


Abstract
Designing effective antisense sequences is a formidable problem. A method for predicting efficacious antisense holds the potential to provide fundamental insight into this biophysical process. More practically, such an understanding increases the chance of successful antisense design as well as saving considerable time, money and labor. The secondary structure of an mRNA molecule is believed to be in a constant state of flux, sampling several different suboptimal states. We hypothesized that particularly volatile regions might provide better accessibility for antisense targeting. A computational framework, GenAVERT was developed to evaluate this hypothesis. GenAVERT used UNAFold and RNAforester to generate and compare the predicted suboptimal structures of mRNA sequences. Subsequent analysis revealed regions that were particularly volatile in terms of intramolecular hydrogen bonding, and thus potentially superior antisense targets due to their high accessibility. Several mRNA sequences with known natural antisense target sites as well as artificial antisense target sites were evaluated. Upon comparison, antisense sequences predicted based upon the volatility hypothesis closely matched those of the naturally occurring antisense, as well as those artificial target sites that provided efficient down-regulation. These results suggest that this strategy may provide a powerful new approach to antisense design.
INTRODUCTION
The ability to manipulate gene expression is one of the most fundamental aspects of biotechnology. It has been accomplished through a variety of methods, including through the use of antisense nucleic acids (DNA and RNA). Since antisense is complementary to a target mRNA, the two strands may hybridize through hydrogen bonding. This double-stranded duplex may hinder ribosomal binding, block ribosomal migration or induce cleavage by an RNase (1,2). In this way, antisense has the potential to be used for numerous applications ranging from metabolic engineering to human gene therapy. Many antisense drugs are in clinical trial for the treatment of a wide variety of diseases, including cancer (3,4).
The process of selecting an antisense sequence that is able to effectively bind to a target mRNA and block protein synthesis is complex and governed by many factors. One of the most important factors is the secondary structure of the target mRNA, which is determined by intramolecular hydrogen bonding that helps to establish a more thermodynamically stable conformation (5). The accepted theory is that this secondary structure would be problematic for antisense-based down-regulation due to the majority of the target mRNA being paired to itself. This intramolecular bonding does not prevent translation because of the ribosome’s ability to unwind mRNA (6), but it greatly decreases accessibility for antisense binding.
There have been many attempts to try and accurately predict the efficacy of antisense sequences to save time, money and labor, all of which are wasted with brute force design and test methods of antisense synthesis. Some approaches involve searching an mRNA sequence for consensus sequences that are present in effective natural and artificial antisense and base their predictions on those motifs (7). Other methods offer the prediction of RNA–RNA interaction mechanisms and may suggest where the target would be in a given mRNA for a specified antisense or small-interfering RNA (siRNA) sequence (8,9). Still other strategies that focus on eukaryotic systems utilize large databases of known species-specific siRNA sequences and predict sequences based on that data. Finally, some methods focus mainly on predicting accessible sites on a target RNA (10,11) or fusing accessibility prediction with hybridization prediction (12–14). There is still much to learn about antisense prediction and the need for more effective strategies remains.
In recent years, the idea that an mRNA strand may not always take the form of a distinct fixed molecular structure has become much more prominent. It is believed that an mRNA molecule may actually be in a state of constant structural fluctuation, transitioning between different conformations near the minimum free energy (MFE) structure, particularly in an ever-changing cellular environment (15–17). Analyzing suboptimal mRNA structures with a thermodynamic stability comparable with that of the MFE structure may reveal that certain regions are more ‘volatile’ than others. Since these regions have the ability to change conformation without significantly altering the Gibbs free energy of the entire molecule, they may have more freedom to alter their hydrogen bonding. Therefore, these regions would likely be the most accessible targets for antisense binding because of their constant formation and breaking of intramolecular hydrogen bonds.
A computational framework, GenAVERT (http://www.rslabs.org), was developed to take advantage of this concept of structural fluctuation to predict the sites on a given strand of mRNA that are most likely to vary in structure within a defined range of free energy. These sites were hypothesized to be superior antisense targets. To test this idea, different types of antisense systems were examined. First, several naturally occurring antisense sequences from prokaryotes were analysed by using GenAVERT. The analysis predicted that the most volatile regions of those mRNAs were essentially the same as those of the natural antisense target sites. Next, genes for which man-made antisense had been designed for down-regulation purposes were analysed. The results of the best antisense compared favorably with GenAVERT predictions, indicating that those antisense exhibiting high levels of down-regulation targeted regions of relatively high volatility.
MATERIALS AND METHODS
Natural and artificial antisense prediction
The ability to predict natural antisense transcripts is a major part of developing a strategy for designing artificial antisense. As noted earlier, many strategies already utilize natural antisense as an indicator of a prediction system’s accuracy (8). Since these antisense expression systems have presumably evolved over millennia, they are thought to result in the most effective inhibitory duplexes possible for a given mRNA. Many of these antisense sequences have been experimentally tested to positively validate their efficiency in the down-regulation or silencing of their corresponding mRNAs.
Perhaps some of the most important and well-studied antisense expression systems are those of toxin–antitoxin systems (18). These systems generally consist of an mRNA that encodes for a ‘suicide protein’ that is extremely toxic to the host cell, as well as a cis-encoded antisense RNA that is transcribed from the same locus but in the opposite direction (as opposed to trans-encoded, where the antisense is encoded at a separate locus). The significance of these toxin–antitoxin systems is in the necessity for efficient inhibition of protein expression to avoid cell death. Translation levels must be brought to an extremely low level or blocked completely for a cell to continue to carry these suicide genes; therefore their corresponding antisense inhibitors must be exceedingly effective. A successful antisense prediction system should be able to predict sequences similar to these RNA antitoxins after analysing their correlating toxin-encoding mRNAs. The volatility hypothesis was evaluated using the following bacterial toxin–antitoxin systems (19): hok/sok (20), pndA/pndB (21), hokC/sokC (22), gef/sof (23), hokA/sokA (22) and ldrA/rdlA (24).
