

Abstract
The essential coenzyme NAD plays important roles in metabolic reactions and cell regulation in all organisms. As such, NAD synthesis has been investigated as a source for novel antibacterial targets. Cross-species genomics-based reconstructions of NAD metabolism in group A streptococci (GAS), combined with focused experimental testing in Streptococcus pyogenes, led to a better understanding of NAD metabolism in the pathogen. The predicted niacin auxotrophy was experimentally verified, as well as the essential role of the nicotinamidase PncA in the utilization of nicotinamide (Nm). PncA is dispensable in the presence of nicotinate (Na), ruling it out as a viable antibacterial target. The function of the “orphan” NadC enzyme, which is uniquely present in all GAS species despite the absence of other genes of NAD de novo synthesis, was elucidated. Indeed, the quinolinate (Qa) phosphoribosyltransferase activity of NadC from S. pyogenes allows the organism to sustain growth when Qa is present as a sole pyridine precursor. Finally, the redundancy of functional upstream salvage pathways in GAS species narrows the choice of potential drug targets to the two indispensable downstream enzymes of NAD synthesis, nicotinate adenylyltransferase (NadD family) and NAD synthetase (NadE family). Biochemical characterization of NadD confirmed its functional role in S. pyogenes, and its potential as an antibacterial target was supported by inhibition studies with previously identified class I inhibitors of the NadD enzyme family. One of these inhibitors efficiently inhibited S. pyogenes NadD (sp.NadD) in vitro (50% inhibitory concentration [IC50], 15 μM), exhibiting a noncompetitive mechanism with a Ki of 8 μM.
INTRODUCTION
Streptococcus pyogenes and S. pneumoniae are among the most important Gram-positive bacterial human pathogens. These species, commonly present in the flora of the respiratory tract, may cause a wide array of diseases, including life-threatening invasive infections, such as streptococcal toxic shock-like syndrome and necrotizing fasciitis (1, 2). The worldwide emergence of antibiotic-resistant streptococci has recently challenged the management of streptococcal infections (3), emphasizing the importance of finding new therapeutics. The current availability of the complete genome sequences of several clinical and nonpathogenic streptococci represents an excellent resource for genomics-based identification of pathogen-specific metabolic pathways. Analysis of such pathways facilitates a deeper comprehension of host-pathogen metabolic interactions, potentially leading to new approaches to the treatment and control of invasive streptococcal diseases.
NAD is an indispensable cofactor functioning both as a coenzyme for numerous hydride transfer-catalyzing enzymes and as a substrate for NAD-hydrolyzing enzymes, such as bacterial DNA ligase and protein deacetylase of the CobB/Sir2 family (4, 5). NAD biosynthesis is an established target pathway for the development of new antibiotics (6–10). Molecular targets of choice within NAD biosynthesis are enzymes participating in the last essential steps that involve phosphorylated intermediates, nicotinate mononucleotide (NaMN) and nicotinate adenine dinucleotide (NaAD). Indeed, most bacterial pathogens, except for some obligatory intracellular parasites, such as Chlamydia (11), cannot take up these intermediates from the host. The two enzymes with such desirable features are NaMN adenylyltransferase (NaMNAT) of the NadD family (for a review, see reference 4) and NAD synthetase of the NadE family (12), which together comprise a two-step conversion of NaMN precursor to NAD (Fig. 1). The development of small-molecule inhibitors targeting representative bacterial NadD and NadE enzymes that suppress the growth of various bacteria supported the choice of these enzymes as prospective broad-spectrum drug targets (6, 13–18).
Fig 1.
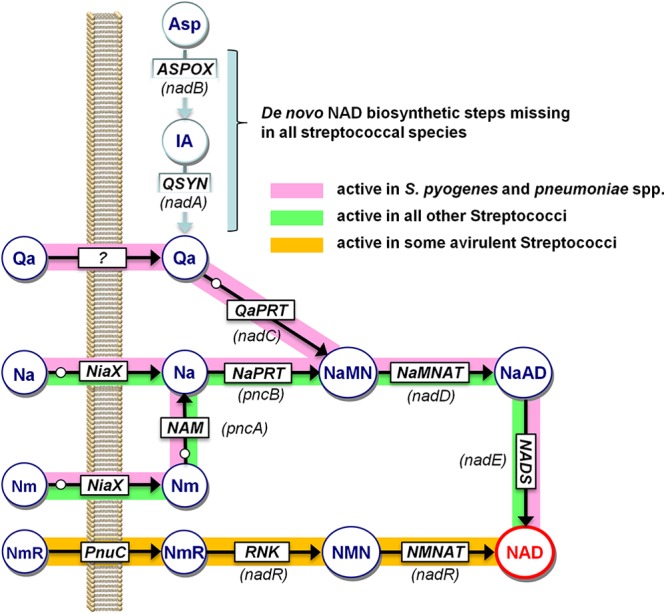
Genomic reconstruction of NAD biosynthesis in Streptococcus spp. Schematic representation of NAD biosynthetic routes as revealed by the in silico genomic reconstruction of NAD metabolism, integrated with experimental mutant analysis and biochemical studies. Predicted enzymes are shown in squares, metabolites are indicated within large circles, and gene names are shown in parentheses. The small white circles indicate coregulated functional roles of the NiaR repressor, as described previously (31). Nonpathogenic streptococcal species with an active nicotinamide riboside salvage pathway are listed in Table S2 in the supplemental material. Metabolites: IA, iminoaspartate; NmR, nicotinamide riboside. Enzymes and transporters: ASPOX, l-aspartate oxidase; QSYN, quinolinic acid synthetase; NaPRT, nicotinic acid phosphoribosyltransferase; NAM, nicotinamide deamidase; NADS, NAD synthetase; RNK, ribosyl nicotinamide kinase; NMNAT, NMN adenylyltransferase; PnuC, family of nicotinamide riboside transporters; NiaX, family of niacin transporters.
