

Abstract
Pseudomonas syringae injects numerous bacterial proteins into host plant cells through a type 3 secretion system (T3SS). One of the first such bacterial effectors discovered, HopA1, is a protein that has unknown functions in the host cell but possesses close homologs that trigger the plant hypersensitive response in resistant strains. Like the virulence factors in many bacterial pathogens of animals, HopA1 depends upon a cognate chaperone in order to be effectively translocated by the P. syringae T3SS. Herein, we report the crystal structure of a complex of HopA1(21–102) with its chaperone, ShcA, determined to 1.56-Å resolution. The structure reveals that three key features of the chaperone-effector interactions found in animal pathogens are preserved in the Gram-negative pathogens of plants, namely, (i) the interaction of the chaperone with a nonglobular polypeptide of the effector, (ii) an interaction centered on the so-called β-motif, and (iii) the presence of a conserved hydrophobic patch in the chaperone that recognizes the β-motif. Structure-based mutagenesis and biochemical studies have established that the β-motif is critical for the stability of this complex. Overall, these results show that the β-motif interactions are broadly conserved in bacterial pathogens utilizing T3SSs, spanning an interkingdom host range.
INTRODUCTION
Type 3 secretion systems (T3SSs) are ancient bacterial injection machines that achieve the translocation of host modulatory proteins across three biological membranes, i.e., the inner and outer membranes of Gram-negative pathogens and the host plasma membrane (1–3). The substrates for these systems modulate eukaryotic cellular biology for the benefit of the infecting organism (4–7). T3SSs are found across a wide spectrum of pathogenic bacteria, including pathogens of animals and plants and pathogens in phytosymbiotic relationships (7–11).
Central to the effective engagement and translocation of many T3SS substrates (often called effectors) are a set of cognate “secretion chaperones” (12, 13). These substrates are typically composed of several domains that can be placed in two general categories: (i) a region at the N terminus that harbors secretion-translocation signals that function within the bacterium and (ii) one or more C-terminal domains that contain the host cell effector activities (12–14). The initial 15 to 20 amino acids are required for secretion and are followed by a small 50- to 100-amino-acid subdomain that binds the secretion chaperones and targets the virulence factors to the T3SS (15). X-ray crystallographic studies of chaperone-effector complexes have shown that the chaperones, which show no ATP binding or hydrolytic activity, share a fold and interact through a conserved structural motif (called the β-motif), which is part of an effector polypeptide that engages the chaperone in an extended nonglobular conformation (16–22).
Different strains of phytopathogenic Pseudomonas syringae are capable of causing disease in different plants, and the pathovars Pseudomonas syringae pv. tomato (a pathogen of tomato and Arabidopsis) and Pseudomonas syringae pv. syringae (a pathogen of bean) have been the most well studied. A T3SS is encoded in both of these strains of P. syringae by the hrp (hypersensitive response and pathogenicity) locus (23, 24). This pathogenicity island has a tripartite mosaic structure with a central conserved gene cluster flanked by a unique exchangeable effector locus (EEL) and a conserved effector locus (23). The Hrp locus contains a large repertoire of effector proteins expressed in different P. syringae strains, although only a few are expressed in all strains (25).
In Pseudomonas syringae pv. syringae strain 61, the gene for the effector HopA1 (previously named HopPsyA or HrmA) is located in the EEL. HopA1 from P. syringae pv. syringae strain 61 is translocated into host Arabidopsis strains, in which it acts as an avirulence protein (Avr) by binding to the resistance protein RPS6 (26). This interaction (the recognition of HopA1 by RPS6) leads to a programmed cell death (PCD) response in the plant and is part of the immune response of the plant to the pathogen. The HopA1 protein from Pseudomonas syringae pv. tomato strain DC3000 is less well characterized but was experimentally demonstrated not to induce PCD (26).
The T3SSs of phytopathogenic Gram-negative bacteria have been less thoroughly studied than their animal counterparts. Phytopathogenic T3SSs must interact with eukaryotic cells that are very different from those of animal pathogens. In particular, the plant cell wall has necessitated adaptations in the T3SS to overcome this substantial barrier. Because of these differences and the evolutionary divergence between animal and plant pathogens, many questions remain concerning the nature of the chaperone-effector complex in phytopathogenic bacteria. In order to understand whether the chaperone-substrate interactions observed in animal pathogenic T3SSs were conserved in phytopathogens, we determined the high-resolution crystal structure of the HopA1-ShcA complex from Pseudomonas syringae pv. tomato strain DC3000.
