2006;8:885–890. doi: 10.1038/ncb1444. [DOI] [PubMed] [Google Scholar]
- 64.Smogorzewska A, Karlseder J, Holtgreve-Grez H, Jauch A, de Lange T. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr. Biol. 2002;12:1635–1644. doi: 10.1016/s0960-9822(02)01179-x. [DOI] [PubMed] [Google Scholar]
- 65.Williams ES, Klingler R, Ponnaiya B, Hardt T, Schrock E, Lees-Miller SP, Meek K, Ullrich RL, Bailey SM. Telomere dysfunction and DNA-PKcs deficiency: characterization and consequence. Cancer Res. 2009;69:2100–2107. doi: 10.1158/0008-5472.CAN-08-2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsiao SJ, Smith S. Sister telomeres rendered dysfunctional by persistent cohesion are fused by NHEJ. J. Cell Biol. 2009;184:515–526. doi: 10.1083/jcb.200810132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Canudas S, Houghtaling BR, Kim JY, Dynek JN, Chang WG, Smith S. Protein requirements for sister telomere association in human cells. Embo. J. 2007;26:4867–4878. doi: 10.1038/sj.emboj.7601903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 69.Dang LH, Chen F, Ying C, Chun SY, Knock SA, Appelman HD, Dang DT. CDX2 has tumorigenic potential in the human colon cancer cell lines LOVO and SW48. Oncogene. 2006;25:2264–2272. doi: 10.1038/sj.onc.1209247. [DOI] [PubMed] [Google Scholar]
- 70.Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260:85–88. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- 71.Kim JS, Crooks H, Dracheva T, Nishanian TG, Singh B, Jen J, Waldman T. Oncogenic beta-catenin is required for bone morphogenetic protein 4 expression in human cancer cells. Cancer Res. 2002;62:2744–2748. [PubMed] [Google Scholar]
- 72.Chamberlain JR, Schwarze U, Wang PR, Hirata RK, Hankenson KD, Pace JM, Underwood RA, Song KM, Sussman M, Byers PH, et al. Gene targeting in stem cells from individuals with osteogenesis imperfecta. Science. 2004;303:1198–1201. doi: 10.1126/science.1088757. [DOI] [PubMed] [Google Scholar]
- 73.Kim JS, Lee C, Foxworth A, Waldman T. B-Raf is dispensable for K-Ras-mediated oncogenesis in human cancer cells. Cancer Res. 2004;64:1932–1937. doi: 10.1158/0008-5472.can-03-3862. [DOI] [PubMed] [Google Scholar]
- 74.Li G, Nelsen C, Hendrickson EA. Ku86 is essential in human somatic cells. Proc. Natl Acad. Sci. USA. 2002;99:832–837. doi: 10.1073/pnas.022649699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ben-Omran TI, Cerosaletti K, Concannon P, Weitzman S, Nezarati MM. A patient with mutations in DNA Ligase IV: clinical features and overlap with Nijmegen breakage syndrome. Am. J. Med. Genet. A. 2005;137A:283–287. doi: 10.1002/ajmg.a.30869. [DOI] [PubMed] [Google Scholar]
- 76.van der Burg M, van Veelen LR, Verkaik NS, Wiegant WW, Hartwig NG, Barendregt BH, Brugmans L, Raams A, Jaspers NG, Zdzienicka MZ, et al. A new type of radiosensitive T-B-NK+ severe combined immunodeficiency caused by a LIG4 mutation. J. Clin. Invest. 2006;116:137–145. doi: 10.1172/JCI26121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Girard PM, Kysela B, Harer CJ, Doherty AJ, Jeggo PA. Analysis of DNA ligase IV mutations found in LIG4 syndrome patients: the impact of two linked polymorphisms. Hum. Mol. Genet. 2004;13:2369–2376. doi: 10.1093/hmg/ddh274. [DOI] [PubMed] [Google Scholar]
- 78.Chistiakov DA. Ligase IV syndrome. Adv. Exp. Med. Biol. 2010;685:175–185. doi: 10.1007/978-1-4419-6448-9_16. [DOI] [PubMed] [Google Scholar]
- 79.Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr. Biol. 1998;8:1395–1398. doi: 10.1016/s0960-9822(98)00021-9. [DOI] [PubMed] [Google Scholar]
