

Abstract
Bacterial morphology imparts physiological advantages to cells in different environments and, judging by the fidelity with which shape is passed to daughter cells, is a tightly regulated characteristic. Surprisingly, only in the past 10 to 15 years has significant headway been made in identifying the mechanisms by which cells create and maintain particular shapes. One reason for this is that the relevant discoveries have relied heavily on the arduous, somewhat subjective process of manual microscopy. Here, we show that flow cytometry, coupled with the sorting capability of fluorescence-activated cell sorting (FACS), can detect, quantify, and enrich bacteria with morphological alterations. The light scattering properties of several highly aberrant morphological mutants of Escherichia coli were characterized by flow cytometry. Cells from a region that overlapped the distribution of normal rod-shaped cells were collected by FACS and reincubated. After 4 to 15 iterations of this enrichment process, suppressor mutants were isolated that returned almost all the population to a near-normal shape. Suppressors were successfully isolated from strains lacking three or four penicillin binding proteins (PBPs) but not from a mutant lacking a total of seven PBPs. The peptidoglycan endopeptidase, AmpH, was identified as being important for the suppression process, as was a related endopeptidase, MepA. The results validate the use of cell sorting as a means for studying bacterial morphology and identify at least one new class of enzymes required for the suppression of cell shape defects.
INTRODUCTION
Bacterial morphology is created by the collaborative processes of cell elongation and division, which synthesize the peptidoglycan wall that provides mechanical strength and imparts to each cell a defined shape (1, 2). Individual species usually exhibit one primary shape, but bacteria as a whole display such a remarkable diversity of shapes that it seems certain that no single morphology is optimum for all environments or conditions (3, 4). Instead, these varied morphologies help bacteria survive in different situations by contributing to the efficiency with which cells take up nutrients, divide, segregate their chromosomes, attach to surfaces, disperse passively or by directed motility, avoid predation and immune responses, and differentiate (reviewed in reference 4). Cell shape also contributes to the virulence of the few pathogens in which the question has been addressed (4–6), including Escherichia coli (7), Helicobacter pylori (8, 9), Campylobacter jejuni (10), and Listeria monocytogenes (11). This accumulating evidence has fueled a growing consensus that morphology is not simply a neutral characteristic but that it plays an important role in bacterial ecology and pathogenesis.
Great headway has been made in describing the mechanisms that create and maintain bacterial morphology (2, 12). In general, these pathways affect either the cell division apparatus (the divisome) as nucleated by the FtsZ protein (13–15) or the cell elongation apparatus as directed by the MreB protein (1, 16, 17). In each case, the resulting morphological effects are best explained by envisioning how the mutations alter the manner or timing of cell wall synthesis. For example, mutations affecting some divisome components alter the shape of the cell and cell pole (14), amino acid replacements in MreB or related proteins alter overall shape or cell diameter (18, 19), and deleting accessory peptidoglycan modifying enzymes creates highly aberrant cells (1, 14, 20, 21). Despite the growing list of mutations that affect cell shape, the molecular events that generate these gross morphological alterations are, for the most part, very poorly understood.
One such mystery that has recently yielded to experiment is the mechanism by which bacteria can alter cell length during their growth cycle. Cells of many species are larger (usually longer, but sometimes also wider) when growing exponentially and become shorter when they reach stationary phase (reviewed in reference 4). Also, individual cells may grow longer when in the presence of certain preferred carbohydrates (22, 23). Researchers spent decades speculating how growth rate and nutritional signals might control cellular morphology (1, 22, 24), but only lately has the first molecular explanation for these phenomena been described. In Bacillus subtilis the presence of sufficient nutrients increases the amount of UDP-glucose, which repositions the UgtP protein so that it can inhibit assembly of the preseptal FtsZ ring (23, 24). This delays the onset of division and explains why cells grown under nutrient-rich conditions become longer than cells grown in nutrient-poor medium. The length of E. coli is influenced by a similar mechanism though the identity and function of the effector protein(s) have yet to be described (23, 25, 26). This conceptually simple method of regulating cell length came as a surprise, as were the identities of the proteins involved. The fact that such a pathway exists indicates that bacteria regulate cell shape at a finer level than has been appreciated, and the unexpected nature of the mechanism suggests that additional morphological control circuits await discovery.
Most proteins and pathways that affect bacterial shape have been discovered accidently while studying something else. A more intentional approach would be better for unearthing mutants that affect shape, given the likely existence of unknown mechanisms for morphological fine-tuning. However, despite what are often dramatic alterations in shape, most known morphological mutants grow at nearly normal rates and suffer few or no negative consequences in laboratory culture. Mutants that deviate only slightly from wild-type morphology have, as expected, growth characteristics even more like their parents. These traits make it difficult to detect, select, and study mutations that alter cell shape, especially when the differences are minor or transient. Searching for such mutants by manual (or even automated) microscopy, the usual and most straightforward approach, is tedious, time-consuming, and fraught with subjective error. Thus, if we are to understand more about how bacteria create, maintain, and modify their shapes, then we need improved tools to identify mutants with modest morphological phenotypes.
One procedure that addresses the above shortcomings is fluorescence-activated cell sorting (FACS). Its most common use in bacteriology has been to determine if organisms are dead or alive, to detect cells bearing specific antigens, or to sort for cells that exhibit certain physiological traits (27–29). For example, FACS has been used to select E. coli cells harboring cloned genes that regulate the production of large amounts of specific peptides (30) or to isolate rifampin-resistant mutants (31). However, FACS and flow cytometry have been used only sparingly to detect morphological differences among bacteria. Exceptions are the use of flow cytometry to measure cell elongation due to the inhibition of cell division (7, 32), to distinguish between spiral and straight forms of Campylobacter jejuni (33), and to investigate the relationship of cell size and DNA replication (26, 34). FACS therefore remains an underutilized but potentially powerful tool for measuring, separating, and collecting individual cells based on differences in fluorescence or morphology.
Previously, we reported that flow cytometry could detect small shape alterations among stained bacteria (35). Here, we demonstrate that flow cytometry can detect morphological differences among unstained live E. coli cells and also show that FACS can capture subpopulations that differ in shape. As proof of principle, we optimized FACS to identify and isolate mutants that suppress the morphological defects of highly aberrant E. coli strains, thus proving that the procedure is a valuable addition to the morphologist's toolbox. In addition, the types of mutants isolated indicate that there must be unexplored mechanisms that modify or regulate bacterial shape. Specifically, our current results indicate that AmpH and MepA, poorly studied peptidoglycan endopeptidases, play an integral morphological role, as do other enzymes of this class.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Bacterial strains are listed in Table 1. The parental strain was E. coli CS109 (W1485F− rpoS rph-1) (36). The shape suppressor strains generated in this work were ALM10 and LCM1 (independently derived from CS315-1K), LCM2 (derived from CS446-1K), and LCM3 (derived from CS449-2K). Deletions marked with kanamycin cassettes flanked by res sites were cured by using the plasmid RP4 ParA resolvase (37). The presence of each deletion was confirmed by diagnostic PCR. Deletions of ampH and mepA were introduced into strains by P1 transduction (40). Plasmid pLP8 was the vector for all plasmids constructed for this work (41) and contains the Plac-lacIq promoter and a kanamycin resistance cassette. The ampH gene was cloned into the multicloning site of pLP8 to create plasmid pLCM1. Site-directed mutagenesis of pLCM1 was employed to insert point mutations in the active site serine of AmpH, thereby creating pMEL1 with an S-to-A change at position 87 encoded by ampH (ampHS87A) and pMEL2 (ampHS87G).