Apart from natural antisense prediction, the prediction of artificial antisense is perhaps just as enlightening. Examining artificial antisense systems that may exhibit a range of gene down-regulation levels provides another platform for measuring the efficacy of identifying volatile regions in mRNA secondary structure. These types of systems represent gene regulation beyond the scope of toxin–antitoxin systems and even those present in various types of bacteria. Three different systems were investigated, with mRNAs of varying lengths, one of which is polycistronic and upwards of 2000 bp. The antisense from these systems demonstrated varying levels of down-regulation and their respective target sites were thus examined for volatility in an attempt to explain the variation in experimental efficiency.
GenAVERT
GenAVERT was developed to test the idea that structurally volatile regions of mRNA made more effective antisense targets. GenAVERT accomplished this objective by generating and comparing suboptimal secondary structures of a given mRNA sequence. Analysis of these comparisons revealed regions that were least ‘similar’ among the set of folds, indicating volatility in intramolecular hydrogen bonding and, according to the proposed hypothesis, accessibility for antisense binding. The program was written in Common Lisp (LispWorks, Cambridge, UK) and calls upon two external programs, UNAFold (http://mfold.rna.albany.edu/) and RNAforester (http://bibiserv.techfak.uni-bielefeld.de/rnaforester/), and a Perl script from the Vienna RNA Package.
GenAVERT functions simply by reading in an mRNA sequence with a given name and then generates a set of potential antisense sequences. Once GenAVERT receives its input, UNAFold is invoked. UNAFold uses the concept of nearest-neighbor thermodynamics to estimate how the bases of an RNA sequence will interact with each other to increase structural stability (25). It outputs the MFE secondary structure for the given mRNA sequence, as well as a set of suboptimal structures with slightly higher Gibbs free energies (26). Since the output from UNAFold is a set of ‘.ct’ files, each of which describes one structure, an external Perl script (Vienna RNA Package) (27) is called to convert each of these .ct files into Vienna bracket format, where periods represent unpaired bases and open and close parentheses represent base pairs. For example, ‘… …((… .))… .(((..)))… .’
Since it is believed, as previously mentioned, that an mRNA molecule is constantly fluctuating in its structure, it is assumed that the range of possible in vivo mRNA structures most likely consists of those that make up the suboptimal range of free energy. If these suboptimal folds are then compared based on structure, they should theoretically point out regions of the mRNA that change more often than others, yet still allow the entire structure’s free energy to remain relatively close to the MFE value. This in turn would indicate that these regions possess superior accessibility and act as likely antisense target sites. To accomplish this goal, the Vienna bracket structures are analysed with the external program, RNAforester.
RNAforester is a comparison program designed for phylogenetic analysis of different RNA molecules (28,29). It takes both sequence and structure into account and compares only two structures at a time. As a result, the basic pairwise input would be two sequences and their corresponding Vienna bracket structures. It then proceeds to generate a homology file showing where the two sequences and structures are similar, using the common method of filling in regions that do not show homology in sequence and structure with ‘gaps.’ In the case of GenAVERT, RNAforester is called to compare the consecutive suboptimal folds that were generated from UNAFold. For example, assume that UNAFold outputs the MFE structure for a specific mRNA sequence as well as three suboptimal folds (1, 2 and 3), for a total of four structural folds. RNAforester would first be called to compare the MFE fold and suboptimal fold 1, then to compare suboptimal fold 1 with suboptimal fold 2, 2 with 3 etc. In this way, there is an artificial sense of transitioning from one structure to another because each file indicates which bases have changed their hydrogen bonding pattern between the two folds (note that all sequences used are that of the given mRNA sequence, while only the structures themselves are changing). Each of the structures is weighted equally. An example of this artificial transitioning is depicted in Figure 1. A more aesthetic method of viewing this transitioning is by simply using the program RNAmovies (15) where the only input required is the mRNA sequence and the set of structures in Vienna bracket format. Whenever GenAVERT is invoked, an RNAmovies input file is created for the user.
Figure 1.
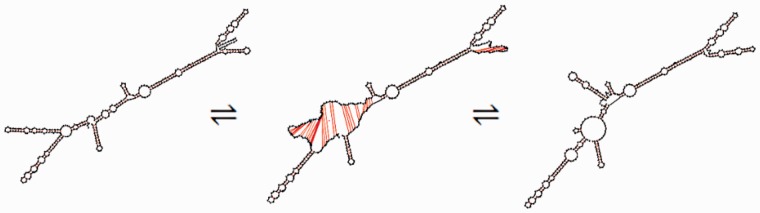
Transition between two possible structures of hok mRNA as predicted by UNAFold and displayed using the interpolating effects of RNAmovies. The large transitioning region (nucleotides 70–150) consists almost entirely of the sok target region (nucleotides 65–131).
GenAVERT then searches all of these RNAforester homology files for any length of mRNA that has changed its conformation (altered hydrogen bonding) between two structures and includes them in a pool of possible volatile regions. It then searches through this pool of possible regions and enumerates the number of times that a certain base shows up. This count indicates the number of times that this base has changed position over the ‘structural transition’ from the MFE fold to the final suboptimal (and least energetically favorable) fold. The number of suboptimal structures is determined by the default settings of the UNAFold window parameter with all kept within 5% of the Gibbs free energy of the MFE structure (26). GenAVERT then uses these values to create a list of every base that meets or exceeds a certain level of volatility. Since this list is not always made of bases that are consecutively located in the mRNA sequence, it may be split into multiple individual volatile regions. The longest of these volatile regions is chosen as the most volatile and therefore the most accessible region on the mRNA strand. The reverse complement of this region is then generated as the antisense sequence that is most likely to down-regulate the expression of the target gene.