However, due to the intrinsic complexity and evolutionary variability of NAD metabolic networks (4), the choice of target enzymes should account for the network specificities of the targeted species. For example, in pathogens from the Haemophilus, Francisella, and Acinetobacter groups, NadD is functionally replaced by the distant homolog NadR (19) or NadM (9), which harbor different substrate preferences (for nicotinamide mononucleotide [NMN]) and are part of an alternative NAD synthesis route (10). Therefore, one goal of this study was to assess the validity of NadD as a prospective drug target for the Streptococcus group of pathogens by reconstructing the NAD network in the group and characterizing the NadD enzyme from S. pyogenes.
Our detailed genomics-based reconstruction of the NAD metabolic subsystem for the genus Streptococcus confirmed that all species in the group lack de novo synthesis, and they are dependent on salvage of exogenous pyridine precursors, nicotinamide or nicotinic acid (vitamin B3) (Fig. 1). This profile, which is also characteristic of Staphylococcus aureus, and the resulting niacin auxotrophy implicate salvage enzymes, PncA and PncB, as potential additional or alternative drug targets. On the other hand, analysis of all sequenced genomes from the group A streptococci (S. pyogenes and S. pneumoniae), but not any other species, revealed a unique feature: the presence of the final gene of the de novo pathway (nadC, encoding quinolinate phosphoribosyltransferase) in the absence of the first two genes (nadB and nadA), responsible for l-aspartate (Asp) conversion to quinolinic acid (Fig. 1). We hypothesized that this may endow such species with the capability to salvage quinolinate, making the PncA-PncB pathway dispensable when quinolinate is present in the medium. Here, we report the experimental verification of this hypothesis by biochemical and genetic methods. The accumulation of quinolinate, a product of tryptophan degradation and a NAD precursor in humans, is associated with certain pathological conditions (20, 21). It is tempting to speculate that the revealed unique metabolic capabilities of the group A streptococci may play a (yet unknown) role in host-pathogen interactions.
MATERIALS AND METHODS
Comparative genome analysis and bioinformatics tools.
Genome analysis and metabolic reconstruction tools implemented in the SEED annotation environment (22) were used to analyze NAD metabolism in 49 sequenced genomes from the Streptococcus group. Regulation of NAD synthesis by the NiaR transcriptional repressor was reconstructed based on the analysis provided in RegPrecise (23). Multiple alignments of analyzed enzyme families were constructed using Clustal Omega (24).
Bacterial strains, media, and growth conditions.
All the bacterial strains and plasmids used in this study are listed in Table 1. For phenotype analysis, S. pyogenes JRS4 and its mutant derivatives were grown in Todd-Hewitt broth (Difco) or defined medium (25) with or without the supplementation of NAD precursors at 30°C under a 5% CO2 atmosphere without agitation. Escherichia coli was grown at 37°C in Luria broth (Fisher) with agitation or on solid LB agar. Erythromycin was used at 1 μg/ml for S. pyogenes and 500 μg/ml for E. coli.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | Used for recombinant DNA methods | Invitrogen |
| BL21(DE3) | Used for recombinant protein expression | Novagen |
| S. pyogenes | ||
| JRS4 (MGAS10394) | Streptomycin-resistant derivative of serotype M6 strain D471 | 44 |
| VDC2488 | JRS4 nadC::erm | This work |
| VDC2576 | JRS4 pncA::erm | This work |
| Plasmids | ||
| pSK-ERM | pBluescript SK derivative; Erm inserted into Amp cassette | 45 |
| pIKB354 | 377-bp nadC inserted into EcoRV site of pSK-Erm | This work |
| pIKB489 | 400-bp pncA inserted into EcoRV site of pSK-Erm | This work |
| pODC | pET-derived vector; IPTG-inducible expression vector; Apr | 26 |
| pET28b | IPTG-inducible expression vector; Apr | Novagen |
| pODC_SPY0281 | S. pyogenes nadD inserted into NcoI and PstI sites of pODC | This work |
| pET28b_SPY0197 | S. pyogenes nadC inserted into NcoI and BamHI sites of pET28b | This work |
Engineering of S. pyogenes gene disruption mutants.
Disruption plasmids were generated by PCR amplifying ∼400 bp of the gene to be disrupted from purified S. pyogenes genomic DNA and by insertion into pSK-Erm, a pBluescript SK (Stratagene) derivative with an erythromycin resistance cassette in place of the ampicillin resistance gene, linearized by EcoRV digestion. The resulting clones were selected for in E. coli with 500 μg/ml erythromycin, and chromosomal integrations were selected for in S. pyogenes using erythromycin at 1 μg/ml (see Fig. S1 in the supplemental material). Integration events were identified with a combination of locus-specific PCRs (see Fig. S2 in the supplemental material), using the primers indicated in Table S1 in the supplemental material. Phenotypic analysis on solid medium was performed by streaking the relevant strains onto defined medium (25) in the presence and absence of 100 μM nicotinamide (Nm), nicotinic acid (Na), or quinolinic acid (Qa). The minimal requirements for these pyridine precursors were tested in the equivalent liquid media supplemented with 0, 0.5, 0.75, 1, 2, 3, 6, 10, 15, or 100 μM Nm, Na, or Qa.
Cloning, heterologous expression, and protein purification.
The nadD gene (locus tag SPY0281; S. pyogenes NadD [sp.NadD]) encoding NaMNAT and the nadC gene (SPY0197; sp.NadC) encoding QaPRT of S. pyogenes JRS4 were amplified by PCR from genomic DNA and cloned into a pET-derived vector (26) and pET28b, respectively, as a fusion with an N-terminal His6 tag. Primers designed for cloning experiments are shown in Table S1 in the supplemental material. All sequences were verified to confirm PCR accuracy. All recombinant proteins were overexpressed in E. coli BL21(DE3) and purified to homogeneity using standard protocols, e.g., as described previously (27). Typically, cells were grown in LB medium to an optical density at 600 nm (OD600) of ∼0.8 to 1.0 at 37°C, induced by 0.2 mM IPTG (isopropyl-β-d-thiogalactopyranoside), and harvested after 12 h of shaking at 20°C. Proteins were purified from 1- to 6-liter cultures by chromatography on an Ni-nitrilotriacetic acid (NTA) agarose column, followed by gel filtration on a HiLoad Superdex 200 16/60 column (Pharmacia) with an AKTA fast protein liquid chromatography (FPLC) system.