Table 1.
Strains
| Strain | Genotype | Reference or source |
|---|---|---|
| CS109 | W1485F− rpoS rph-1 | 36 |
| ALM10-1K | CS315-1K, selected for cell shape suppression | This work |
| ALM10 | ALM10-1K with dacB::res | This work |
| CS15-3 | CS109 ampH::kan | 37 |
| CS315-1K | CS109 dacA::res pbpG::res dacB::kan | 37 |
| CS315-1 | CS109 dacA::res pbpG::res dacB::res | 37 |
| CS446-1K | CS109 dacA::res pbpG::kan dacB::res dacC::res | 37 |
| CS448-3K | CS109 dacA::res pbpG::kan dacB::res ampH::res | 37 |
| CS449-2K | CS109 dacA::res pbpG::kan dacB::res ampC::res | 37 |
| CS456-1K | CS109 dacA::res pbpG::res dacB::res mrcA::kan | 38 |
| CS534-1K | CS109 dacA::res pbpG::res dacB::kan ampC::res ampH::res | 37 |
| CS545-1K | CS109 dacA::res pbpG::res dacB::res dacC::res mrcA::kan | 38 |
| CS703-1K | CS109 mrcA::Kan1-862 ampC::res459-2 dacC::res520-1 pbpG::res501-1 ampH::res480-2 dacA::res512-1 dacB::res516-2 | 38 |
| EC100 | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara leu)7697 galU galK λ− rpsL nupG | Epicentre |
| LCM1-K | CS315-1K cell shape suppressor | This work |
| LCM1 | LCM1-K dacB::res | This work |
| LCM2-K | CS446-1K cell shape suppressor | This work |
| LCM2 | LCM2-K pbpG::res | This work |
| LCM3-K | CS449-2K cell shape suppressor | This work |
| LCM3 | LCM3-K pbpG::res | This work |
| LCM5-K | ALM10 ampH::kan | This work |
| LCM5 | ALM10 ampH::res | This work |
| LCM10 | ALM10 mrcA/yrfE/yrfF::Res460-1 | This work |
| LCM10-K | ALM10 mrcA/yrfE/yrfF::Kan460-1 | This work |
| MC1061 ΔmepA | araD139 Δ(ara leu)7697 ΔlacX74 galU galK hsrR hsrM rpsL mepA::kan [hsr− hsm+] | 39 |
| MEL1 | ALM10 mepA::kan | This work |
| MEL2 | ALM10 ampH::kan mepA::kan (from LCM5) | This work |
Media and growth conditions.
Bacteria were incubated at 37°C in Luria-Bertani (LB) agar plates or broth, M9 medium (ATCC medium 1281), or Trypticase soy (T-soy) broth (Difco). Before use, T-soy broth was cleared by filtration through a 0.22-μm-pore-size filter (GP Express Plus, Millipore, Billerica, MA). Prior to FACS analysis, cells were washed three times in phosphate-buffered saline (PBS) (Sigma Chemical Co., St. Louis, MO) that had been clarified by filtration, after which the cells were resuspended in 1× PBS. Kanamycin (40 μg/ml final), chloramphenicol (20 μg/ml), and ampicillin (100 μ/ml) (Sigma) were added when appropriate. Aztreonam (5 μg/ml final) (Bristol Myers Squibb) was added to inhibit cell division, forcing cells to grow as filaments so that minor shape defects could be accentuated for microscopic analysis. When required, glucose (0.4%) was added to the medium to suppress gene expression from plasmids.
FACS and flow cytometry.
Because each FACS machine has different capabilities (42), the settings for analysis and sorting must be determined empirically. Here, cell sorting was performed by using a BD FACSAria machine (Becton, Dickinson, Franklin Lakes, NJ). FACSDiva software was used to analyze the results. Flow cytometry of cells without sorting was performed on a BD FACSCalibur machine. The results were analyzed with CELLQUEST software. The FACS machine was run for 5 min with filtered distilled water to remove residual sodium azide before the sorting and analytical parameters were set. The machine was sterilized with 10% bleach for 5 min prior to and after each use, and bleach was removed by running filtered distilled water through the machine for 5 to 10 min afterwards.
FACS procedure for isolating E. coli suppressor strain ALM10.
A single colony of CS315-1K from an LB plate was inoculated into 5 ml of filtered LB broth containing kanamycin (40 μg/ml) and incubated overnight at 37°C. The overnight culture was diluted 1:100 into 30 ml of filtered LB broth with kanamycin and incubated at 37°C until the culture reached an optical density at 600 nm (OD600) of 0.35 to 0.40. Cells were washed three times by centrifugation in filtered PBS and diluted into filtered PBS to give a final concentration of ∼106 cells/ml; this solution was used for FACS and flow cytometry. For determining shape parameters, 20,000 to 50,000 cells were analyzed for each sample. The resulting dot plot for the parental strain E. coli CS109 was used to define the sorting gate that most closely represented the spread of normally shaped cells. When normally shaped cells were enriched from mutants, 104 to 106 cells were collected from within the gate, the cells were pelleted by centrifugation, the supernatant was removed, and the cells were resuspended in 5 ml of filtered LB broth containing kanamycin. Kanamycin was included in the growth medium to select for CS315-1K, so as to prevent contamination by the unmarked CS109 parent. Sorted cells were incubated overnight at 37°C, diluted 1:100 into 30 ml of filtered LB broth, and incubated at 37°C until the culture reached an OD600 of 0.35 to 0.40. Simultaneously, an overnight culture of the strain being enriched but which had not passed through FACS was diluted 1:100 into filtered LB broth containing kanamycin and incubated in the same way; this culture served as a control to be sure that the FACS procedure and not simply prolonged growth was the source of any suppressors. The enrichment protocol was repeated once per day for 5 to 20 enrichment iterations. Each iteration began with the culture that had been collected and incubated the previous day.
FACS procedure for isolating E. coli suppressor strains LCM1, LCM2, and LCM3.
Cells were incubated overnight at 37°C in 4 ml of LB broth with appropriate antibiotics. The cultures were then diluted 1:100 into 4 ml of LB broth and incubated at 37°C until the OD600 reached 0.4 to 0.5. Cells (1 ml) were washed twice with filtered PBS and resuspended in 1 ml of filtered PBS. These suspensions were normalized to an OD600 of 0.4, diluted 1:10 in PBS, and analyzed with a BD FACSCalibur machine or sorted by using a BD FACSAria machine (University of Arkansas for Medical Sciences [UAMS] Flow Cytometry Core Facility). Detection voltages were adjusted until the background signal (generated by particles smaller than bacterial cells) was minimized so that only bacterial cells were being measured. Preliminary analysis of 20,000 cells was performed using the BD FACSCalibur. Sorting parameters were defined by comparing the dot plot of the CS109 population distribution with that of strains containing PBP deletions. Cells with normal rod shapes were defined by gate P1 because this area represented ∼40 to 60% of cells in the wild-type population but included only ∼10 to 17% of cells from the mutant populations. This difference formed the basis for enriching more normally shaped cells from the mutant populations.