MATERIALS AND METHODS
Protein expression and purification.
The gene for HopA1 was PCR amplified from chromosomal DNA of Pseudomonas syringae pv. tomato (American Type Culture Collection number BAA-871D-5) and was cloned into a modified pGEX-2T vector at the HindIII and NotI restriction sites to produce an N-terminal glutathione S-transferase (GST) fusion followed by a rhinovirus 3C protease cleavage site. Shorter constructs of HopA1, including HopA1(21-102), were subcloned from this construct or directly from genomic DNA. The ShcA chaperone was also PCR amplified from P. syringae chromosomal DNA but cloned with a rhinovirus 3C protease-removable N-terminal 2×His6 fusion (two hexahistidine sequences separated by a 7-amino-acid linker) into the pCDFDuet-1 vector (EMD Chemicals Inc., Gibbstown, NJ). All constructs were verified by DNA sequencing.
The plasmids HopA1/pGEX and ShcA-pCDFDuet-1 were cotransformed into BL21(DE3) cells (Stratagene, La Jolla, CA), and expression was induced at 20°C with 1 mM isopropyl 1-thio-β-d-galactopyranoside (IPTG) for 16 h (full-length wild-type or mutant proteins) or 0.1 mM IPTG for 2 days [HopA1(21-102)]. After the induction period, cells were harvested by centrifugation, the pellet was dissolved in a buffer consisting of 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, 5 mM imidazole (pH 8.0), and 1 mM phenylmethylsulfonyl fluoride (PMSF), and cells were lysed using an Emulsiflex C-5 cell homogenizer (Avestin Inc., Ottawa, Ontario, Canada).
The lysate was centrifuged at 16,000 rpm for 20 min at 4°C, and the supernatant was loaded onto a gravity Ni-nitrilotriacetic acid (NTA) column (Qiagen), washed with the same buffer with 50 mM imidazole, and eluted from the resin with a buffer containing 300 mM imidazole. The eluate was then loaded onto a glutathione-Sepharose column, washed with several column volumes of buffer containing 25 mM Tris (8.0), 500 mM NaCl, and 2 mM dithiothreitol (DTT), eluted with buffer containing 25 mM Tris-HCl, 200 mM NaCl, 2 mM DTT, and 5 mM glutathione, and digested with 3C protease at 4°C for at least 16 h. Full-length HopA1-ShcA complexes were separated from affinity tags by fast protein liquid chromatography with a MonoS ion-exchange column (GE Healthcare) (in the case of the full-length complex) or a MonoQ column [in the case of the HopA1(21-102)-ShcA complex], concentrated by ultrafiltration, and then applied to a Superdex 200 gel filtration column (GE Healthcare). All protein complexes were eluted from the gel filtration column and stored in a final buffer containing 25 mM Tris-HCl, 200 mM NaCl, and 2 mM DTT. Screening of the different crystallization conditions was performed with the HopA1(21-102)-ShcA complex at a concentration of 20 mg/ml in gel filtration buffer.
For selenomethionine (SeMet) labeling of the protein complex, the GST-HopA1(21-102)/pGEX and 2×His6-ShcA/pCDFDuet-1 plasmids were cotransformed into the methionine auxotrophic strain B834 (Novagen), and cells were grown overnight in LB medium. Cells were pelleted, resuspended in minimal medium with methionine replaced with l-(+)-selenomethionine (Acros Organics), and grown at 37°C until the culture reached an optical density at 600 nm (OD600) of 0.6. This culture was transferred to fresh minimal medium with SeMet (12 liters in total), grown at 37°C to an OD600 value of 0.5, and subsequently treated with 1 mM IPTG overnight at 20°C.
Purification of SeMet-containing protein was performed as for the native protein, except that all solutions used for purification contained 10 mM DTT and 2 mM EDTA. Structure-based single and double mutants were constructed by site-directed mutagenesis using complementary mutagenesis primers and subsequent digestion of wild-type DNA by the DpnI nuclease and were purified on a Ni-NTA column as described above for the wild-type constructs.
Limited proteolysis experiments were performed with 5 μg of protein complex, to which were added increasing amounts of subtilisin (from 0 to 5% subtilisin/protein complex ratio [wt/wt]). The reactions were allowed to proceed for 15 min on ice and then were stopped by adding SDS loading buffer and boiling the sample for 5 min at 92°C. N-terminal sequences of proteolysis-resistant fragments were obtained at the Proteomics Resource Center at The Rockefeller University.
Crystallization and structural determination.