However, this first antisense sequence may not always be a practical choice and it may be necessary to continue to collect a set of potential antisense sequences. For example, consider a hypothetical set of five consecutive bases that alter in hydrogen bonding 20 times over the entire structural transition, with no other bases coming close to that level of volatility. As a result, an antisense sequence complementary to those five bases would be predicted as the optimal. However, such a short antisense is unlikely to be viable. As a result, the process is continued for bases that alter their bonding at the next highest level, for example, 15 times, 14 times, 12 times, etc., essentially providing a set of high ranking antisense possibilities of varying length. Therefore, GenAVERT continues to generate the optimal antisense sequence for each level of volatility until no significant difference between bases can be detected. A flowchart of the processes that make up GenAVERT is shown in Figure 2.
Figure 2.

Flowchart depicting GenAVERT algorithm.
Some of the predicted results were considered too small to be viable. We therefore implemented a heuristic criterion to only select sequences that were at least 35 bp in length. Thus, although the program will generate antisense sequences less than 35 bp (see Supplementary Dataset 1), discussion is restricted to those sequence of 35 bp or greater as predicted by GenAVERT.
Comparison with Sfold
As a benchmark, the results from GenAVERT were then compared with that of a currently available program that has a similar goal of predicting inhibitory RNA sequences for prokaryotes. The program used for comparison was the Soligo partition of Sfold (http://sfold.wadsworth.org/cgi-bin/index.pl) (12–14). Sfold is designed to predict antisense sequences with the caveat that the user must declare a pre-determined length for the antisense sequence. Therefore, different values for the antisense length were used with Sfold to present a wider range of the program’s capabilities. Lengths of 35 bp, 50 bp and the exact target length were used. The authors have no knowledge of any other de novo antisense prediction software available at this time and none that requires as little input from the user as that of GenAVERT.
RESULTS
Natural antisense prediction
hok/sok
The hok (host killing) gene is located on plasmid R1 of Escherichia coli and encodes for a protein of 52 amino acids that is toxic to the host cell. A naturally occurring antisense transcript found at the same locus but encoded in the opposite direction, is denoted sok (suppression of killing). The overall function of the hok/sok expression system is for plasmid stabilization. Cells that lose the plasmid carrying the locus are killed due to translation of stable hok mRNA that remains behind in the cell, while the more quickly degraded sok RNA is unable to further inhibit protein synthesis (18,20,22).
When GenAVERT was used to evaluate hok mRNA, the highest ranking antisense sequence greater than 35 bp was the 70 bp antisense sequence shown in Figure 3. This sequence overlapped with the sok target region (sokT) by almost 93% and only had an overhang of 8 bases (Figure 3). All three of the other lengths of antisense predicted by Sfold were designed to target sites near the 3′-end of the mRNA, far from the naturally occurring target site. Table 1 summarizes these results, as well as all of the remaining results.
Figure 3.
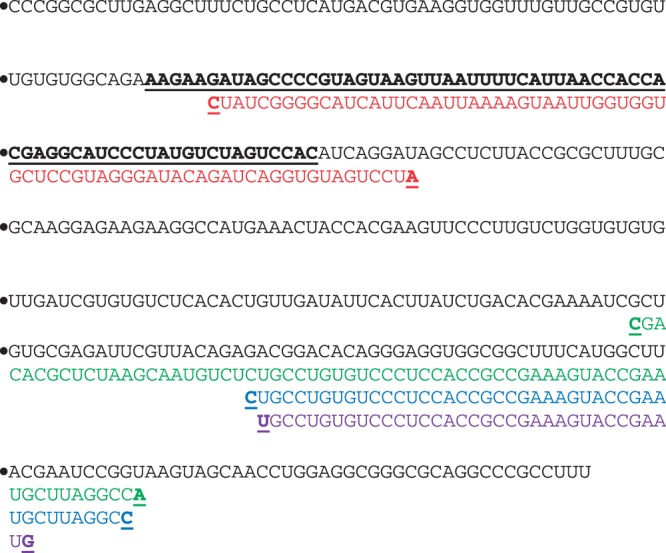
hok mRNA sequence with predicted antisense from GenAVERT (red), Sfold (35 bp) (purple), Sfold (50 bp) (blue) and Sfold (exact target length) (green). The sok target as well as the beginning and end of each antisense sequence are bold and underlined.
Table 1.
Natural antisense prediction summary table. The percent of overlapping base pairs of the naturally occurring target with antisense predicted by GenAVERT and by Sfold (with differing target lengths) are shown
| mRNA | Sfold (35 bp) (%) | Sfold (50 bp) (%) | Sfold (exact target length) (%) | GenAVERT (%) |
|---|---|---|---|---|
| hok | 0 | 0 | 0 | 92.5 |
| pndA | 0 | 14.3 | 14.3 | 87.3 |
| ldrA | 44.8 | 0 | 0 | 92.5 |
| hokC | 47.3 | 60 | 76.4 | 81.8 |
| gef | 52.5 | 0 | 0 | 77 |
| hokA | 40.4 | 32.7 | 75 | 100 |
Note that some sequences are smaller than others and that some may have overhanging base pairs. Also, unlike Sfold, GenAVERT does not require a priori specification of antisense length by the user.
pndA/pndB
The pndA gene is located on plasmid R483 of E. coli. It is, in fact, a hok-homologue and has many of the same functional characteristics of the hok family genes, including a tac (translational activation) sequence, mok (method of killing) reading frame, as well as having a cis-encoded antisense denoted pndB. It also, unsurprisingly, encodes for a toxic protein (21).
Figure 4 displays the various antisense predictions, including antisense generated from GenAVERT that overlapped with ∼87% of the pndB target region (pndBT) with an overhang of 11 bases. The Sfold antisense with the exact target site length, as well as the 50 bp length both overlapped with pndBT by only about 14.3%. The 35 bp antisense from Sfold was designed to target the complete opposite end of the mRNA, clearly illustrating the variability in Sfold predictions, even when antisense length is changed only by 15 bp.