Activity assays and steady-state kinetic analysis.
The NaMNT activity of the recombinant purified NadD was measured by in situ conversion of the NaAD product to NAD (by an excess of Bacillus anthracis NAD synthetase [13]) coupled to the reduction of NAD to NADH (by alcohol dehydrogenase) with spectrophotometric monitoring at 340 nm, as described previously (10). The Qa phosphoribosyltransferase (QaPRT) activity of NadC was assessed by monitoring changes in UV absorption at 266 nm, which reflects conversion of Qa to NaMN (28). Steady-state kinetic parameters were evaluated by fitting initial rates to the standard Michaelis-Menten equation using GraphPad Prism. Selected small-molecule compounds (a kind gift from ChemDiv, Inc., San Diego, CA), chemical analogs of previously described inhibitors of bacterial NadD, were tested for inhibition of sp.NadD at 20 μM using the malachite green assay in 96-well plate format, as described previously (13). The 50% inhibitory concentration (IC50) was determined as described previously (13). The apparent Ki was determined by fitting the kinetic data to the equation for noncompetitive inhibition (29).
RESULTS
An “orphan” NadC enzyme is a unique feature of the genomic reconstruction of the NAD biosynthetic network in group A streptococci.
We used genome context analysis techniques, including chromosomal clustering, gene occurrence profiles, and regulon analysis (30), to reconstruct NAD metabolism in the genus Streptococcus (Fig. 1 and Table 2). The results of this analysis across multiple sequenced genomes covering 11 distinct Streptococcus species were captured in the SEED database (available at http://theseed.uchicago.edu/FIG/subsys.cgi within the NAD and NADP cofactor biosynthesis subsystem). This analysis showed that all analyzed species share the following key features (Fig. 1): (i) the absence of Qa de novo synthesis; (ii) reliance on the salvage of exogenous Nm or Na via a recently characterized high-affinity ECF-type transporter, NiaX (31, 32), followed by PncA-PncB-driven conversion to NaMN; (iii) transcriptional regulation of the salvage pathway by an ortholog of the previously described NiaR repressor (31); (iv) universal two-step conversion of NaMN to NAD by the downstream NadD-NadE pathway. Given this network topology, nearly all the enzymes listed above could be indispensable and thus considered potential drug targets. Here, we aimed to test this possibility by physiological and genetic experiments.
Table 2.
Functional roles of key NAD biosynthetic genes in pathogenic and nonpathogenic streptococcia
| Streptococcus species (no. of strains) | Virulence trait(s) | Presence/clusteringb |
||||||
|---|---|---|---|---|---|---|---|---|
|
De novo |
Salvage |
Downstream |
||||||
| nadA | nadB | nadC | pncA | pncB | nadD | nadE | ||
| S. pyogenes (12) | Major human pathogen | + | + | + | + | + | ||
| S. pneumoniae (21) | Major human pathogen | + | + | + | + | + | ||
| S. mutans (1) | Oral flora; major player in cavity formation | + | + | + | + | |||
| S. agalactiae (3) | Gastrointestinal flora; opportunistic pathogen | + | + | + | + | |||
| S. gordonii (1) | Mouth flora; opportunistic pathogen (endocarditis) | + | + | + | + | |||
| S. mitis (1) | Mouth flora; opportunistic pathogen (endocarditis) | + | + | + | + | |||
| S. sanguinis (1) | Mouth flora; opportunistic pathogen (endocarditis) | + | + | + | + | |||
| S. equi (2) | Not a human pathogen | + | + | + | + | |||
| S. suis (3) | Not a human pathogen | + | + | + | + | |||
| S. thermophilus (3) | Probiotic | + | + | + | + | |||
| S. uberis (1) | Environmental | + | + | + | + | |||
A full version of this table that includes the distribution of genes involved in the nicotinamide riboside salvage pathway in some nonpathogenic streptococci is presented in Table S2 in the supplemental material.
The presence and clustering of genes are indicated by + and shading, respectively.
A striking distinctive feature of S. pyogenes and S. pneumoniae compared to all other Streptococcus species is the presence of a putative ortholog of the NadC enzyme. This genomic feature is unique among hundreds of other analyzed microbial species that either have NadC along with a full complement of other genes of de novo synthesis from Asp (or, more rarely, from Trp [33]) or completely lack NadC and other de novo biosynthetic genes (as in other Streptococcus species). Such an out-of-context occurrence of a metabolic enzyme often points to an incorrect homology-based functional assignment of the respective gene, which may, in fact, play a different functional role. Likewise, proteins encoded by pseudogenes that have lost their metabolic context as a result of deletion of other pathway components typically lose their functional activity due to rapid divergence in the absence of selective pressure. To account for these possibilities, we aimed to clone the NadC homolog from S. pyogenes (sp.NadC) and assess the predicted QaPRT activity of the purified recombinant protein overexpressed in E. coli.
Conversely, the observed conservation of the key active-site residues in sp.NadC (see Fig. S3 in the supplemental material) argued in favor of its functional assignment as an active QaPRT enzyme. Moreover, its likely involvement in NAD metabolism is additionally supported by the identification of an NiaR-binding motif in the upstream region of nadC in both S. pyogenes and S. pneumoniae (31) (Fig. 1). However, these considerations led to another open question about a possible source of Qa substrate for an otherwise “orphan” QaPRT in these species. As mentioned above, neither of the two known biochemical pathways of Qa synthesis (from Asp or from Trp) is present in Streptococcus species, suggesting that Qa can either be produced by a third, yet unknown, pathway or, more likely, it can be scavenged from the host. The only known precedent for the latter was reported in some highly virulent isolates of Shigella with multiple mutations inactivating nadB and nadA, but not nadC, genes (34, 35). In this study, we combined gene disruption/growth phenotype approaches to discern these two possibilities and elucidate the physiological role of NadC in group A streptococci.
sp.PncA is essential for the salvage of nicotinamide, but it is dispensable for growth of S. pyogenes when nicotinic acid is present in the medium.