Cells from gate P1 were sorted into a conical tube until 106 cells were collected. The cells were pelleted by centrifugation for 10 min at 26,000 × g, resuspended in 3 ml of LB broth containing kanamycin (40 μg/ml), and incubated overnight at 37°C. This culture was diluted 1:100 into 4 ml of LB broth and incubated at 37°C until the OD600 reached 0.4 to 0.5. Cultures were prepared for FACS and sorted as described above. This sequential process of sorting, incubation, and enrichment was continued until a cell population was generated that exhibited a near-normal population distribution as defined by that of the parent strain CS109. FACS results were analyzed by using WinMIDI, version 2.9, software and FlowJo for Windows, version 7.6.3.
Microscopy.
Cells were visualized and photographed as described previously (41). Briefly, cells in mid-exponential phase were immobilized on microscopic slides covered with a thin layer of 1% agarose and viewed by using a Zeiss Axio Imager.Z1 microscope fitted with a 100× EC Plan-Neofluar oil immersion objective (numerical aperture, 1.3). Cells were photographed with an Axiocam MRm or SenSys cold charge-coupled-device (CCCD) camera. Cells were categorized as follows: (i) normal if they deviated little or not at all from the uniform rod shape of wild-type cells; (ii) abnormal if they displayed bends, kinks, bumps, or small bulbous protrusions; or (iii) branched if they had one or more elongated protrusions that accounted for one-third or more of the total cell volume.
DNA purification and PCR.
DNA was extracted by using a Qiagen DNeasy Tissue Kit (Qiagen) and was stored at −20°C. DNA primers (Table 2) were synthesized by MWG Biotech and used to determine the presence or absence of genes encoding penicillin binding proteins (PBPs) 4, 5, 6, 6b, or 7 (dacA, dacB, dacC, dacD, or pbpG, respectively) by diagnostic PCR. PCR was performed by using a Failsafe PCR kit (EpiCentre) and a 2400 Gene Amp thermal cycler (Perkin-Elmer). Incubation temperatures and duration were matched to individual melting points of each primer, and the resulting products were analyzed by gel electrophoresis. PCR products were purified from gel slices by using a Qiagen QIAquick Gel Extraction Kit and were sequenced by the UAMS DNA Core Sequencing facility.
Table 2.
Primers
| Primer | Sequence |
|---|---|
| dacA 5′ | GTAGTATGACGGCTCGATTCC |
| dacA 3′ | GCCGGGGTTTCTTTTTGACT |
| dacB 5′ | GCTTAGTATATGGGGACGGA |
| dacB 3′ | CACGGATGTCGTAGTTCA |
| dacC 5′ | GTGCGTTATTAATCACCA |
| dacC 3′ | TTTCGCGCCATCCGGTTA |
| dacD 5′ | CCAGTCTGCTGCAAAAGA |
| dacD 3′ | TCCGTAAAAAGAACAGCT |
| pbpG 5′ | CAGAGTATCGCCATCCGA |
| pbpG 3′ | GCGCGTGAACCACTATCT |
| ampH_HindIII_REV | CAGAGAAGCTTTCAGGACGCGGGGATAACCA |
| ampH_EcoRI_FWD | CTTCAGAATTCAGGAGGACACCACCGTTGAAACGTAGTCTGCT |
| ampH_ala_Fwd | CGGTCGTGCGTATTGCTGCACTCACCAAGCTAATGACC |
| ampH_ala_Rev | GGTCATTAGCTTGGTGAGTGCAGCAATACGCACGACCG |
| ampH_gly_Fwd | CGGTCGTGCGTATTGCTGGTCTCACCAAGCTAATGACC |
| ampH_gly_Rev | GGTCATTAGCTTGGTGAGACCAGCAATACGCACGACCG |
Plasmid construction.
Plasmid pLCM1 was created as follows. The wild-type ampH gene was amplified using the primers ampH_EcoRI_FWD and ampH_HindIII_REV (Table 2). The resulting PCR product and plasmid pLP8 were digested with EcoRI and HindIII (New England BioLabs). The fragments were purified by agarose gel extraction using a QIAquick gel extraction kit and were ligated to one another by using New England BioLabs Quick ligase. Ligation reaction products were transformed into chemically competent EC100 cells, and transformants were selected on LB agar plates containing kanamycin. The ampH gene in plasmids obtained from selected transformants was sequenced to rule out the presence of unwanted mutations.
Plasmids pMEL1 and pMEL2 were created by site-directed mutagenesis of pLCM1. Point mutations in the active site of ampH were constructed using a QuikChange Lightning Site-directed Mutagenesis kit (Agilent Technologies). Primers were designed to replace the serine at position 87 with either alanine or glycine (Table 2) (ampH_ala_Fwd, ampH_ala_Rev, ampH_gly_Fwd, and ampH_gly_Rev). pLCM1 was amplified according to the manufacturer's instructions for a 9-min extension time. Following PCR amplification, plasmids were digested with DpnI for 1 h at 37°C and transformed into chemically competent EC100 cells, and transformants were selected on LB agar plates containing kanamycin (40 μg/ml) and glucose (0.4%). The presence of point mutations was verified by sequencing.
RESULTS
FACS protocol to isolate cell shape suppressors.
Previously, we established that flow cytometry could detect and quantify cell shape differences between the rod-shaped cells of E. coli CS109 and the seriously deformed cells of E. coli CS315-1K, a mutant lacking PBPs 4, 5, and 7 (35). However, those experiments were performed with cells that had been stained with a fluorescent dye and fixed before analysis. We wished to develop and apply a fluorescence-activated cell sorting (FACS) procedure to sort and recover unstained live cells to enrich bacterial mutants having altered shapes.
We first confirmed that flow cytometry detected shape differences between the two E. coli strains described previously (35). When E. coli CS109 and CS315-1K were grown in filtered LB broth and analyzed in the absence of added fluorophore, the two populations could be distinguished from one another by examining the distribution of forward scattered light (Fig. 1A; see also Fig. S1A in the supplemental material). (Although side scatter is said to reflect shape complexity [43], we found that this measure varied less than forward scatter among E. coli strains, so the results presented here consist only of forward scatter.) Cells of the parent strain CS109 clustered in a symmetrical peak on a three-dimensional graph (data not shown) and produced a tight symmetrical peak on the dot array of the two-dimensional scatter plot, with few cells falling outside this range (Fig. 1A; see also Fig. S1A). In contrast, the CS315-1K population was skewed to the right on the x axis (forward-scattered light), indicating that the cells were aberrantly shaped and possibly larger. Approximately 30% of CS315-1K cells fell outside the curve defined by the CS109 parent population. Also, a larger portion of the CS315-1K population displayed a difference in side-scattered light (the y axis) (Fig. 1D and 1E; see also Fig. S1A), presumably due to less uniform light scattering from branched, curved, and aberrant cells. These flow cytometry results correlated with our visual evaluation that 50 to 60% of CS315-1K cells were abnormal or branched. The difference in the two percentages of aberrantly shaped cells may be due to the addition of aztreonam to the samples we observed by microscopy, a treatment that inhibits cell division and exaggerates otherwise minor deformities so that odd shapes can be observed more clearly. When cells were counted without the addition of aztreonam, the population of aberrantly shaped cells was between 30 and 40% (data not shown), similar to the flow cytometry results. Microscopically, the aberrant CS315-1K cells were longer and wider than average and displayed one or more protrusions or branches (Fig. 1B and C). In addition, many cells, while having nearly normal morphologies, were not perfectly symmetrical or uniform though minor differences were sometimes hard to distinguish visually.