Figure 4.
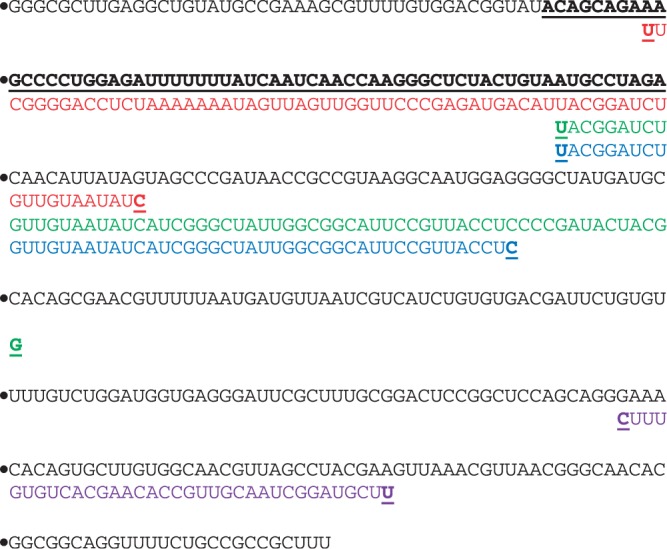
pndA mRNA sequence with predicted antisense from GenAVERT (red), Sfold (35 bp) (purple), Sfold (50 bp) (blue) and Sfold (exact target length) (green). The pndB target as well as the beginning and end of each antisense sequence are bold and underlined.
ldrA/rdlA
The ldrA (long direct repeat A) gene is found on the E. coli K-12 genome (as opposed to being located on a plasmid like hok or pndA) along with its own cis-encoded antisense transcript known as rdlA (regulator detected in ldrA). ldrA is analogous (not homologous) to hok and is part of a different gene family. However, it also encodes for a toxic protein lethal to the host cell, consisting of only 35 amino acids. rdlA was shown to effectively inhibit the translation of ldrA by Kawano et al. (24). Actual sequences were taken from the EcoCyc Database (http://www.ecocyc.org) (30) using the putative transcription start and end sites of the homologous and well-characterized ldrD gene.
GenAVERT predicted an antisense sequence of 95 bp, which overlaps with 92% of the 67 bp rdlA target region (rdlAT). However, it also has an overhang that is not complementary to rdlAT of about 34 bases. Despite this, when GenAVERT’s sequence is compared with those produced by Sfold, only one of the predicted Sfold sequences shared ∼45% overlap with rdlAT, as shown in Supplementary Figure S1. Again, the 50 and 67 bp Sfold antisense strands target the 3′-end, while the 35 bp strand targets the 5′-end, showing the same variability as before.
hokC/sokC
The hokC gene (also homologous to hok) was found to be carried by E. coli ECOR24, and its transcript is believed to be ∼330 nucleotides long. Its cis-encoded antisense was named sokC (22). After using GenAVERT to predict a potential antisense inhibitor for the hokC mRNA sequence, it was able to predict an antisense sequence complementary to 82% of the sokC target (sokCT) sequence with an overhang of 28 bases. Sfold predictions were much more accurate in this case than in other examples with percent overlaps of 47%, 60% and 76% to sokCT for 35 bp, 50 bp, and exact target lengths respectively (Supplementary Figure S2).
gef/sof
The gef (gene expression fatal) gene encodes for a 50 amino acid cell-killing protein and is found on the E. coli K-12 genome. It is essentially the same as hokC, with a cis-encoded antisense almost exactly that of sokC, denoted sof (suppression of fatality). Unlike hokC, however, there is an IS186 insertion sequence located downstream of the coding region, a sequence which is 1338 bp long (31) and is thought to disrupt the usual hokC fbi (fold-back inhibition) sequence and usual mRNA processing. However, despite the presence of this insertion, it has been shown that it is still active on a transcriptional level and is being regulated by sof RNA. The length of the gef mRNA in this case is thought to be about 644 nucleotides long and terminates within the insertion sequence, 200 bp downstream of the stop codon (22,23,32).
GenAVERT predicted an antisense sequence that was complementary to 77% of the sof target region (sofT). However, it also targeted a portion of the mRNA outside of the sofT region, as can be seen in Supplementary Figure S3. Comparing this data with the Sfold predictions revealed only one sequence that was predicted to overlap with 53% of the natural target. The other top scoring antisense sequences for 50 bp and for the exact target length did not overlap the natural target in any way. Again, the variability with small changes in sequence length is exhibited in this case.
hokA/sokA
Like pndA and hokC, hokA is also a hok homologue and displays the same characteristics but was found in E. coli C instead of E. coli K-12 or E. coli ECOR24. Its cis-encoded transcript, sokA regulates the expression of the toxic HokA protein in the usual manner (22).
It can be seen from Supplementary Figure S4 that all of the predicted antisense sequences included at least some complementary bases with the hokA target region (hokAT). GenAVERT’s sequence contained a sequence that is complementary to all of hokAT (100%) with a short overhang of 13 bases. Sfold predicted a 35 bp region that exhibited 40% overlap with the target, whereas the antisense with the exact target length exhibited 75% overlap. However, even with the exact target length (52 bp) antisense showing such a high percent overlap, changing the antisense length by only 2 bp to 50 bp, radically shifted the sequence to display only 33% overlap.
As mentioned previously, Table 1 provides a summary of the results for all of the natural antisense prediction.