The niacin (vitamin B3) auxotrophy observed in this study is consistent with our genomic reconstruction and previous reports (36). Wild-type S. pyogenes could not grow on defined medium in the absence of a pyridine precursor (Fig. 2). Both Nm and Na supplementation sustained growth with comparable minimal requirements (∼3 μM) for both amidated and deamidated forms of niacin (Fig. 3). Of note, the presence of NAD, which was historically included in the chemically defined medium (32), is not required for S. pyogenes growth.
Fig 2.
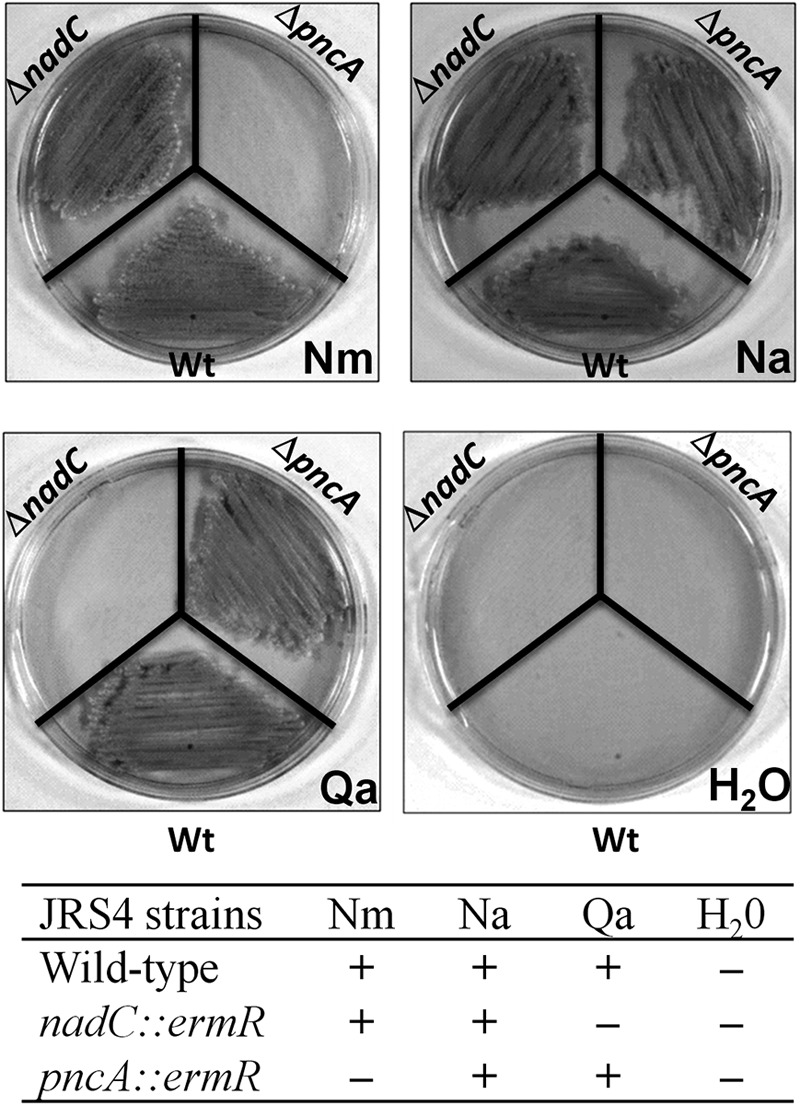
Growth phenotype of S. pyogenes JRS4 on pyridine NAD precursor. The wild-type (Wt), nadC, and pncA strains were streaked on defined media supplemented with 100 μM Na, Nm, Qa, or H2O (negative control) as indicated. The growth phenotypes are also summarized as follows: +, growth; −, no growth.
Fig 3.
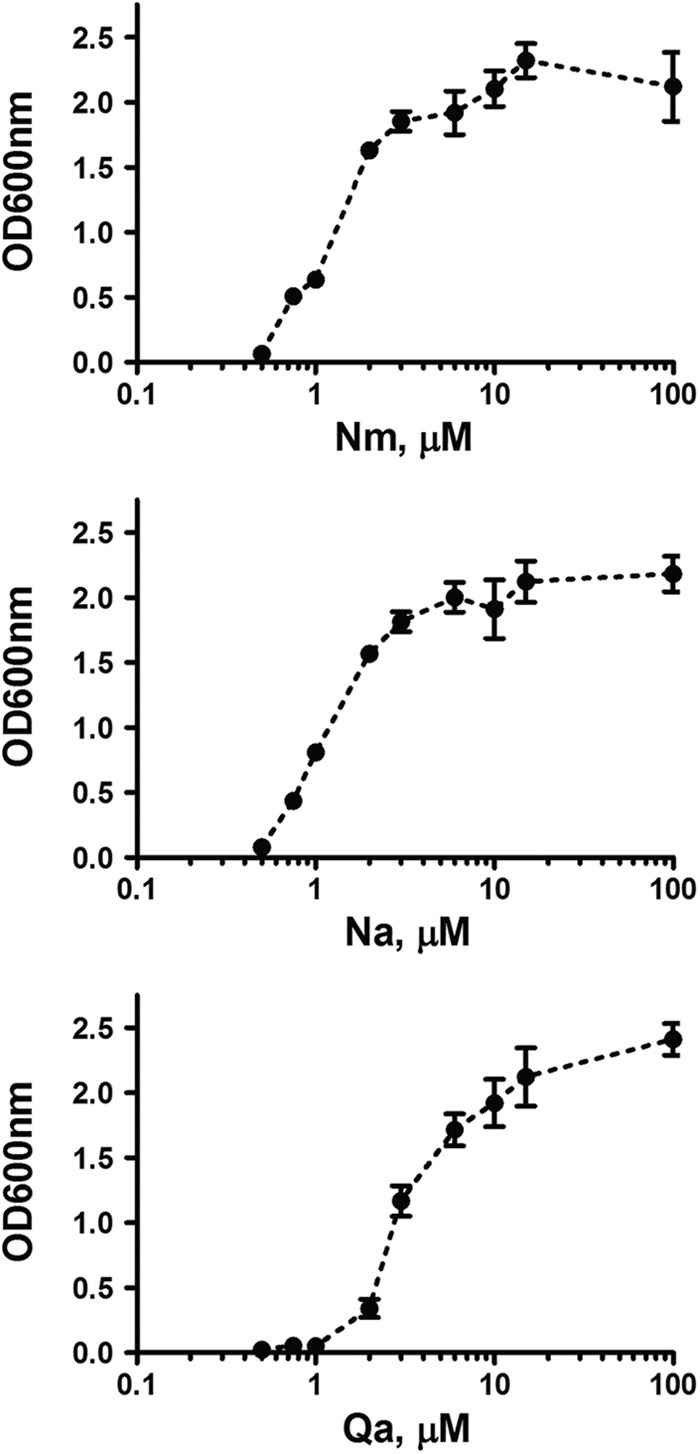
S. pyogenes minimal requirements for vitamin B3 and quinolinate. The culture endpoint optical densities (OD600) of S. pyogenes JRS4 are plotted by pyridine precursor supplement concentration (log scale). Each point represents the mean of three independent repeats. The error bars represent the standard deviations.