Crystals were grown through vapor diffusion, using hanging drops formed by mixing a 1:1 volume ratio of the HopA1(21–102)-ShcA protein complex and equilibration buffer consisting of 3.0 to 3.1 M sodium formate at 23°C. For cryoprotection, crystals were transferred directly into a buffer containing 3.6 M ammonium sulfate and 10% glycerol, flash-cooled in liquid nitrogen, and maintained at −160°C during data collection. Selenomethionine-labeled protein crystals were obtained and cryopreserved in the same fashion.
Data were collected for SeMet-substituted protein crystals at Brookhaven National Synchrotron Light Source beamline X3A, at the selenium absorption edge, and were processed by using HKL2000 (20). The crystals belonged to space group P6522 with the following unit cell parameters: a = b = 67.832 Å and c = 182.887 Å. There was a single heterodimer of ShcA-HopA1 in the asymmetric unit; the canonical secretion chaperone dimer was reproduced along a crystallographic 2-fold axis of symmetry. Phases were determined using SHELX (27), and 90% of the final model was built by using ARP/wARP (28). This build was used to initiate a refinement against the higher-resolution native data set. Cycles of manual building with Coot (29) and refinement with REFMAC5 (30) resulted in a model with R and Rfree values of 20.9% and 22.9%, respectively, to 1.56-Å resolution. The crystallographic statistics are summarized in Table 1.
Table 1.
Crystallographic statistics
| Parameter | Native protein statistica | Selenomethionine-substituted protein statistic |
|---|---|---|
| Data collection | ||
| Space group | P6522 | P6522 |
| Cell dimensions | ||
| a (Å) | 67.83 | 68.14 |
| b (Å) | 67.83 | 68.14 |
| c (Å) | 182.89 | 183.00 |
| α (°) | 90 | 90 |
| β (°) | 90 | 90 |
| γ (°) | 120 | 120 |
| Wavelength (Å) | 1.0809 | 0.97869 |
| Resolution (Å) | 58.74–1.56 | 50.00–1.95 |
| No. of reflections | 2,195,374 | 1,714,379 |
| No. of unique reflections | 36,473 | 34,285 |
| Rmerge(%)b | 2.9 (51.8) | 6.1 (42.3) |
| I/σ(I) | 45.9 (2.1) | 42.3 (5.0) |
| Completeness (%) | 100.0 (100.0) | 98.6 (88.8) |
| Redundancy (%) | 23.4 (23.2) | 18.3 (10.8) |
| Refinement | ||
| Resolution (Å) | 58.74–1.56 | |
| No. of reflections | 34,556 | |
| Rwork (%)c | 20.9 | |
| Rfree (%)c | 22.9 | |
| No. of atoms | ||
| All | 1,687 | |
| Protein | 1,572 | |
| Water | 115 | |
| Average B-factor (Å2) | 40.6 | |
| Protein | 30.4 | |
| Solvent | 37.2 | |
| Root mean square deviation | ||
| Bond lengths (Å) | 0.018 | |
| Bond angles (°) | 1.86 | |
| Ramachandran plot | ||
| Favored regions (%) | 93.6 | |
| Allowed regions (%) | 6.4 | |
| Outliers (%) | 0 |
Mutagenesis and binding assays.
Structure-based point mutants were assayed for binding by following protocols identical to those followed for the purification of wild-type constructs. The soluble fraction (supernatant) of the cell lysate from the coexpression was passed over a Ni-NTA column to isolate the His-tagged chaperone ShcA; GST-tagged HopA1 was present at near-stoichiometric levels when a stable complex was formed. Binding was scored as positive when both the chaperone and effector were present at wild-type levels after elution from the nickel affinity resin and was scored as negative when protein was not present in the eluate (but was present in the whole-cell fraction, indicating expression). Wild-type ShcA was mostly insoluble when expressed alone but was solubilized at high levels through coexpression with HopA1 containing the chaperone-binding domain (CBD). Reversion of the bulk of ShcA to the insoluble fraction in the mutants was interpreted as impairing complex formation and therefore preventing the rescue of the uncomplexed proteins.
The lack of HopA1 pulldown in the β-motif mutants was not due to detrimental effects on the expression or solubility of HopA1. GST-HopA1 β-motif mutants expressed in isolation or coexpressed with ShcA were produced in equivalent amounts and were equivalently soluble with respect to wild-type GST-HopA1 (data not shown).
Protein structure accession number.
The coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under accession number 4G6T.
RESULTS
Overall structure of the complex.