Antisense design applications
Regulation of acetoacetate decarboxylase in Clostridium acetobutylicum
The adc gene of C. acetobutylicum ATCC 824 encodes for acetoacetate decarboxylase (AADC) and is a major component of the acetone formation pathway. The adc mRNA is 859 bp long (33,34). Tummala et al. (35) designed plasmids expressing three different antisense strands of varying lengths in an effort to try and alter acetone formation through the down-regulation of AADC. The first antisense RNA shared complementary base pairs with the first 38% of the adc mRNA, the second shared complementary base pairs with the first 68% of the adc mRNA, while the third shared complementary base pairs with 100% of the adc mRNA. The percent down-regulation of AADC was reported to be greater than 80% for all three strains containing the three different antisense plasmids in both transitional and stationary growth phases. The level of AADC expression was found to be too low for quantification of down-regulation in the late exponential phase.
Figure 5 depicts the volatility profile for the adc mRNA based on nucleotide position. The target for the first antisense sequence (38%) is from nucleotides 1 to 328, whereas the target for the second (68%) is from nucleotides 1 to 560. GenAVERT first predicts that the optimal antisense target region is from nucleotides 261 to 322. This 62 bp region is targeted by all three of the expressed antisense RNAs. The second best antisense target as predicted by GenAVERT targets nucleotides 252–488, again, a region with parts encompassed by all three. The next three subsequently scoring target sequences were predicted to be from nucleotides 252–504, 244–504 and 237–504, staying generally in the same volatile region on the mRNA (not depicted). The targeting of all three of the expressed antisense RNAs for this highly volatile region (>80% volatility within a 5% free energy range of the MFE structure) may be an explanation for their ability to induce efficient downregulation.
Figure 5.

Volatility profile for adc mRNA. Target regions are indicated on the plot. The nucleotides complementary to the two top scoring antisense sequences predicted by GenAVERT are represented by numbered horizontal lines. The lower the percent volatility, the more conserved the predicted secondary structure in that region.
Regulation of phosphotransbutyrylase and butyrate kinase in C. acetobutylicum
The ptb and buk genes of C. acetobutylicum ATCC 824 encode for phosphotransbutyrylase (PTB) and butyrate kinase (BK), respectively. They are transcribed from the same operon and therefore the two genes are encoded for on one polycistronic mRNA that is 2128 bp long (36). PTB and BK are both essential parts of the butyrate production pathway and they were targeted in an effort to alter the primary metabolism, specifically, the solventogenesis pathways. Desai and Papoutsakis (37) designed two different plasmids, each expressing antisense RNAs that targeted either the ptb region of the mRNA or the buk region of the mRNA. Strains expressing the ptb antisense resulted in about a 70% decrease in the peak level of PTB compared with the control, with peak levels of BK being ∼80% less than that of the control. Strains expressing the buk antisense resulted in about an 85% decrease in the peak level of BK compared with the control strain, while also showing about a 45% decrease in the peak level of PTB.
Figure 6 illustrates the volatility profile for the ptb-buk mRNA, as well the first eight predicted volatile regions from GenAVERT. The most volatile region is 10 bp and falls within the ptb target region (nucleotides 25–577) but is probably not viable. The region ranked as second is 65 bp and also falls within the ptb target site. The third top scoring volatile region of 267 bp overlapped with the buk target region (nucleotides 973–1018). The following three top scoring regions were all generally from nucleotides 1503–2059, indicating there was perhaps down-regulation potential by targeting sites nearer the 3′-end. Amazingly, volatile region number eight overlapped with almost the entire ptb target region, encompassing nucleotides 5–568. It therefore seems likely that the two target regions on this polycistronic mRNA coincided with two highly volatile areas in the secondary structure, allowing for accessible binding and subsequent down-regulation of these two enzymes. When GenAVERT was restricted to searching for potential target sites in only the first 75% of the mRNA sequence, every single predicted site overlapped with the actual target sites (Supplementary Figure S5).
Figure 6.

Volatility profile for the ptb-buk polycistronic mRNA. The red portion of the profile indicates the ptb target sequence, while the green portion the profile indicates the buk target. The nucleotides complementary to the top 8 scoring antisense sequences predicted by GenAVERT are indicated by numbered horizontal lines. The lower the percent volatility, the more conserved the predicted secondary structure in that region.
Regulation of the σ32 transcription factor in E. coli
The rpoH gene of E. coli encodes for the σ32 transcription factor that is required for the transcription of specific genes. Upregulation of σ32 can be induced under multiple circumstances including ethanol shock, heat shock or the overexpression of recombinant proteins. Srivastava et al. (38) designed a plasmid to express an antisense RNA that would target rpoH mRNA under heat shock, ethanol shock and expression of organophosphorus hydrolase (OPH). However, it has been reported that under various conditions and with multiple strains there may be up to six possible rpoH mRNAs present in vivo due to different promoters under various regulation as noted in the EcoCyc Database (30). It is believed that the rpoHp1 (promoter 1 transcript) mRNA is present under most physiological conditions and is the primary transcript (39).
It was reported by Srivastava et al. (38) that under ethanol shock, control cultures showed a 10-fold increase in σ32 expression, while cultures with induced antisense expression showed only an initial 3-fold increase, which then fell to a 2-fold increase. Likewise, the σ32-regulated GroEL chaperone protein showed a significant decrease in expression during the first hour after ethanol shock when antisense was expressed. However, the GroEL expression levels were comparable in both antisense-expressing and control cultures beyond the 2 hour time point. Under heat shock conditions, GroEL levels dropped 30% in antisense-expressing cultures after the first 5 minutes. Finally, under conditions of OPH expression, OPH levels should theoretically have been higher with antisense expression. However, it was instead observed that OPH was actually higher in the control cultures without antisense expression.