The inability of a ΔpncA knockout mutant to grow on Nm (Fig. 2) confirms the role of the encoded enzyme as a nicotinamidase required for the utilization of the amidated form of niacin. However, the growth of the mutant on a plate in the presence of Na suggests that PncA enzyme could be dispensable under physiological conditions in the host (where Na is readily available), thus eliminating its value as a potential bacterium-specific drug target even if it is the only enzyme in the network that does not have even distant homologs in the human genome.
sp.NadC is a functional QaPRT enzyme that allows S. pyogenes to salvage quinolinate from the medium, bypassing the requirement for niacin salvage.
The enzymatic properties of the recombinant purified sp.NadC enzyme (see Fig. S4 in the supplemental material) are fully consistent with its homology-based assignment as QaPRT. Indeed, its steady-state kinetic parameters for quinolinate (Km, 10 μM; kcat, 0.1 s−1) (Table 3) are comparable to the values reported for the enzyme from other sources (37, 38). This functional assignment is also consistent with the established capability of S. pyogenes to grow on Qa as a single pyridine precursor (Fig. 2). The minimal requirement for Qa (∼10 μM) (Fig. 3) is slightly higher than for Nm or Na but in the range of the QaPRT affinity for Qa. The absolute requirement of S. pyogenes for a pyridine precursor (Nm, Na, or Qa) in the medium also rules out the existence of an endogenous source of Qa in streptococci.
Table 3.
Steady-state kinetic parameters of S. pyogenes NadD and NadC enzymes and comparison with human counterparts
| Enzymea | Variable substrate | Fixed substrateb | Km (μM)c | kcat (s−1)c | kcat/Km (s−1 mM−1) | Source or reference |
|---|---|---|---|---|---|---|
| sp.NadDd | NaMN | ATP | 103 ± 18 | 17.4 ± 0.7 | 1.7 × 102 | This study |
| NMN | ATP | ND | ND | This study | ||
| sp.NadC | Qa | PRPP | 10 ± 3 | 0.1 ± 0.01 | 10 | This study |
| hsPNAT-1e | NaMN | ATP | 68 ± 2 | 43 ± 1 | 6.3 × 103 | 46 |
| NMN | ATP | 22 ± 3 | 54 ± 3 | 2.4 × 103 | 46 | |
| hsQaPRTg | Qa | PRPP | 22 ± 3 | 1.19 ± 0.05f | 38 |
Initial rates were measured by coupled (sp.NadD) or direct (sp.NadC) spectrophotometrical assays.
For fixed substrates, concentrations were 0.25 mM PRPP and 2 mM ATP.
The kinetic parameters Km and kcat are apparent values (±standard deviations) determined at fixed (saturating) concentrations of cosubstrates. ND, not detectable.
The NMN adenylyltransferase activity of sp.NadD was not detected under the assay conditions that ensured a maximal rate for NaMN.
Note that for hsPNAT, data were reported only for human PNAT isoform 1 (hsPNAT-1), the most abundant and ubiquitous of the three human PNAT isoforms. The kinetic properties of hsPNAT-2 and hsPNAT-3 are similar to those of hsPNAT-1 (46, 47).
Expressed in μM min−1.
hsQaPRT, human quinolinate phosphoribosyltransferase.
Finally, the observation that the S. pyogenes ΔnadC mutant no longer grows on Qa while sustaining normal growth on Nm or Na confirms the physiological role of sp.NadC in Qa salvage. Although Qa, an intermediate of human NAD de novo synthesis from Trp, may accumulate in the host under certain pathological conditions (20), it is not expected to be nearly as abundant as the dietary niacin. Therefore, the actual physiological role of this pathway and its potential association with virulence have yet to be clarified. The Qa uptake mechanism and whether it is mediated by NiaX or another, specialized, transporter also remains to be determined. Indirect evidence, such as the higher minimal requirement for Qa than for niacin and our failure to obtain a viable ΔpncB mutant even in the presence of excess Qa, suggests that the Qa salvage pathway may be less robust than the Nm/Na salvage routes. Although our failure to inactivate the pncB gene may be due to technical reasons, the possibility that the apparent essentiality of PncB might be due to its hypothesized role in nicotinamide recycling (mediated by NAD glycohydrolase and nicotinamidase activities) cannot be excluded.
sp.NadD is a functional NaMNAT enzyme and a promising drug target in S. pyogenes.