A minimal stable complex of HopA1 and ShcA (31) was identified by an iterative trial-and-error combination of protease footprinting and sequence analysis (Fig. 1A and C). Well-diffracting crystals of HopA1 bound to ShcA were grown (Fig. 1B and C), phases were determined with the selenomethionine-substituted protein using single-wavelength anomalous dispersion (SAD), and the complex structure was refined to 1.56-Å resolution against a native data set (Table 1) (see Materials and Methods). Although limited proteolysis demarcated HopA1 residues 21 to 102, only residues 24 to 94 were visible in the electron density maps.
Fig 1.

Identification of the chaperone-binding domain of HopA1. (A) SDS-PAGE analysis (with Coomassie blue staining) of limited proteolytic digestion of the HopA1-ShcA complex. Lane St, protein standard markers for assigning molecular weight; lane 8, undigested purified complex of full-length proteins. For lanes 1 to 7, increasing amounts of protease were added to the reaction mixture (subtilisin/protein [wt/wt], for lanes 1 to 7: 0.0001, 0.0025, 0.05, 0.1, 1, 5, and 10%, respectively). (B) Well-diffracting crystals of the HopA1(21-102)-ShcA complex. (C) Domain delineation of the effector HopA1. SS, secretion signal. (D) Final refined (2Fo − Fc) model-phased electron density maps contoured at 1σ (gray).
The overall structure confirms that the nature of the protein-protein interactions between virulence factor T3SS substrates and their cognate chaperones that are observed in animal pathogens is conserved in the interactions of their counterparts from plant pathogens. To begin, the chaperone-binding domain (CBD) of HopA1 adopts a nonglobular structure that wraps itself around the ShcA chaperone (Fig. 2), and the chaperone itself possesses a highly conserved fold and similar dimeric assembly (Fig. 2B and C and 3). The chaperone-effector interaction (see below for details) centers on the formation of an intermolecular β-sheet that inserts a 3-amino-acid “plug” (the so-called β-motif) into a large hydrophobic patch in the chaperone (Fig. 4 and 5). In addition, as in numerous chaperone-effector complexes from animal pathogens, a second large hydrophobic patch produced by a concave face of the β-sheet of the chaperone partially encircles an α-helix of the effector (Fig. 2 and 4).
Fig 2.

Overall structure. (A) Overall fold shown as a cartoon diagram in two orientations related by a 90° rotation about the vertical axis. (B) Topology diagrams of the HopA1 and ShcA polypeptides in the structure, generated with PDBSum (31). (C) Secondary structure and sequence of the HopA1 and ShcA polypeptides in the structure. Blue H, helices; red β, strands; pink β, β turns; pink γ, γ turns; red ⊃, β hairpins.
Fig 3.
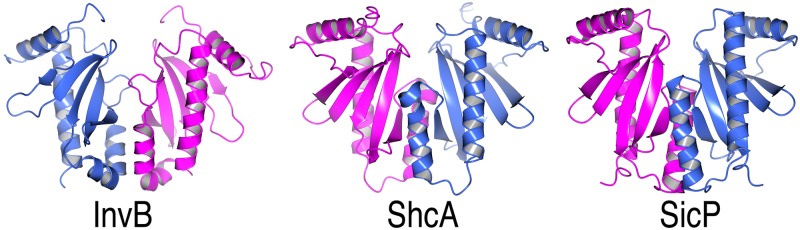
ShcA crystallographic dimer. Shown are ribbon diagrams of the SicP and InvB effector chaperones of Salmonella (PDB accession numbers 1JYO and 2FM8, respectively) and the reconstructed crystallographic ShcA dimer. Each monomer in the dimers is colored either blue or pink.
Fig 4.

HopA1-ShcA interactions. (A) Hydrophobic patches in ShcA (yellow) that bind the HopA1 polypeptide (shown as a ribbon cartoon in salmon); (B) residues making contact between the S2 strand (β-motif) of HopA1 (salmon and orange) and ShcA (purple and blue); (C) contacts between the HopA1 H2 helix and the ShcA helix-binding groove.
Fig 5.