The volatility profile for rpoHp1 is shown in Figure 7 along with the first five predicted volatile regions from GenAVERT. It is clear from this profile, that the most conserved secondary structure of rpoHp1 is encompassed by the designated target site (nucleotides 90–369). GenAVERT ranks the top scoring volatile region as nucleotides 380–439, followed by the second ranked region of nucleotides 317–514. Regions ranked as fourth and fifth also show some overlap with the target region of ∼52 and 73 bp, respectively. Volatility charts for rpoHp2, rpoHp3, rpoHp4, rpoHp5 and rpoHp6 show the same trend, if not more pronounced conserved secondary structure in their respective target regions (Supplementary Figure S6. through Figure S10 for volatility profiles).
Figure 7.

Volatility profile for rpoHp1 mRNA. The red portion of the profile indicates the target site. The nucleotides complementary to the top five scoring antisense sequences predicted by GenAVERT are represented by numbered horizontal lines. The lower the percent volatility, the more conserved the predicted secondary structure in that region.
The level of regulation of σ32 that was detailed earlier using this target region indicates that there was perhaps a greater potential for superior down-regulation if the target had been chosen elsewhere, despite rationale of choosing the location of the Shine-Dalgarno sequence. This may be particularly true because of the presence of multiple mRNAs with very stable secondary structure in the target region. The yield of OPH could have perhaps been higher, while GroEL and σ32 levels could have been much lower under both heat shock and ethanol shock.
DISCUSSION AND CONCLUSION
The ability to determine accessible regions on a strand of mRNA for antisense binding has been the goal of many researchers for decades and continues to be a puzzling problem to this day. The proposed hypothesis that structurally volatile regions of mRNA make the best antisense targets may provide insight into how natural antisense transcripts evolved to be most effective in regulating the expression of their corresponding gene. It is particularly important to analyse cis-encoded antisense systems because they most closely resemble artificial antisense sequences and reveal information about mechanisms through which completely complementary sequences are utilized in nature. This is opposed to the potentially less informative trans-encoded antisense that often have unusual binding mechanisms or even multiple binding sites, many of which are difficult to predict in silico (40).
All of the natural antisense systems described here are toxin–antitoxin systems, where an mRNA encodes for a host-killing protein and a shorter strand of antisense RNA that blocks the translation of this protein-encoding mRNA. This is a particularly interesting aspect of our findings because it is assumed that these antisense have evolved to be particularly effective, minimizing leaky translation so that a cell carrying such a ‘suicide protein’ gene will not only survive but will suffer the least amount of growth inhibition possible. The gef/sof example is of particular interest because even after the insertion sequence disrupted the gene and lengthened the mRNA sequence by more than 300 nucleotides, most of the volatile region was still overlapping with almost all of the sof target sequence. It is surprising that this extreme lengthening did not alter the structural free energy calculations in such a way that another target region might be more volatile. The longer an RNA sequence is, the more options UNAFold has to predict suboptimal structures within the allowed deviance from the minimum free energy. Even if sof did not initially evolve to regulate the significantly longer mRNA, the cells carrying the insertion may still have been able to survive because the volatile region on the mRNA remained relatively intact at the sof target site. As a result, antisense continued to effectively bind to the same region.
The fact that the predicted sequences from GenAVERT most closely matched naturally occurring antisense when sequence length was restricted to 35 bp is very intriguing. In most cases, the antisense sequence length jumped from lengths of about 19, 32 or 34 bp up to lengths of 60, 70 or 90 bp at the next (and best) ranking. This indicates that there may be an underlying fundamental design aspect through evolution where these lengths may provide a greater antisense efficiency by maximizing specificity and thermodynamic hybridization capabilities while still minimizing antisense secondary structure.
In terms of benchmarks, Sfold does not recommend an antisense length and it simply defaults to a length of 20 and searches through the entirety of the given mRNA for accessibility and hybridization potential. The results show that the top scoring antisense sequences are clearly not predicted in the same way as GenAVERT’s, and their predicted target sites may be scattered all over an mRNA sequence. This is evident in all of the natural antisense examples except for that of hokC. GenAVERT was meant to overcome this problem and provide a better direction towards what the best antisense sequence would be without forcing a guess at a ‘preferred sequence length.’
In examining the mRNAs that had been previously targeted with artificial antisense, the volatility profiles provided extensive insight into their potential mechanisms of action. The most volatile regions on adc mRNA and ptb-buk matched up well with the antisense target sites, offering a possible explanation for their high level of down-regulation of the encoded proteins. The predicted antisense from GenAVERT for ptb-buk mRNA was particularly striking when only the first 75% of the mRNA was included as sequence search space. In this case, all of the top eight scoring antisense sequences encompassed or included the majority of the antisense target sites. In addition, after examining the set of rpoH mRNAs, it is apparent that the majority of the artificial antisense target sites on each of these sequences may have possessed a high level of conserved secondary structure and thus lack of volatility. The experimental results did indicate some protein down-regulation, which lines up with the fact that some shorter lengths of the target region exhibited high volatility. However, GenAVERT predicted regions of even higher volatility elsewhere, indicating that there was a much greater potential to inhibit σ32 expression at those locations away from the target region. With the long lengths (800–2000 bp) of these mRNAs, it is apparent that GenAVERT has the potential to have an impact and be applicable in current research.
Since only bacterial examples have been examined, any benefits of using GenAVERT to design antisense sequences for eukaryotic mRNA remains unknown. Many eukaryotic mRNA sequences are extraordinarily long compared with the mRNA sequences of bacteria, and their structures may not, in fact, be accurately predicted by UNAFold. Likewise, making comparisons of sequences and structures that are thousands upon thousands of bases long with RNAforester exponentially increases the program’s runtime. However, the concept of volatility in mRNA secondary structure may still be a useful and applicable approach to blocking eukaryotic gene expression. This could perhaps be accomplished by breaking up long mRNA sequences into smaller overlapping parts and analyzing each individually for potential volatile regions. For the moment, 2500 bp may be a reasonable absolute limit for the sequence length that GenAVERT can handle because of runtime and the potential lack of accuracy in secondary structure prediction in RNAs longer than this.