Of the indispensable downstream enzymes of NAD biosynthesis of the NadD and NadE families, NadD seems a more promising antibacterial target, as it is structurally and functionally (by substrate specificity) more distant from its human counterpart (4). All previously described members of the bacterial NadD family have a strong (100- to 10,000-fold) preference for NaMN over an alternative substrate, NMN. This is consistent with their exclusive roles in the two-step deamidated pathway: NaMN→NaAD→NAD. On the other hand, the distantly homologous human pyridine nucleotide adenylyltransferase (PNAT) shows nearly equal catalytic efficiency with either the NaMN or NMN substrate, which is consistent with its involvement in both the universal two-step pathway from NaMN and a relatively rare one-step pathway from NMN (which is generated in humans by the enzyme NMPRT directly from Nm [39]).
The enzymatic properties of the purified recombinant sp.NadD (Table 3) are consistent with those of other members of the NadD family, particularly from close homologs in Gram-positive bacteria (13, 40, 41), showing at least ∼500-fold preference for NaMN over NMN (based on the limit of detection of NaMN or NMN adenylyltransferase activity under the assay conditions). To further assess the potential of sp.NadD as a target enzyme, we tested its inhibition by analogs of previously identified NadD inhibitors (13, 14). Several compounds previously shown to be active on Gram-positive NadD enzymes (L. Sorci and A. Osterman, unpublished data) were tested. One compound, K891-0166 (ChemDiv Inc.) displayed inhibitory properties in the low micromolar range, with an IC50 of 15 ± 3 μM (Fig. 4A). Kinetic analysis indicated that K891-0166 is a noncompetitive inhibitor of sp.NadD, with a Ki of 7.9 ± 0.6 μM (Fig. 4B). These observations provide additional support for selection of NadD as a target for the development of new antibiotics against Gram-positive pathogens, including group A streptococci.
Fig 4.
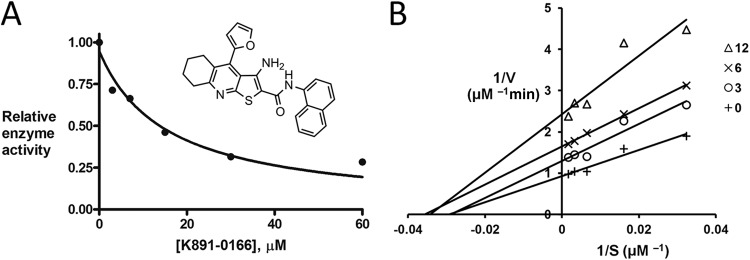
K891-0166 inhibition activity of sp.NadD. (A) K891-0166 (ChemDiv) (inset), a chemical analog of the previously identified NadD small-molecule inhibitor 3_05 (13), efficiently inhibits sp.NadD in vitro (IC50, 15 μM). (B) Double-reciprocal (Lineweaver-Burk) plot of initial rates (V) as a function of the NaMN substrate concentration (S) measured at a fixed ATP concentration (0.1 mM) in the presence of various concentrations of the K891-0166 inhibitor (0 to 12 μM).
DISCUSSION
Targeting essential enzymes involved in the biosynthesis of NAD, the indispensable redox cofactor, has been recognized as a promising strategy for the development of novel antibiotics. However, the rational choice of actual drug targets requires better understanding of pathways that comprise NAD metabolic networks, which may feature notable variations even between closely related species. The most variable and redundant parts of NAD metabolism pertain to the upstream de novo and salvage routes, while the most conserved parts, and potentially richer in drug targets, are found in the downstream pathway.
In this study, we combined a comparative genomics-based reconstruction with focused experimental verification to assess NAD biosynthetic networks in the genus Streptococcus and potential drug targets therein. We focused on S. pyogenes as a representative of the most virulent species of GAS. This analysis yielded a number of conclusions important for a general understanding of NAD metabolism and, particularly, for selection of drug targets in these species.
The presence of a stand-alone but functionally active and NiaR-regulated nadC gene is one of the most unexpected findings of this study. It is clearly a unique feature of GAS species, not only among other Streptococcus species, but among all other analyzed bacterial genomes. A striking correlation between the presence of orphan NadC and the human pathogenicity phenotype of GAS strains might indicate a potential role of NadC in virulence (Table 2). Indeed, a recent study on the gene content evolution of S. pyogenes identified nadC as one of genes that have been acquired during the strict human host adaptation (42).
The conjecture of a possible link between nadC gene preservation and virulence in GAS species is indirectly supported by a similar case described for Shigella flexneri, a very different bacterial pathogen (35). Indeed, in this pathogen, the loss of function by mutations and deletions of de novo nadB and nadA genes, along with the presence of nadC, was implicated in virulence (35). Moreover, quinolinate was demonstrated to be a strong inhibitor of Shigella virulence (34). Given a relatively low abundance of Qa (compared to Nm and Na) in the human body, the preservation of QaPRT function in Shigella and Streptococcus is more likely driven by its scavenging function rather than an actual contribution to NAD biogenesis. Thus, an inflammatory response to bacterial infection is known to induce indoleamine 2,3-dioxygenase (IDO), which is involved in tryptophan degradation and may lead to accumulation of Qa (an intermediate of mammalian NAD de novo synthesis from tryptophan [43]). As with Shigella, quinolinate might inhibit the virulence of group A streptococci, as well. Therefore, the ability to consume Qa mediated by NadC may act as a “detoxification” process during inflammation and may potentially contribute to the virulence of Shigella and GAS pathogens. Further mechanistic exploration of such phenomena and elucidation of the actual role of Qa scavenging would lead to better understanding of host-pathogen interactions and potentially translate into new approaches to diagnostics, prevention, and eradication of deadly GAS infections.
While the clinical implications of these findings have yet to be explored, the enzymatic and inhibitory characterization of sp.NadD by one of the NadD inhibitors that was accomplished provides a starting point and guidelines for pursuing this enzyme as a target for the development of novel antibiotics against GAS pathogens.
Supplementary Material
ACKNOWLEDGMENTS
This work was partly supported by the Montalcini International Program grant through the Italian Ministry of Education, University, and Research to L.S.; by National Institutes of Health (NIH) grant 1R03MH095597 to A.L.O.; and by NIH grant RO1 AI066244 to V.D.C.-L.
We thank Kelly Rice for critical reading of the paper.
Footnotes
Published ahead of print 30 November 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02002-12.