Fig 1.

Flow cytometry reveals shape defects of CS315. (A) Histograms of forward scatter for cells of CS109 (gray-filled peak) and CS315-1K (black line). (B and C) Phase-contrast microscopy of cells from CS109 (B) and CS315-1K (C) cultures that were forced to elongate in the presence of aztreonam to accentuate shape defects. (D) Scatter plot of 50,000 CS109 cells. The gray box (P1) delineates the region selected for sorting and contained ∼40% of the population. (E) Scatter plot of CS315-1K. The gray box (P1) corresponds to the selected population in panel D. Cells were collected from this gate to enrich for mutants that had suppressed the shape defects of CS315-1K.
Conditions for analyzing and sorting bacterial cells by flow cytometry and FACS were optimized by varying the growth medium, temperature, and stage at which cells were harvested (see Fig. S1 and S2 in the supplemental material). The optimum procedure is described in Materials and Methods. Briefly, the largest shape differences among mutants were detected when cells were incubated at 37°C in filtered LB broth until the culture reached exponential phase and an OD600 of 0.35 to 0.40 (see Fig. S1).
Isolation of shape suppressor mutant ALM10.
We surmised that normally shaped cells might arise in a population of shape-defective E. coli due to the accumulation of one or more suppressor mutations. The nature of these mutations should help define the pathways that influence shape determination in rod-shaped bacteria. Wild-type E. coli CS109 and mutant CS315-1K cells were analyzed by FACS, and the shape distributions were superimposed. A FACS gate was drawn to encompass the CS109 plot area that had a high density of cells but that was also most distinct from the plot of CS315-1K (Fig. 2). Cells falling into this zone (gate) should have normal or near-normal rod shapes. CS315-1K cells were sorted by FACS, and 106 cells were collected from within this “normal” gate and amplified by overnight growth. On each subsequent day the culture was subjected to one additional round of FACS, with enrichment of those cells falling into the normal gate. Such sequential cultures were sorted and recovered until a new subpopulation arose that displayed a shape distribution nearly identical to that of wild-type E. coli CS109. Depending on how the normal gate was selected (see below), this enrichment procedure required 4 to 15 iterations. At each time point we treated a parallel culture of CS315-1K similarly except that the cells were never sorted by FACS. This served as a control to ensure that any changes in the shape distribution were due to FACS enrichment and did not arise as an artifact of repeated rounds of growth in liquid culture.
Fig 2.

Selection of the region to be used for sorting wild-type-shaped cells from a population of CS315-1K. The dot plots of the parental strain CS109 (A) and the PBP mutant CS315-1K (B) were compared. Side scatter was plotted on the y axis, and forward scatter was plotted on the x axis. Each point represents a single cell. A gate (black box) was drawn around a subpopulation in the lower left quadrant on the dot plot of CS109. CS315-1K cells that fell into this area were sorted and collected to enrich for normally shaped cells.
After the first round of sorting and regrowth, the dot plot distribution of the first subpopulation of E. coli CS315-1K (referred to as CS315-1K-1F, for 1 FACS sort) became less uniform, and the histogram of the population shifted much further to the right (see Fig. S3 in the supplemental material). Thus, the first generation of sorted cells displayed a shape distribution that was more abnormal than the original mutant from which it was derived. This was counterintuitive because we expected the enrichment procedure to produce cells that were shaped more like those of the CS109 parent. However, this phenomenon repeated itself for every misshapen strain we sorted by FACS, indicating that it was a natural consequence of the sorting process (see also Fig. S3 in the supplemental material). We cannot, as yet, explain this phenomenon.
In contrast to the effects observed after the first iteration, cell populations subjected to further sorting steps became more homogeneous, and the histogram moved leftward, indicating that the shapes of an increasing proportion of CS315-1K cells were becoming more like those of the wild-type parent. This shape change became most apparent after four or five enrichment cycles, and it was at this point that the shape distribution began to appear less aberrant than that of the original CS315-1K mutant (see Fig. S3 in the supplemental material). However, at this point it was difficult to verify the existence of a definitive shape change by visual microscopic examination (data not shown).
After 15 rounds of FACS, the enriched population (CS315-1K-15F) displayed a shape histogram that was very close to that of the wild-type strain (Fig. 3; see also Fig. S3). Microscopically, the majority of the cells had normal rod shapes that were comparable to those of CS109 cells (Fig. 3F). The uniformity of cell shapes in the CS315-1K-15F population was most obvious when the cells were filamented by the addition of aztreonam to accentuate small shape deformities; very few cells were branched, bent, or kinked (Fig. 3F), as would be expected for cells that had regained a normal rod shape. Although the CS315-1K-15F population still contained a small minority of branched or misshapen cells, which were rare or nonexistent in CS109, the process had enriched a subpopulation of cells having one or more mutations that suppressed the aberrant shapes of CS315-1K.
Fig 3.

Enrichment of normally shaped cells from E. coli CS315-1K after 15 rounds of FACS sorting. (A to C) Histograms of forward light scatter CS109 (A), CS315-1K (B), and CS315-1K-15F (C). This was the population distribution after 15 rounds of enrichment sorting, and it was from this population that E. coli ALM10 was isolated. (D to F) Phase-contrast microscopy of cells treated with aztreonam (5 μg/ml) filaments the cells and accentuates any abnormalities: CS109 (D), CS315-1K (E), and CS315-1K-15F (F). The lack of branching in panel F indicates that the sorted population was enriched for normal rod-shaped cells.
In tandem with the stages of FACS enrichment, a control CS315-1K culture was subjected to the same sequence of procedures but was not sorted. Surprisingly, when these tandem cultures were analyzed by flow cytometry, not only did each population not regain a wild-type morphology, but also with each passage the cell shapes became progressively and dramatically more irregular than those of the original CS315-1K strain (see Fig. S4 in the supplemental material). This verified that the near-normal shapes of the sorted cell population had originated during enrichment by FACS and did not arise simply because the more normally shaped cells had overgrown the mutant strain during continued incubation in liquid culture.