Also, simply because GenAVERT predicts a certain volatile region on an mRNA sequence, does not mean that this entire region is the absolute optimal antisense binding site. Other factors may also play a significant role in antisense down-regulation. Thus, in the future, it may be helpful to incorporate hybridization algorithms to pinpoint a subsequence within a larger volatile region that would maximize hybridization. It has also been shown that perhaps different antisense sequences are more effective at different in vivo concentrations. As a result, it may be possible to search this volatile region for optimum hybridization while also utilizing equilibrium concentration data to identify the utmost effective antisense sequence. Programs such as Ensemble_Calc (http://mfold.rna.albany.edu/?q=DINAMelt/Ensemble-calc) (41) may aid in this task.
Finally, since each individual base is crucial in the structural thermodynamic calculations and since so many different genes exhibit such intriguing patterns in volatility, it is almost certain that more antisense examples have yet to be found. By leveraging the idea that an mRNA molecule is not static, the hypothesis presented here may provide a new strategy in rational antisense design by predicting which sites on an mRNA strand are truly accessible for antisense targeting.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–10 and Supplementary Dataset 1.
FUNDING
Funding for open access charge: University of Connecticut Summer Undergraduate Research Fund; University of Connecticut Presidential Scholars Enrichment Award.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors express their appreciation for the input and suggestions of the two referees which helped to improve this article.
REFERENCES
- 1.Lee LK, Roth CM. Antisense technology in molecular and cellular bioengineering. Curr. Opin. Biotechnol. 2003;14:505–511. doi: 10.1016/s0958-1669(03)00115-0. [DOI] [PubMed] [Google Scholar]
- 2.Darfeuille F, Unoson C, Vogel J, Wagner EGH. An antisense RNA inhibits translation by competing with standby ribosomes. Mol. Cell. 2007;26:381–392. doi: 10.1016/j.molcel.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 4.Chi KN, Eisenhauer E, Fazli L, Jones EC, Goldenberg SL, Power J, Tu D, Gleave ME. A phase I pharmacokinetic and pharmacodynamic study of OGX-011, a 2′-methoxyethyl antisense oligonucleotide to clusterin, in patients with localized prostate cancer. J. Nat. Cancer Inst. 2005;97:1287–1296. doi: 10.1093/jnci/dji252. [DOI] [PubMed] [Google Scholar]
- 5.Walton SP, Stephanopoulos GN, Yarmush ML, Roth CM. Prediction of antisense oligonucleotide binding affinity to a structured RNA target. Biotechnol. Bioeng. 1999;65:1–9. [PubMed] [Google Scholar]
- 6.Wen J, Lancaster L, Hodges C, Zeri A, Yoshimura SH, Noller HF, Bustamante C, Tinoco I. Following translation by single ribosomes one codon at a time. Nature. 2008;452 doi: 10.1038/nature06716. 10.1038/nature06716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matveeva OV, Tsodikov AD, Giddings M, Freier SM, Wyatt JR, Spiridonov AN, Shabalina SA, Gesteland RF, Atkins JF. Identification of sequence motifs in oligonucleotides whose presence is correlated with antisense activity. Nucleic Acids Res. 2000;28:2862–2865. doi: 10.1093/nar/28.15.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eggenhofer F, Tafer H, Stadler PF, Hofacker IL. RNApredator: fast accessibility-based prediction of sRNA targets. Nucleic Acids Res. 2011;39 doi: 10.1093/nar/gkr467. 10.1093/nar/gkr467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith C, Heyne S, Richter AS, Will S, Backofen R. Freiburg RNA Tools: a web server integrating IntaRNA, ExpaRNA and LocaRNA. Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkq316. 10.1093/nar/gkq316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patzel V, Steidl U, Kronenwett R, Haas R, Sczakiel G. A theoretical approach to select effective antisense oligodeoxyribonucleotides at high statistical probability. Nucleic Acids Res. 1999;27:4328–4334. doi: 10.1093/nar/27.22.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bo X, Wang S. TargetFinder: a software for antisense oligonucleotide target site selection based on MAST and secondary structures of target mRNA. Bioinformatics. 2005;21:1401–1402. doi: 10.1093/bioinformatics/bti211. [DOI] [PubMed] [Google Scholar]
- 12.Ding Y, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res. 2003;31:7280–7301. doi: 10.1093/nar/gkg938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding Y, Chan CY, Lawrence CE. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 2004;32:10. doi: 10.1093/nar/gkh449. 1093/nar/gkh449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding Y, Chan CY, Lawrence CE. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA. 2005;11:1157–1166. doi: 10.1261/rna.2500605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evers D, Giegerich R. RNA movies: visualizing RNA secondary structure spaces. Bioinformatics. 1999;15:32–37. doi: 10.1093/bioinformatics/15.1.32. [DOI] [PubMed] [Google Scholar]
- 16.Giegerich R, Voss B, Rehmsmeier M. Abstract shapes of RNA. Nucleic Acids Res. 2004;32:4843. doi: 10.1093/nar/gkh779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huthoff H, Berkhout B. Two alternating structures of the HIV-1 leader RNA. RNA. 2001;7:143. doi: 10.1017/s1355838201001881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerdes K, Wagner EGH. RNA antitoxins. Curr. Opin. Microbiol. 2007;10:117–124. doi: 10.1016/j.mib.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Gerdes K, Gultayaev AP, Franch T, Pedersen K, Mikkelsen ND. Antisense RNA-regulated programmed cell death. Annu. Rev. Genet. 1997;31:1–31. doi: 10.1146/annurev.genet.31.1.1. [DOI] [PubMed] [Google Scholar]