REFERENCES
- 1. Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13: 470–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gillespie SH, Balakrishnan I. 2000. Pathogenesis of pneumococcal infection. J. Med. Microbiol. 49: 1057–1067 [DOI] [PubMed] [Google Scholar]
- 3. Morens DM, Folkers GK, Fauci AS. 2004. The challenge of emerging and re-emerging infectious diseases. Nature 430: 242–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sorci L, Kurnasov O, Rodionov DA, Osterman AL. 2010. Genomics and enzymology of NAD biosynthesis, p 213–257 In Lew M, Hung-Wen L. (ed), Comprehensive natural products II. Elsevier, Oxford, United Kingdom [Google Scholar]
- 5. Gazzaniga F, Stebbins R, Chang SZ, McPeek MA, Brenner C. 2009. Microbial NAD metabolism: lessons from comparative genomics. Microbiol. Mol. Biol. Rev. 73: 529–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boshoff HI, Xu X, Tahlan K, Dowd CS, Pethe K, Camacho LR, Park TH, Yun CS, Schnappinger D, Ehrt S, Williams KJ, Barry CE., III 2008. Biosynthesis and recycling of nicotinamide cofactors in Mycobacterium tuberculosis. An essential role for NAD in nonreplicating bacilli. J. Biol. Chem. 283: 19329–19341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gerdes SY, Scholle MD, D'Souza M, Bernal A, Baev MV, Farrell M, Kurnasov OV, Daugherty MD, Mseeh F, Polanuyer BM, Campbell JW, Anantha S, Shatalin KY, Chowdhury SA, Fonstein MY, Osterman AL. 2002. From genetic footprinting to antimicrobial drug targets: examples in cofactor biosynthetic pathways. J. Bacteriol. 184: 4555–4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Osterman AL, Begley TP. 2007. A subsystems-based approach to the identification of drug targets in bacterial pathogens. Prog. Drug Res. 64: 131, 133–170. [DOI] [PubMed] [Google Scholar]
- 9. Sorci L, Blaby I, De Ingeniis J, Gerdes S, Raffaelli N, de Crecy Lagard V, Osterman A. 2010. Genomics-driven reconstruction of acinetobacter NAD metabolism: insights for antibacterial target selection. J. Biol. Chem. 285: 39490–39499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sorci L, Martynowski D, Rodionov DA, Eyobo Y, Zogaj X, Klose KE, Nikolaev EV, Magni G, Zhang H, Osterman AL. 2009. Nicotinamide mononucleotide synthetase is the key enzyme for an alternative route of NAD biosynthesis in Francisella tularensis. Proc. Natl. Acad. Sci. U. S. A. 106: 3083–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haferkamp I, Schmitz-Esser S, Linka N, Urbany C, Collingro A, Wagner M, Horn M, Neuhaus HE. 2004. A candidate NAD+ transporter in an intracellular bacterial symbiont related to Chlamydiae. Nature 432: 622–625 [DOI] [PubMed] [Google Scholar]
- 12. De Ingeniis J, Kazanov MD, Shatalin K, Gelfand MS, Osterman AL, Sorci L. 2012. Glutamine versus ammonia utilization in the NAD synthetase family. PLoS One 7: e39115 doi:10.1371/journal.pone.0039115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sorci L, Pan Y, Eyobo Y, Rodionova I, Huang N, Kurnasov O, Zhong S, MacKerell AD, Jr, Zhang H, Osterman AL. 2009. Targeting NAD biosynthesis in bacterial pathogens: structure-based development of inhibitors of nicotinate mononucleotide adenylyltransferase NadD. Chem. Biol. 16: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang N, Kolhatkar R, Eyobo Y, Sorci L, Rodionova I, Osterman AL, Mackerell AD, Zhang H. 2010. Complexes of bacterial nicotinate mononucleotide adenylyltransferase with inhibitors: implication for structure-based drug design and improvement. J. Med. Chem. 53: 5229–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moro WB, Yang Z, Kane TA, Zhou Q, Harville S, Brouillette CG, Brouillette WJ. 2009. SAR studies for a new class of antibacterial NAD biosynthesis inhibitors. J. Comb. Chem. 11: 617–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moro WB, Yang Z, Kane TA, Brouillette CG, Brouillette WJ. 2009. Virtual screening to identify lead inhibitors for bacterial NAD synthetase (NADs). Bioorg. Med. Chem. Lett. 19: 2001–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Velu SE, Mou L, Luan CH, Yang ZW, DeLucas LJ, Brouillette CG, Brouillette WJ. 2007. Antibacterial nicotinamide adenine dinucleotide synthetase inhibitors: amide- and ether-linked tethered dimers with alpha-amino acid end groups. J. Med. Chem. 50: 2612–2621 [DOI] [PubMed] [Google Scholar]
- 18. Plata G, Hsiao TL, Olszewski KL, Llinas M, Vitkup D. 2010. Reconstruction and flux-balance analysis of the Plasmodium falciparum metabolic network. Mol. Syst. Biol. 6: 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kurnasov OV, Polanuyer BM, Ananta S, Sloutsky R, Tam A, Gerdes SY, Osterman AL. 2002. Ribosylnicotinamide kinase domain of NadR protein: identification and implications in NAD biosynthesis. J. Bacteriol. 184: 6906–6917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reinhard JF, Jr, Erickson JB, Flanagan EM. 1994. Quinolinic acid in neurological disease: opportunities for novel drug discovery. Adv. Pharmacol. 30: 85–127 [DOI] [PubMed] [Google Scholar]
- 21. Schwarcz R. 2004. The kynurenine pathway of tryptophan degradation as a drug target. Curr. Opin. Pharmacol. 4: 12–17 [DOI] [PubMed] [Google Scholar]