Even though in the majority, the population of putative suppressor mutants was evidently still contaminated by at least a few abnormally shaped cells. To isolate a pure culture of one suppressor candidate, CS315-1K-15F cells were streaked for isolation, isolated colonies were inoculated into filtered LB broth, and their shape distributions were determined by flow cytometry. Microscopic examination of the CS315-1K-15F population indicated that about two-thirds of the cells were morphologically normal rods, leading us to expect that a similar fraction of purified colonies would produce FACS distributions nearly indistinguishable from those of CS109. In fact, the shape distributions of about two-thirds of the screened colonies were similar to those of CS109 (see Fig. S5 in the supplemental material), with some distributions even more compact and uniform than those of CS109 (see, Fig. S5, colony 1). A single colony of this type was chosen as a representative suppressor mutant and was renamed E. coli ALM10. The remaining third of the colonies yielded populations whose shape distributions were virtually identical to those of the shape-defective mutant CS315-1K (see, Fig. S5, colony 2), indicating that the enriched CS315-1K-15F population was not pure but still contained some misshapen cells.
The putative shape suppressor E. coli ALM10 was inspected by microscopy after growth in the presence of aztreonam to amplify shape defects. ALM10 formed straight filaments of uniform diameter like those formed by strain CS109. In contrast, the unsorted parent, CS315-1K, formed filaments with irregular diameters and branches (Fig. 3; see also Fig. S6 in the supplemental material). The results confirmed that ALM10 cells had regained a morphologically normal rod shape. The growth rate of ALM10 was more similar to that of CS315 than to that of CS109 (see Fig. S7) though none of these growth rates differed significantly from one another. To ensure that ALM10 was not a contaminant, such as might occur if the original parent CS109 had been reintroduced during FACS enrichment, diagnostic PCR was performed, and we confirmed that ALM10 retained the three mutations (ΔdacB, ΔdacA, and ΔpbpG) present in CS315-1K. Thus, the mutations responsible for the shape defects of CS315-1K were still present in ALM10, indicating that the latter strain must carry one or more suppressor mutations that can reverse the morphological effects caused by the absence of PBPs 4, 5, and 7.
Isolation of shape suppressor mutants LCM1, LCM2, and LCM3.
The procedure described above required 15 consecutive rounds of FACS before a population arose that was highly enriched for normally sized, rod-shaped cells. To isolate suppressors in fewer enrichment steps, we maximized the selection of normally shaped cells and minimized contamination by aberrantly shaped cells by using a more circumscribed collection gate. This gate, P1, was located in the upper left portion of the dot plot (Fig. 1D and E). As before, cells falling within the gate were enriched by sequential rounds of sorting and growth.
After the first sorting the population distribution shifted to the right on the x axis, indicating that cell shape was more diverse in this first subpopulation (Fig. 4B). However, during subsequent sorts the histogram shifted rapidly leftward and became more like that generated by wild-type cells (Fig. 4C to E). By using the revised selection gate, only four rounds of FACS enrichment were required to bring the population distribution of CS315-1K into line with that of CS109 (Fig. 4; Table 3, compare experiment 2 with experiment 1), indicating that the population was dominated by cells with more normal shapes. More than half of the colonies isolated from this population produced cells whose shape distributions were virtually identical to those of CS109 (Fig. 5A, C, and E), while cells from the remaining colonies exhibited the abnormal shape distribution of the CS315-1K parent (Fig. 5B and D). Cells with CS109-like distributions had normal rod shapes upon visual inspection (data not shown). Also, when putative suppressor mutants were filamented in the presence of aztreonam, the cells produced uniform filaments with no (or very few) branches or protrusions, like those of similarly treated CS109 cells (Fig. 6B and J). In contrast, cells from the nonsorted CS315-1K control were highly abnormal and displayed extreme shape defects (data not shown; see also Fig. S8 in the supplemental material). One suppressor candidate was chosen for further analysis and designated E. coli LCM1. Using the more restrictive sorting gate, two additional enrichments were performed by starting with E. coli CS446-1K (Fig. 6G and 7A) and CS449-2K (Fig. 6H and 7B). After only four enrichment steps, CS446-1K produced the suppressor mutant LCM2 (Fig. 6C and K and 7A; Table 3, experiment 5) and enrichment from CS449-2K produced the suppressor LCM3 (Fig. 6D and L and 7B; Table 3, experiment 7). All three suppressor strains (LCM1, LCM2, and LCM3) grew at essentially the same rates as their parents (see Fig. S7A to C in the supplemental material), and the generation times did not vary significantly from the CS109 generation time (see Fig. S7D), indicating that changes in growth rate were not responsible for shape suppression.
Fig 4.
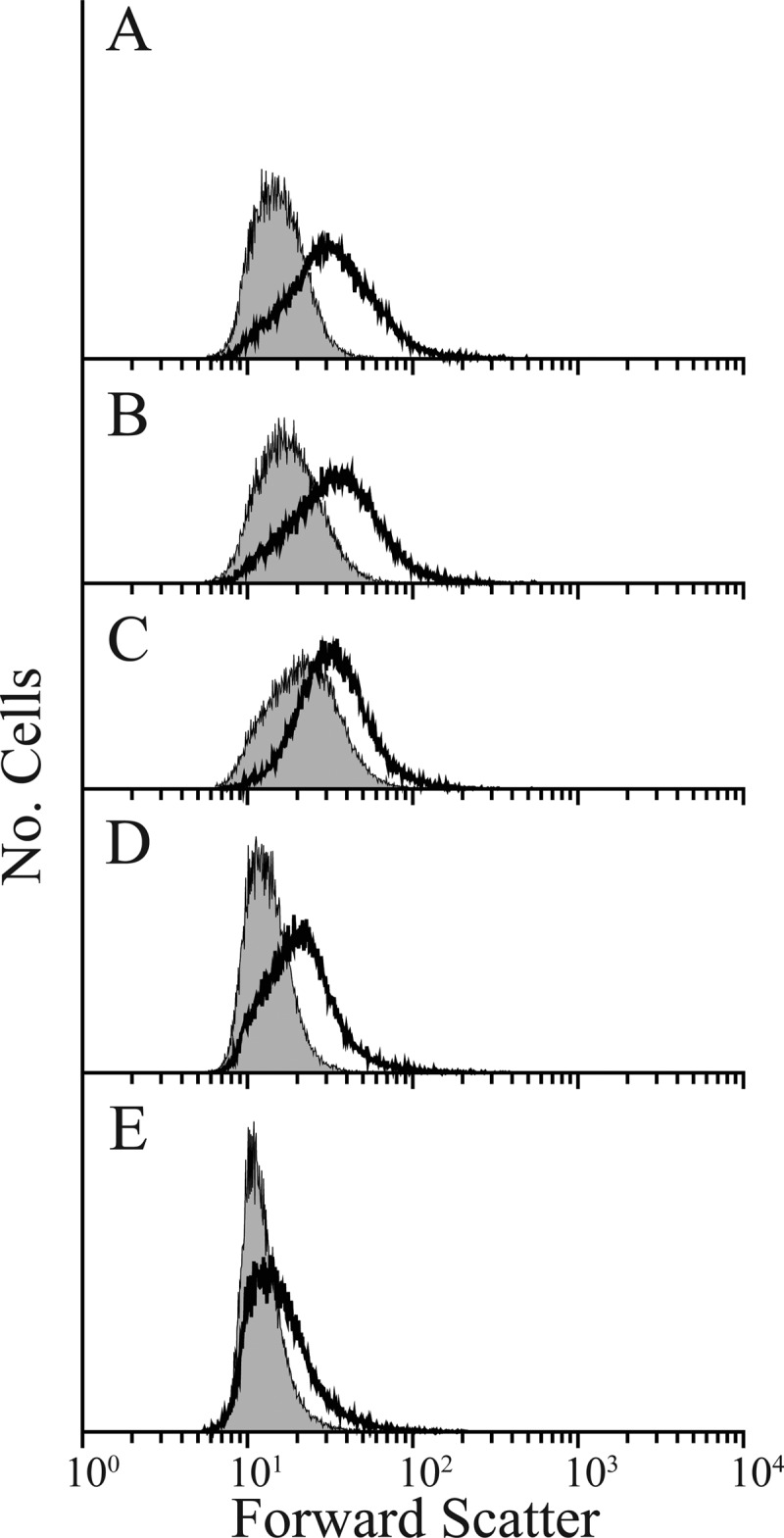
Enrichment of mutant LCM1, which suppresses abnormal cell shape after four iterations of FACS. The histogram distribution of forward light scatter from wild-type E. coli CS109 is represented on every graph (gray-filled peak). The black line in each graph reflects the shape distribution of E. coli CS315-1K at the start of the experiment (A) and after one (B), two (C), three (D), and four (E) enrichments by FACS.