HopA1 possesses a β-motif analogous to virulence factors of animal pathogens. (A) Comparison of T3SS effector nonglobular polypeptides from several complexes with chaperones (alignment was generated by aligning the complexes based on the chaperone-conserved folds). The effector polypeptides include Salmonella SipA (PDB accession number 2FM8) (red) and SptP (PDB accession number 1JYO) (purple), Yersinia YopN (PDB accession number 1XKP) (yellow) and YopE (PDB accession number 1L2W) (cyan), and Pseudomonas syringae HopA1 (green). A surface rendering of ShcA, with the hydrophobic patches shown in yellow, is presented to orient the image. (B) Alignment of several β-motifs from animal and plant effectors. (C) Alignment of β-motif binding pockets in chaperones from animal and plant pathogens. Left, three key elements of the structure in all chaperones characterized to date, consisting of six highly conserved hydrophobic amino acids which closely superimposed and frequently make contacts with β-motif residues. Right, space-filling representation of ShcA, with the six conserved hydrophobic residues of the chaperone pocket shown in yellow and indicated by arrows. The main chain trace of HopA1 is drawn as a tube in salmon, and the β-motif is indicated.
Truncation of the effector HopA1 (which was required to obtain crystals) allowed for the same N-terminal portion of the virulence factor to bind to the hydrophobic patch in each monomer of the chaperone dimer, as seen previously in the structure of SycH-YscM2 (19). This is in contrast to most chaperone-effector complexes in which two different regions of the effector insert into these patches. The truncations in HopA1 that were required to obtain crystals may have removed a second binding site in the effector CBD. However, the standard dimer interface that is observed in numerous secretion chaperones of this family was present in the crystal along a crystallographic 2-fold axis of symmetry (Fig. 3). This phenomenon was also observed in crystals of the Yersinia chaperone SycH with both YscM2 (19) and YopH (PDB accession number 4GF3), which suggests that the chaperone dimer is not a product of crystal packing.
Structural details of the interaction.
Due to the nonglobular nature of the interaction, the interface of ShcA with HopA1(24–94) is extensive and significantly hydrophobic, burying approximately 2,000 Å2 of surface area. For comparison, the dimerization interface of the chaperone dimer buries approximately 1,200 Å2 of surface area. The interactions between the chaperone and the effector are centered on two primary contact regions.
The first contact region involves the formation of a seven-stranded intermolecular β-sheet between the proteins. The edge of the five-stranded β-sheet of ShcA is extended by two strands; the S2 polypeptide from HopA1 stacks directly with the sheet, interacting with S1 of ShcA. Both strands of HopA1 insert residues into a large predominantly hydrophobic pocket of ShcA (Fig. 4A). Three residues of HopA1 S1, i.e., Gln29, Leu30, and Val32, make significant contacts with ShcA, primarily using hydrophobic and van der Waals contacts to interact with hydrophobic residues in the effector binding patch of the chaperone, such as ShcA residues Phe13, Leu17, the aliphatic portions of K109 and Arg110, and Ala 113 (Fig. 4B).
Of significant interest is the S2 strand, which contains the 3-amino-acid β-motif, like its counterparts in the effectors of animal pathogens, and is situated in a very similar fashion with respect to the interactions seen in chaperone-effector complexes of animal pathogens. The HopA1 β-motif interactions are analyzed more below; in brief, they involve the motif residues Tyr37, Ile39, and Ala41 of HopA1, which insert into the ShcA hydrophobic patch, contacting residues Leu17, Met19, Leu22, Met32, Ile33, Ile34, Met112, Ala113, Leu116, and Arg120 of the chaperone (Fig. 4C).
The second critical contact between these proteins occurs on an adjacent face of the chaperone. As the polypeptide of HopA1 winds around ShcA, it inserts a helix into the “helix-binding groove” of the chaperone. This concave groove, formed by the β-sheet of ShcA, exposes a significant hydrophobic patch, and these residues are sequestered from solvent by the helix of the effector. This interaction is centered on the insertion of two bulky hydrophobic residues from the H2 helix of HopA1 (Phe61 and Phe62) and is augmented by numerous other contacts, with the residues Gln54 and Lys57 contributing most significantly (Fig. 4C).
The β-motif is conserved in phytopathogenic T3SSs.
In all of the chaperone-effector interactions characterized through structural biology, there is an analogue to the intermolecular β-sheet formation observed in the HopA1-ShcA complex. In each of those interactions, a strand from the effector in the position of the HopA1 S2 strand extends the chaperone β-sheet and inserts three residues (predominantly hydrophobic) into a hydrophobic patch in the chaperone (Fig. 5A). This so-called β-motif, first recognized in the SipA-InvB effector-chaperone complex of Salmonella (16), also is present in the plant effector HopA1, indicating that this structural motif is conserved from animal to plant pathogens. The residues Y37, I39, and A41 constitute the HopA1 β-motif, insert into a ShcA hydrophobic pocket, and also align well with the motif residues in animal pathogen effectors (Fig. 5B).