- 22. Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang Cohoon H-YM, de Crecy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Ruckert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V. 2005. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33: 5691–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Novichkov PS, Laikova ON, Novichkova ES, Gelfand MS, Arkin AP, Dubchak I, Rodionov DA. 2010. RegPrecise: a database of curated genomic inferences of transcriptional regulatory interactions in prokaryotes. Nucleic Acids Res. 38: D111–D118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van de Rijn I, Kessler RE. 1980. Growth characteristics of group A streptococci in a new chemically defined medium. Infect. Immun. 27: 444–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Osterman AL, Lueder DV, Quick M, Myers D, Canagarajah BJ, Phillips MA. 1995. Domain organization and a protease-sensitive loop in eukaryotic ornithine decarboxylase. Biochemistry 34: 13431–13436 [DOI] [PubMed] [Google Scholar]
- 27. Daugherty M, Vonstein V, Overbeek R, Osterman A. 2001. Archaeal shikimate kinase, a new member of the GHMP-kinase family. J. Bacteriol. 183: 292–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cao H, Pietrak BL, Grubmeyer C. 2002. Quinolinate phosphoribosyltransferase: kinetic mechanism for a type II PRTase. Biochemistry 41: 3520–3528 [DOI] [PubMed] [Google Scholar]
- 29. Lyon RP, Atkins WM. 2002. Kinetic characterization of native and cysteine 112-modified glutathione S-transferase A1-1: reassessment of nonsubstrate ligand binding. Biochemistry 41: 10920–10927 [DOI] [PubMed] [Google Scholar]
- 30. Osterman A, Overbeek R. 2003. Missing genes in metabolic pathways: a comparative genomics approach. Curr. Opin. Chem. Biol. 7: 238–251 [DOI] [PubMed] [Google Scholar]
- 31. Rodionov DA, Li X, Rodionova IA, Yang C, Sorci L, Dervyn E, Martynowski D, Zhang H, Gelfand MS, Osterman AL. 2008. Transcriptional regulation of NAD metabolism in bacteria: genomic reconstruction of NiaR (YrxA) regulon. Nucleic Acids Res. 36: 2032–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rodionov DA, Hebbeln P, Eudes A, ter Beek J, Rodionova IA, Erkens GB, Slotboom DJ, Gelfand MS, Osterman AL, Hanson AD, Eitinger T. 2009. A novel class of modular transporters for vitamins in prokaryotes. J. Bacteriol. 191: 42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kurnasov O, Goral V, Colabroy K, Gerdes S, Anantha S, Osterman A, Begley TP. 2003. NAD biosynthesis: identification of the tryptophan to quinolinate pathway in bacteria. Chem. Biol. 10: 1195–1204 [DOI] [PubMed] [Google Scholar]
- 34. Prunier AL, Schuch R, Fernandez RE, Mumy KL, Kohler H, McCormick BA, Maurelli AT. 2007. nadA and nadB of Shigella flexneri 5a are antivirulence loci responsible for the synthesis of quinolinate, a small molecule inhibitor of Shigella pathogenicity. Microbiology 153: 2363–2372 [DOI] [PubMed] [Google Scholar]
- 35. Prunier AL, Schuch R, Fernandez RE, Maurelli AT. 2007. Genetic structure of the nadA and nadB antivirulence loci in Shigella spp. J. Bacteriol. 189: 6482–6486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fujiwara S, Kobayashi S, Nakayama H. 1978. Development of a minimal medium for Streptococcus mutans. Arch. Oral Biol. 23: 601–602 [DOI] [PubMed] [Google Scholar]
- 37. Bhatia R, Calvo KC. 1996. The sequencing expression, purification, and steady-state kinetic analysis of quinolinate phosphoribosyl transferase from Escherichia coli. Arch. Biochem. Biophys. 325: 270–278 [DOI] [PubMed] [Google Scholar]
- 38. Liu H, Woznica K, Catton G, Crawford A, Botting N, Naismith JH. 2007. Structural and kinetic characterization of quinolinate phosphoribosyltransferase (hQPRTase) from Homo sapiens. J. Mol. Biol. 373: 755–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O, Andris F. 2002. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur. J. Immunol. 32: 3225–3234 [DOI] [PubMed] [Google Scholar]
- 40. Lu S, Smith CD, Yang Z, Pruett PS, Nagy L, McCombs D, Delucas LJ, Brouillette WJ, Brouillette CG. 2008. Structure of nicotinic acid mononucleotide adenylyltransferase from Bacillus anthracis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64: 893–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Olland AM, Underwood KW, Czerwinski RM, Lo MC, Aulabaugh A, Bard J, Stahl ML, Somers WS, Sullivan FX, Chopra R. 2002. Identification, characterization, and crystal structure of Bacillus subtilis nicotinic acid mononucleotide adenylyltransferase. J. Biol. Chem. 277: 3698–3707 [DOI] [PubMed] [Google Scholar]
- 42. Lefebure T, Richards VP, Lang P, Pavinski-Bitar P, Stanhope MJ. 2012. Gene repertoire evolution of Streptococcus pyogenes inferred from phylogenomic analysis with Streptococcus canis and Streptococcus dysgalactiae. PLoS One 7: e37607 doi:10.1371/journal.pone.0037607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carlin JM, Ozaki Y, Byrne GI, Brown RR, Borden EC. 1989. Interferons and indoleamine 2,3-dioxygenase: role in antimicrobial and antitumor effects. Experientia 45: 535–541 [DOI] [PubMed] [Google Scholar]
- 44. Scott JR, Guenthner PC, Malone LM, Fischetti VA. 1986. Conversion of an M- group A streptococcus to M+ by transfer of a plasmid containing an M6 gene. J. Exp. Med. 164: 1641–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Biswas I, Scott JR. 2003. Identification of rocA, a positive regulator of covR expression in the group A streptococcus. J. Bacteriol. 185: 3081–3090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sorci L, Cimadamore F, Scotti S, Petrelli R, Cappellacci L, Franchetti P, Orsomando G, Magni G. 2007. Initial-rate kinetics of human NMN-adenylyltransferases: substrate and metal ion specificity, inhibition by-products and multisubstrate analogues, and isozyme contributions to NAD+ biosynthesis. Biochemistry 46: 4912–4922 [DOI] [PubMed] [Google Scholar]
- 47. Berger F, Lau C, Dahlmann M, Ziegler M. 2005. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 280: 36334–36341 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.