Table 3.
Principle PBP mutants and the number of FACS iterations required to isolate cell shape suppressors
| Expt no.a | Strain | PBPs deleted | No. of FACS iterations | Suppressor isolatedb |
|---|---|---|---|---|
| 1 | CS315 | 4, 5, 7 | 15 | + (ALM10) |
| 2 | CS315 | 4, 5, 7 | 4 | + (LCM1) |
| 3 | CS315 | 4, 5, 7 | 6 | + |
| 4 | CS315 | 4, 5, 7 | 5 | + |
| 5 | CS446 | 4, 5, 6, 7 | 4 | + (LCM2) |
| 6 | CS448 | 4, 5, 7, AmpH | 7 | − |
| 7 | CS449 | 4, 5, 7, AmpC | 4 | + (LCM3) |
| 8 | CS456 | 1A, 4, 5, 7 | 6 | − |
| 9 | CS534 | 4, 5, 7, AmpC, AmpH | 9 | − |
| 10 | CS545 | 1A, 4, 5, 6, 7 | 9 | − |
| 11 | CS703 | 1A, 4, 5, 6, 7, AmpC, AmpH | 4 | − |
| 12 | CS703 | 1A, 4, 5, 6, 7, AmpC, AmpH | 15 | − |
Each experiment is independent experiment. The least restrictive sorting gate was used in experiment 1. All other experiments were performed with the more circumscribed and more efficient sorting gates described in the text.
+, shape suppressors were generated; −, shape suppressors were not generated. Strain names for suppressor mutants discussed in the text are given in parentheses.
Fig 5.
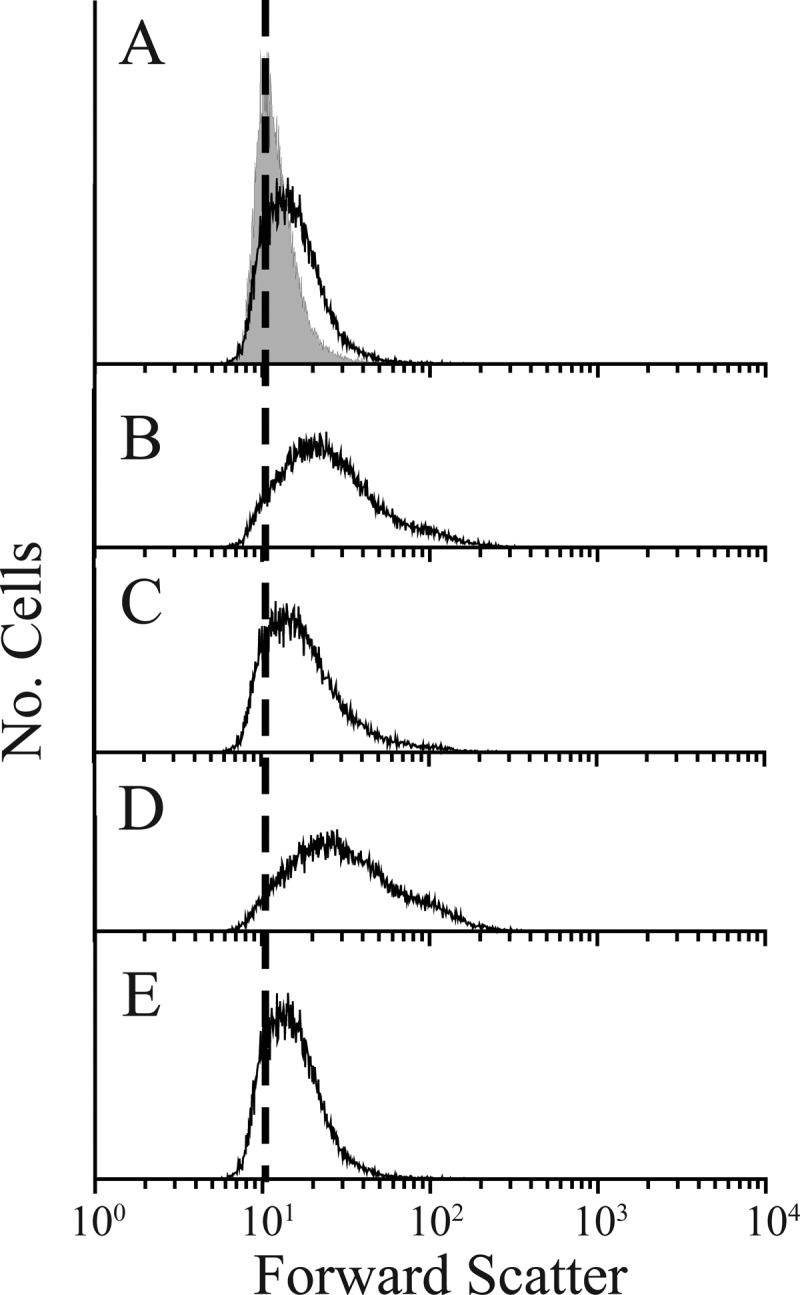
Cell shape distributions of individual isolates after enrichment for shape suppressor mutants from CS315-1K. These suppressor strains were suppressor 311 (A), suppressor 312 (B), suppressor 315 (C), suppressor 316 (D), and suppressor LCM1 (E). Overall, approximately half of the tested colonies recovered normal rod cell shape (exemplified by the strains depicted in panels A, C, and E). The S312 and S316 isolates remained abnormally shaped when grown in isolation.
Fig 6.

Cell shape distribution and microscopy of cell shape suppressor mutants isolated after FACS enrichment. (A to D) The gray-filled peaks represent the shape distribution of E. coli CS109 cells. The unfilled black line in each graph is the shape distribution of the following suppressor mutants: ALM10 (A), LCM1 (B), LCM2 (C), and LCM3 (D). (E to L) Phase-contrast microscopy of the following suppressor mutants (cell division was inhibited by adding 5 μg/ml aztreonam, to enhance shape abnormalities): CS109 (E), CS315-1 (F), CS446-1 (G), CS449-2 (H), ALM10 (I), LCM1 (J), LCM2 (K), and LCM3 (L).