Further extending the similarities to the chaperone-effector complexes in animals is the conserved nature of the ShcA hydrophobic pocket that binds the β-motif (Fig. 5C). Six positions harboring hydrophobic residues in animal pathogen chaperones that contribute significantly to the pocket also possess counterparts in positions very similar to those in the ShcA chaperone (Fig. 5C), indicating that the corresponding “receptor” for the β-motif “ligand” is also highly conserved in plant-pathogenic bacteria.
Structure-based mutagenesis.
These similarities suggest that the β-motif in HopA1 would be critical to the chaperone-effector association, in analogy to the complexes of animal pathogens. Therefore, a series of structure-based mutagenesis experiments was performed to ascertain the importance of the β-motif interactions with ShcA. Key β-motif residues of HopA1 (or other nearby residues that also make contacts with ShcA) were mutated to glycine, and the constructs were coexpressed in a manner identical to that of the wild-type constructs and assayed in an affinity pulldown experiment (in the manner in which the wild-type complex was isolated, with a dodecahistidine tag on ShcA). Table 2 summarizes the results.
Table 2.
Structure-based mutagenesis of HopA1
Complex formation was assayed as described in Materials and Methods on the basis of recombinant bacterial coexpression and nickel affinity pull-down of untagged full-length GST-tagged HopA1 with dodecahistidine-tagged ShcA chaperone. This purification scheme was used to isolate milligram quantities of the complex used in crystallization studies. +, robust stable complex formation in affinity pull-down and size exclusion chromatography analyses; +/−, a significantly destabilized complex in all assays; −, a stable complex could not be isolated.
Residue in the β-motif.
In brief, β-motif mutations, even single loss-of-contact point mutations in the context of a surface area of protein-protein interactions of several thousand square angstroms, were disruptive enough to significantly reduce the pulldown of the chaperone-effector complex. Double mutations in the β-motif abolished binding. In contrast, mutations of nearby residues that were involved in contacting ShcA but not part of the β-motif (e.g., Leu30 and Val32) did not disrupt the complex, even in the double loss-of-contact L30G V32G mutant. These data strongly support the hypothesis that the chaperone-effector complex of ShcA relies heavily on the β-motif interaction.
DISCUSSION
T3SSs are found across pathogens of animals and plants and those in phytosymbiotic relationships, conferring on bacteria the ability to deliver bacterial proteins into eukaryotic cells. An interaction that is often important for the proper translocation of effectors from the bacterium to a host cell is that between the secretion chaperone and the secretion substrate, i.e., the so-called chaperone-effector interaction. A flurry of activity initially characterized several such complexes from bacterial pathogens of animals, and the structures of plant secretion chaperones indicated that they were very similar to those found in animals (18). However, there has been no characterization of the complex of a plant effector and its cognate chaperone. Because the cell walls of plants have necessitated divergent adaptations for the T3SSs of phytopathogenic Gram-negative bacteria, it has been unclear whether the chaperone-effector complex in those organisms is conserved.
The ShcA protein of P. syringae strain 61 was the first phytopathogenic T3SS secretion chaperone discovered (32). Here, we report the structure of its homolog from Pseudomonas syringae pv. tomato strain DC3000 in a complex with the N terminus of the effector HopA1, which is, to our knowledge, the first structure of a phytopathogenic chaperone-effector complex reported.
The X-ray crystal structure of HopA1-ShcA reveals that, at least for some chaperone-effector complexes of phytopathogens, there is significant conservation in the interactions, compared with those of animal-pathogenic bacteria. In particular, the structural β-motif present in all known complexes is also present in the HopA1-ShcA interaction. The three key aspects of the β-motif found in animal pathogens are present also in HopA1: (i) the β-motif strand extends the β-sheet of the chaperone, (ii) three predominantly hydrophobic residues, located in conserved positions in three-dimensional space, interact with a hydrophobic patch in the chaperone, and (iii) the hydrophobic patch of the chaperone is located in a conserved structural location and is composed largely of six hydrophobic residues that align well three-dimensionally with counterparts in the chaperones of bacteria that infect animals. Mutagenesis studies echo those with animal effectors; loss-of-contact mutations in the β-motif prove disruptive to the formation and stability of the chaperone-effector complex.