- 80.Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell. 2000;5:993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- 81.Jeppesen DK, Bohr VA, Stevnsner T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 2011;94:166–200. doi: 10.1016/j.pneurobio.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Massaad CA, Klann E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 2011;14:2013–2054. doi: 10.1089/ars.2010.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miller DG, Trobridge GD, Petek LM, Jacobs MA, Kaul R, Russell DW. Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. J. Virol. 2005;79:11434–11442. doi: 10.1128/JVI.79.17.11434-11442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Simsek D, Brunet E, Wong SY, Katyal S, Gao Y, McKinnon PJ, Lou J, Zhang L, Li J, Rebar EJ, et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 2011;7:e1002080. doi: 10.1371/journal.pgen.1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Adachi N, Ishino T, Ishii Y, Takeda S, Koyama H. DNA ligase IV-deficient cells are more resistant to ionizing radiation in the absence of Ku70: implications for DNA double-strand break repair. Proc. Natl Acad. Sci. USA. 2001;98:12109–12113. doi: 10.1073/pnas.201271098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hochegger H, Dejsuphong D, Fukushima T, Morrison C, Sonoda E, Schreiber V, Zhao GY, Saberi A, Masutani M, Adachi N, et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. Embo. J. 2006;25:1305–1314. doi: 10.1038/sj.emboj.7601015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y, Hefferin ML, Chen L, Shim EY, Tseng HM, Kwon Y, Sung P, Lee SE, Tomkinson AE. Role of Dnl4-Lif1 in nonhomologous end-joining repair complex assembly and suppression of homologous recombination. Nat. Struct. Mol. Biol. 2007;14:639–646. doi: 10.1038/nsmb1261. [DOI] [PubMed] [Google Scholar]
- 90.Han L, Yu K. Altered kinetics of nonhomologous end joining and class switch recombination in ligase IV-deficient B cells. J. Exp. Med. 2008;205:2745–2753. doi: 10.1084/jem.20081623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Budman J, Kim SA, Chu G. Processing of DNA for nonhomologous end-joining is controlled by kinase activity and XRCC4/ligase IV. J. Biol. Chem. 2007;282:11950–11959. doi: 10.1074/jbc.M610058200. [DOI] [PubMed] [Google Scholar]
- 92.Grob P, Zhang TT, Hannah R, Yang H, Hefferin ML, Tomkinson AE, Nogales E. Electron microscopy visualization of DNA-protein complexes formed by Ku and DNA ligase IV. DNA Repair (Amst) 2012;11:74–81. doi: 10.1016/j.dnarep.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bae NS, Baumann P. A RAP1/TRF2 complex inhibits nonhomologous end-joining at human telomeric DNA ends. Mol. Cell. 2007;26:323–334. doi: 10.1016/j.molcel.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 94.Mao Z, Seluanov A, Jiang Y, Gorbunova V. TRF2 is required for repair of nontelomeric DNA double-strand breaks by homologous recombination. Proc. Natl Acad. Sci. USA. 2007;104:13068–13073. doi: 10.1073/pnas.0702410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bombarde O, Boby C, Gomez D, Frit P, Giraud-Panis MJ, Gilson E, Salles B, Calsou P. TRF2/RAP1 and DNA-PK mediate a double protection against joining at telomeric ends. Embo. J. 2010;29:1573–1584. doi: 10.1038/emboj.2010.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hsu HL, Gilley D, Galande SA, Hande MP, Allen B, Kim SH, Li GC, Campisi J, Kohwi-Shigematsu T, Chen DJ. Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev. 2000;14:2807–2812. doi: 10.1101/gad.844000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Song K, Jung D, Jung Y, Lee SG, Lee I. Interaction of human Ku70 with TRF2. FEBS Lett. 2000;481: 81–85. doi: 10.1016/s0014-5793(00)01958-x. [DOI] [PubMed] [Google Scholar]
- 98.Fink LS, Lerner CA, Torres PF, Sell C. Ku80 facilitates chromatin binding of the telomere binding protein, TRF2. Cell Cycle. 2010;9:3798–3806. doi: 10.4161/cc.9.18.13129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang H, Rosidi B, Perrault R, Wang M, Zhang L, Windhofer F, Iliakis G. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005;65:4020–4030. doi: 10.1158/0008-5472.CAN-04-3055. [DOI] [PubMed] [Google Scholar]
- 100.Capper R, Britt-Compton B, Tankimanova M, Rowson J, Letsolo B, Man S, Haughton M, Baird DM. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007;21:2495–2508. doi: 10.1101/gad.439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.