- 20.Thisted T, Gerdes K. Mechanism of post-segregational killing by the hok/sok system of plasmid R1. J. Mol. Biol. 1992;223:41–54. doi: 10.1016/0022-2836(92)90714-u. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen AK, Gerdes K. Mechanism of post-segregational killing by hok-homologue pnd of plasmid R483: two translational control elements in the pnd mRNA. J. Mol. Biol. 1995;249:270–282. doi: 10.1006/jmbi.1995.0296. [DOI] [PubMed] [Google Scholar]
- 22.Pedersen K, Gerdes K. Multiple hok genes on the chromosome of Escherichia coli . Mol. Microbiol. 1999;32:1090–1102. doi: 10.1046/j.1365-2958.1999.01431.x. [DOI] [PubMed] [Google Scholar]
- 23.Poulsen LK, Refn A, Molin S, Andersson P. The gef gene from Escherichia coli is regulated at the level of translation. Mol. Microbiol. 1991;5:1639–1648. doi: 10.1111/j.1365-2958.1991.tb01911.x. [DOI] [PubMed] [Google Scholar]
- 24.Kawano M, Oshima T, Kasai H, Mori H. Molecular characterization of long direct repeat (LDR) sequences expressing a stable mRNA encoding for a 35-amino-acid cell-killing peptide and a cis-encoded small antisense RNA in Escherichia coli . Mol. Microbiol. 2002;45:333–349. doi: 10.1046/j.1365-2958.2002.03042.x. [DOI] [PubMed] [Google Scholar]
- 25.SantaLucia J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl Acad. Sci. USA. 1998;95:1460. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. In: M. Keith Jonathan., editor. Bioinformatics. Vol. II: Structure, Function and Applications. 2008. Humana Press (a part of Springer Science + Business Media), Totowa, NJ. book doi: 10.1007/978-1-60327-429-6. [DOI] [PubMed] [Google Scholar]
- 27.Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh Chem. 1994;125:167–188. [Google Scholar]
- 28.Hochsmann M. The tree alignment model: algorithms, implementations and applications for the analysis of RNA secondary structures (Doctoral Dissertation). Universitat Bielefeld,Bielefeld. 2005 [Google Scholar]
- 29.Hochsmann M, Voss B, Giegerich R. Pure multiple RNA secondary structure alignments: a progressive profile approach. IEEE Trans. Comput. Biol. Bioinformatics. 2004;1:53–62. doi: 10.1109/TCBB.2004.11. [DOI] [PubMed] [Google Scholar]
- 30.Keseler IM, Collado-Vides J, Santos-Zavaleta A, Peralta-Gil M, Gama-Castro S, Muniz-Rascado L, Bonavides-Martinez C, Paley S, Krummenacker M, Altman T, et al. EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res. 2010;39 doi: 10.1093/nar/gkq1143. 10.1093/nar/gkq1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sengstag C, Iida S, Hiestand-Nauer R, Arber W. Terminal inverted repeats of prokaryotic transposable element IS186 which can generate duplications of variable length at an identical target sequence. Gene. 1986;49:153–156. doi: 10.1016/0378-1119(86)90395-1. [DOI] [PubMed] [Google Scholar]
- 32.Poulsen LK, Larsen NW, Molin S, Andersson P. A family of genes encoding a cell-killing function may be conserved in all Gram-negative bacteria. Mol. Microbiol. 1989;3:1463–1472. doi: 10.1111/j.1365-2958.1989.tb00131.x. [DOI] [PubMed] [Google Scholar]
- 33.Gerischer U, Durre P. mRNA Analysis of the adc Gene Region of Clostridium acetobutylicum during the Shift to Solventogenesis. J. Bacteriol. 1992;174:426–433. doi: 10.1128/jb.174.2.426-433.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peterson DJ. Characterization of the acetone production pathway genes from Clostridium acetobutylicum ATCC 824 (Doctoral Thesis) Rice University. 1991 Houston, TX. [Google Scholar]
- 35.Tummala SB, Welker NE, Papoutsakis ET. Design of antisense RNA constructs for downregulation of the acetone formation pathway of Clostridium acetobutylicum . J. Bacteriol. 2003;185:1923–1934. doi: 10.1128/JB.185.6.1923-1934.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walter KA, Nair RV, Cary JW, Bennett GN, Papoutsakis ET. Sequence and arrangement of two genes of the butyrate-synthesis pathway of Clostridium acetobutylicum ATCC 824. Gene. 1993;134:107–111. doi: 10.1016/0378-1119(93)90182-3. [DOI] [PubMed] [Google Scholar]
- 37.Desai RP, Papoutsakis ET. Antisense RNA strategies for metabolic engineering of Clostridium acetobutylicum . Appl. Environ. Microbiol. 1999;65:936–945. doi: 10.1128/aem.65.3.936-945.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srivastava R, Cha HJ, Peterson MS, Bentley WE. Antisense downregulation of σ32 as a transient metabolic controller in Escherichia coli: effects on yield of active organophosphorus hydrolase. Appl. Environ. Microbiol. 2000;66:4366–4371. doi: 10.1128/aem.66.10.4366-4371.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagai H, Yano R, Erickson JW, Yura T. Transcriptional regulation of the heat shock regulatory gene rpoH in Escherichia coli: involvement of a novel catabolite-sensitive promoter. J. Bacteriol. 1990;172:2710–2715. doi: 10.1128/jb.172.5.2710-2715.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Argaman L, Altuvia S. fhlA repression by OxyS RNA: kissing complex formation at two sites results in a stable antisense-target RNA complex. J. Mol. Biol. 2000;300:1101–1112. doi: 10.1006/jmbi.2000.3942. [DOI] [PubMed] [Google Scholar]
- 41.Ragan C, Zuker M, Ragan M. Quantitative prediction of miRNA-mRNA interaction based on equilibrium concentrations. PLoS Comp. Biol. 2011;7 doi: 10.1371/journal.pcbi.1001090. e1001090.10.1371/journal.pcbi.1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.