The essential role of the N terminus of HopA1 (residues 21 to 102) in binding to ShcA was also observed in experiments with homologs from P. syringae pv. syringae strain 61. Those experiments showed that the 166 N-terminal amino acids of the effector strongly interacted with ShcA, in contrast to a C-terminal construct (residue 120 to the end) that failed to bind to the chaperone (32). The high degree of sequence conservation in the N-terminal portion of homologous HopA1 proteins from different strains, as well as the central role of this region in binding to the secretion chaperone, supports the hypothesis that HopA1 from P. syringae pv. syringae strain DC3000, like Avr HopA1 from P. syringae pv. syringae strain 61, is translocated into plant cells. This also suggests that the middle portion of the protein, which is the least conserved region of the avirulent and virulent HopA1 variants, may be an important determinant in interactions with the resistance protein RPS6.
These results provide evidence that the β-motif is an ancient binding signal in T3SSs, present prior to the divergence of the pathogenic bacteria that infect animals and plants. The strongly conserved nature of this signal suggests that it is likely present in the majority of chaperone-effector complexes and thereby might present an attractive target for broad-spectrum antibacterial drug development.
ACKNOWLEDGMENTS
We thank D. Oren of Rockefeller University and W. Shi of Brookhaven National Synchrotron Light Source beamlines X3A and X29 for access to and assistance with crystallographic equipment.
This work was funded in part by NIH grant R01AI052182 (to C.E.S.).
Footnotes
Published ahead of print 30 November 2012
REFERENCES
- 1. Galan JE, Wolf-Watz H. 2006. Protein delivery into eukaryotic cells by type III secretion machines. Nature 444:567–573 [DOI] [PubMed] [Google Scholar]
- 2. Cornelis GR. 2006. The type III secretion injectisome. Nat. Rev. Microbiol. 4:811–825 [DOI] [PubMed] [Google Scholar]
- 3. Horn M, Collingro A, Schmitz-Esser S, Beier CL, Purkhold U, Fartmann B, Brandt P, Nyakatura GJ, Droege M, Frishman D, Rattei T, Mewes HW, Wagner M. 2004. Illuminating the evolutionary history of chlamydiae. Science 304:728–730 [DOI] [PubMed] [Google Scholar]
- 4. Stebbins CE, Galan JE. 2001. Structural mimicry in bacterial virulence. Nature 412:701–705 [DOI] [PubMed] [Google Scholar]
- 5. Barbieri JT, Riese MJ, Aktories K. 2002. Bacterial toxins that modify the actin cytoskeleton. Annu. Rev. Cell Dev. Biol. 18:315–344 [DOI] [PubMed] [Google Scholar]
- 6. Stebbins CE. 2004. Structural insights into bacterial modulation of the host cytoskeleton. Curr. Opin. Struct. Biol. 14:731–740 [DOI] [PubMed] [Google Scholar]
- 7. Galan JE. 2009. Common themes in the design and function of bacterial effectors. Cell Host Microbe 5:571–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cunnac S, Lindeberg M, Collmer A. 2009. Pseudomonas syringae type III secretion system effectors: repertoires in search of functions. Curr. Opin. Microbiol. 12:53–60 [DOI] [PubMed] [Google Scholar]
- 9. Parsot C. 2009. Shigella type III secretion effectors: how, where, when, for what purposes? Curr. Opin. Microbiol. 12:110–116 [DOI] [PubMed] [Google Scholar]
- 10. McGhie EJ, Brawn LC, Hume PJ, Humphreys D, Koronakis V. 2009. Salmonella takes control: effector-driven manipulation of the host. Curr. Opin. Microbiol. 12:117–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Poueymiro M, Genin S. 2009. Secreted proteins from Ralstonia solanacearum: a hundred tricks to kill a plant. Curr. Opin. Microbiol. 12:44–52 [DOI] [PubMed] [Google Scholar]
- 12. Stebbins CE, Galan JE. 2003. Priming virulence factors for delivery into the host. Nat. Rev. Mol. Cell Biol. 4:738–743 [DOI] [PubMed] [Google Scholar]
- 13. Journet L, Hughes KT, Cornelis GR. 2005. Type III secretion: a secretory pathway serving both motility and virulence. Mol. Membr. Biol. 22:41–50 [DOI] [PubMed] [Google Scholar]
- 14. Parsot C, Hamiaux C, Page AL. 2003. The various and varying roles of specific chaperones in type III secretion systems. Curr. Opin. Microbiol. 6:7–14 [DOI] [PubMed] [Google Scholar]