Fig 7.

Enrichment of mutants that suppress abnormal cell shape in strains lacking different combinations of PBPs. The upper graph in each column is the shape distribution of the parental strain CS109 (gray-filled peak) and of the mutant to be sorted (black line), as determined by flow cytometry on the first day of the experiment, prior to sorting. Distribution curves below the first graph in each column represent the shape distributions for each mutant as sorting progressed. The number attached to each line denotes the number of times the mutant had been sorted before being analyzed by flow cytometry. In each column, the vertical dotted line corresponds to the location of the distribution peak of the parent, CS109. The following mutants were sorted (PBPs deleted from each strain are in parentheses): CS446-1K (PBPs 4, 5, 6, and 7) (A), CS449-2K (PBPs 4, 5, and 7 and AmpC) (B), CS703-1K (PBPs 1A, 4, 5, 6, and 7, AmpC, and AmpH) (C), CS456-1K (PBPs 1A, 4, 5, and 7) (D), and CS448-3K (PBPs 4, 5, and 7 and AmpH) (E). During this experiment, the shape distribution of the CS109 wild-type strain varied somewhat from the peak position shown on day 1 (which is the reason we run this control every time). Therefore, to illustrate the failure of CS448-3K to return to normal shape, the CS109 distribution (gray-filled peak) is presented for the third and sixth sorts.
Thus, the revised procedure reduced the number of required enrichment steps from 15 to 4 rounds of sorting for isolating candidate suppressor mutants and generated three additional independent shape suppressor mutants.
Shape suppression is limited to certain PBP mutants.
Shape suppressor mutants were generated first from E. coli CS315-1K, which lacks PBPs 4, 5, and 7. However, removing additional PBPs from this strain produces mutants with greater numbers of morphological abnormalities (37). We hypothesized that three scenarios might occur if we attempted to enrich for shape suppressors by starting with a mutant lacking additional PBPs. First, the same suppressor mutation(s) might be isolated in each case (e.g., if the suppressors bypassed the need for low-molecular-weight [LMW] PBPs by creating an alternate mechanism for controlling cell shape). Second, new mutations might be isolated (e.g., if the suppressors created different bypass mechanisms). Third, by deleting increasing numbers of PBPs, there might come a point at which no further suppressor mutations could be isolated (e.g., if one or more LMW PBPs was required for shape suppression).
To determine which of these scenarios might apply, we attempted to use FACS to enrich shape suppressor mutants from E. coli CS703-1K, which lacks seven PBPs (the high-molecular-weight [HMW] PBP1A, the two d,d-carboxypeptidase PBPs 5 and 6, the three endopeptidase PBPs 4 and 7 and AmpH, and the class C β-lactamase AmpC). No near-normal suppressor mutants were isolated from CS703-1K even after 15 consecutive rounds of FACS enrichment (Fig. 7C; Table 3, experiments 11 and 12). In contrast, as described above, suppressors were isolated from CS315-1K after only four rounds of enrichment. The results suggested that CS703-1K was unable to compensate for the loss of one or more of the four PBPs beyond those absent from CS315-1K. We also failed to isolate suppressors from strains CS534-1K and CS545-1K, which lacked five of these PBPs in different combinations (Table 3, experiments 9 and 10). Thus, one of four PBPs (PBP1A, PBP6, AmpC, or AmpH) appeared to be involved in the shape suppression pathway. We therefore attempted to enrich suppressor mutants from strains lacking one additional PBP in addition to the PBPs absent from CS315-1K. In two cases, E. coli CS446-1K (ΔPBP6) (Table 3, experiment 5) and CS449-2K (ΔAmpC) (Table 3, experiment 7) shape suppressors were isolated easily after five rounds of sorting (Fig. 7A and B, respectively). Thus, neither PBP6 nor AmpC was involved in generating shape suppressors. In contrast, in each of two other strains, E. coli CS456-1K (ΔPBP1A) (Table 3, experiment 6) and CS448-3K (ΔAmpH) (Table 3, experiment 8), no suppressor mutants were isolated after four to six rounds of FACS enrichment (Fig. 7D and E, respectively).
We noted that the shape distribution of cells derived from CS456-1K (ΔPBP1A) shifted slightly toward the parental distribution after the final enrichment step (Fig. 7D). However, every colony isolated from this enriched population was composed of cells that were as aberrantly shaped as the starting PBP mutant (n = 15) (see Fig. S9 in the supplemental material for a representative example). Cells from strains lacking PBP1A are slightly smaller than those of the wild-type E. coli (44; also S. Kannan and K. D. Young, unpublished results), and this reduced cell size may explain why cells with deformed shapes had light-scattering properties similar to those of wild-type E. coli. In any event, the results support the contention that PBP1A and AmpH are active in the shape-suppressing pathway.
d,d-Endopeptidase activity is important for suppressing cell shape aberrations.
The first candidate that might play a role in shape suppression, PBP1A, is a bifunctional HMW PBP peptidoglycan polymerase and could perhaps alter bacterial morphology during the normal course of cell elongation. However, when we deleted mrcA, which encodes PBP1A, from ALM10 and the other suppressor strains, there was little effect on cell shape other than an ∼5% decrease in average cell width or length (data not shown; see also Fig. S10 in the supplemental material). These data are consistent with PBP1A being required during the generation of shape suppressors but not in the shape suppression pathway per se.
AmpH, the second candidate, is closely related to the well-characterized β-lactamase AmpC, and both proteins play some role in the genesis or maintenance of bacterial cell shape (45). When ampH was deleted from the suppressor strain ALM10, cells of the resulting strain, LCM5-K, were slightly more aberrant as measured by flow cytometry (see Fig. 9A; see also Fig. S10 in the supplemental material), indicating that the loss of AmpH partially reduced shape suppression in this strain. The morphological effect was more obvious when cells were treated with aztreonam for one mass doubling (Fig. 8), but the abnormalities were difficult to confirm by microscopy alone because the number of abnormalities was about the same in each strain (data not shown). The same subtle increase in cell shape abnormalities was also observed when ampH was deleted from the suppressor strains LCM1, LCM2, and LCM3 (data not shown). Even though the morphological effects of removing AmpH were fairly small, shape suppression was restored in LCM5 by expressing ampH in trans (Fig. 8B and C; see also Fig. S10C and D in the supplemental material). AmpH could exert its morphological effect by playing either an enzymatic or structural role. To distinguish between these alternatives, we replaced the conserved active-site serine and found that the inactive protein no longer suppressed shape defects when expressed in trans in LCM5 (Fig. 8D; see also Fig. S10E). This confirmed that AmpH was required for full shape suppression and indicated that the mechanism of suppression depended on the ability of AmpH to act on peptidoglycan side chains, either as an endopeptidase or d,d-carboxypeptidase.
Fig 9.