- 15. Lee SH, Galan JE. 2004. Salmonella type III secretion-associated chaperones confer secretion-pathway specificity. Mol. Microbiol. 51:483–495 [DOI] [PubMed] [Google Scholar]
- 16. Lilic M, Vujanac M, Stebbins CE. 2006. A common structural motif in the binding of virulence factors to bacterial secretion chaperones. Mol. Cell 21:653–664 [DOI] [PubMed] [Google Scholar]
- 17. Schubot FD, Jackson MW, Penrose KJ, Cherry S, Tropea JE, Plano GV, Waugh DS. 2005. Three-dimensional structure of a macromolecular assembly that regulates type III secretion in Yersinia pestis. J. Mol. Biol. 346:1147–1161 [DOI] [PubMed] [Google Scholar]
- 18. Singer AU, Desveaux D, Betts L, Chang JH, Nimchuk Z, Grant SR, Dangl JL, Sondek J. 2004. Crystal structures of the type III effector protein AvrPphF and its chaperone reveal residues required for plant pathogenesis. Structure 12:1669–1681 [DOI] [PubMed] [Google Scholar]
- 19. Phan J, Tropea JE, Waugh DS. 2004. Structure of the Yersinia pestis type III secretion chaperone SycH in complex with a stable fragment of YscM2. Acta Crystallogr. D Biol. Crystallogr. 60:1591–1599 [DOI] [PubMed] [Google Scholar]
- 20. Evdokimov AG, Tropea JE, Routzahn KM, Waugh DS. 2002. Three-dimensional structure of the type III secretion chaperone SycE from Yersinia pestis. Acta Crystallogr. D Biol. Crystallogr. 58:398–406 [DOI] [PubMed] [Google Scholar]
- 21. Stebbins CE, Galan JE. 2001. Maintenance of an unfolded polypeptide by a cognate chaperone in bacterial type III secretion. Nature 414:77–81 [DOI] [PubMed] [Google Scholar]
- 22. Vogelaar NJ, Jing X, Robinson HH, Schubot FD. 2010. Analysis of the crystal structure of the ExsC·ExsE complex reveals distinctive binding interactions of the Pseudomonas aeruginosa type III secretion chaperone ExsC with ExsE and ExsD. Biochemistry 49:5870–5879 [DOI] [PubMed] [Google Scholar]
- 23. Alfano JR, Charkowski AO, Deng WL, Badel JL, Petnicki-Ocwieja T, van Dijk K, Collmer A. 2000. The Pseudomonas syringae Hrp pathogenicity island has a tripartite mosaic structure composed of a cluster of type III secretion genes bounded by exchangeable effector and conserved effector loci that contribute to parasitic fitness and pathogenicity in plants. Proc. Natl. Acad. Sci. U. S. A. 97:4856–4861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Collmer A, Badel JL, Charkowski AO, Deng WL, Fouts DE, Ramos AR, Rehm AH, Anderson DM, Schneewind O, van Dijk K, Alfano JR. 2000. Pseudomonas syringae Hrp type III secretion system and effector proteins. Proc. Natl. Acad. Sci. U. S. A. 97:8770–8777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lindeberg M, Salmond GP, Collmer A. 1996. Complementation of deletion mutations in a cloned functional cluster of Erwinia chrysanthemi out genes with Erwinia carotovora out homologues reveals OutC and OutD as candidate gatekeepers of species-specific secretion of proteins via the type II pathway. Mol. Microbiol. 20:175–190 [DOI] [PubMed] [Google Scholar]
- 26. Kim SH, Kwon SI, Saha D, Anyanwu NC, Gassmann W. 2009. Resistance to the Pseudomonas syringae effector HopA1 is governed by the TIR-NBS-LRR protein RPS6 and is enhanced by mutations in SRFR1. Plant Physiol. 150:1723–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sheldrick GM. 2008. A short history of SHELX. Acta Crystallogr. A 64:112–122 [DOI] [PubMed] [Google Scholar]
- 28. Langer G, Cohen SX, Lamzin VS, Perrakis A. 2008. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3:1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255 [DOI] [PubMed] [Google Scholar]
- 31. Laskowski RA, Chistyakov VV, Thornton JM. 2005. PDBsum more: new summaries and analyses of the known 3D structures of proteins and nucleic acids. Nucleic Acids Res. 33:D266–D268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Dijk K, Tam VC, Records AR, Petnicki-Ocwieja T, Alfano JR. 2002. The ShcA protein is a molecular chaperone that assists in the secretion of the HopPsyA effector from the type III (Hrp) protein secretion system of Pseudomonas syringae. Mol. Microbiol. 44:1469–1481 [DOI] [PubMed] [Google Scholar]