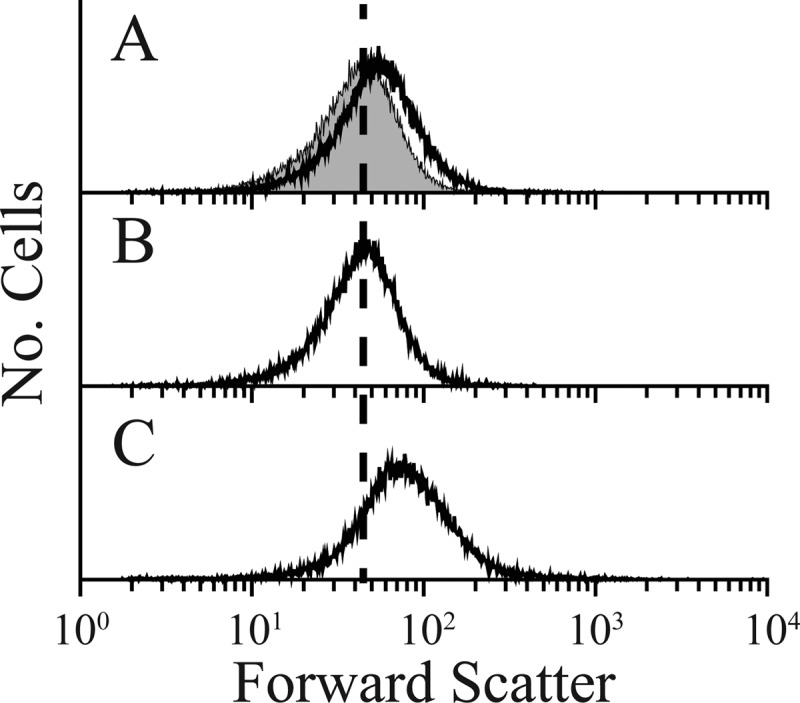
Simultaneous removal of the AmpH and MepA endopeptidases increases cell shape abnormalities. (A) Gray-filled histogram, ALM10 shape distribution; black curve, LCM5 (ALM10 ΔampH) shape distribution. (B) MEL1 (ALM10 ΔmepA) shape distribution. (C) MEL2 (ALM10 ΔampH ΔmepA) shape distribution. In all panels, the vertical dashed line marks the peak of the ALM10 cell shape distribution.
Fig 8.
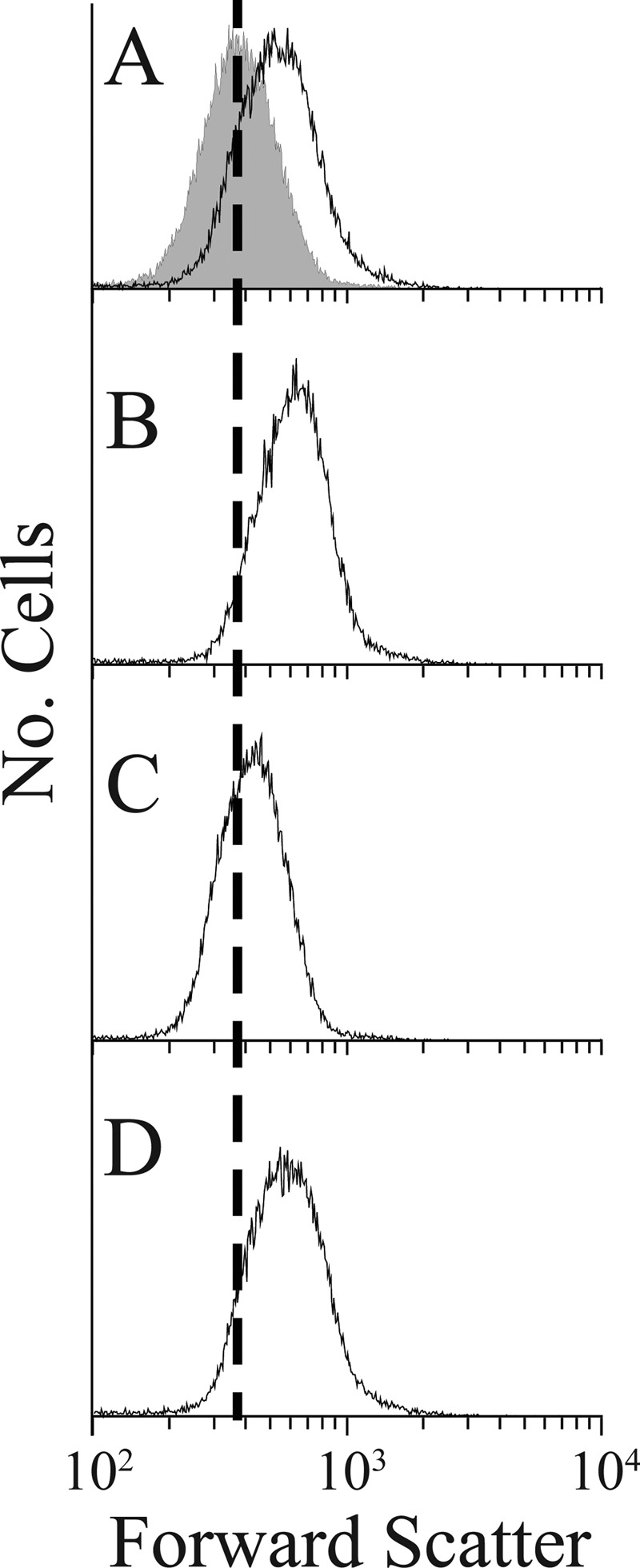
The absence of AmpH from the suppressor strain ALM10 increases cell shape deformities. In each graph, the gray-filled curve represents the normal shape distribution of strain ALM10, and the black unfilled curve represents shape distribution of strain LCM5 (ALM10 ΔampH) with or without additional cloned genes. The vertical dashed line marks the peak of the ALM10 distribution in each graph. In panel B, ALM10 also contains the empty plasmid vector pLP8. For each strain, cell division was inhibited by adding aztreonam (5 μg/ml) for one mass doubling prior to flow cytometry to accentuate any shape differences. Strains were as follows: ALM10 and LCM5 (A), LCM5 pLP8 (vector) (B), LCM5 pLCM1 (expressing wild type AmpH) (C), and LCM5 pMEL1 (expressing active-site mutant AmpHS87A) (D).
AmpH is a dual-function d,d-carboxypeptidase/d,d-endopeptidase, with the endopeptidase activity predominating, and its activity and substrate requirements are similar to those of the endopeptidases PBP4 and MepA (46). This suggested that shape suppression might depend on the presence of at least one of these endopeptidases. Of the four known peptidoglycan endopeptidases in E. coli (PBP4, PBP7, AmpH, and MepA), only MepA remained in strain LCM5 (ALM10 ampH), and its shape distribution was slightly more aberrant than that of its suppressor parent, ALM10 (in which both MepA and AmpH were active) (Fig. 8A), implying that AmpH had some effect on cell shape. On the other hand, the shape distribution of strain MEL1 (ALM10 mepA), in which AmpH remained active, was approximately the same as that of ALM10 and slightly less aberrant than that of LCM5, which lacked AmpH (compare Fig. 9A and B). These findings were supported by microscopy: MEL1 (ALM10 mepA) exhibited ∼8% abnormally shaped cells, whereas ALM10 and LCM5 (ALM10 ampH) contained ∼13% imperfectly shaped cells (data not shown). When both AmpH and MepA were removed from ALM10, the resulting strain (MEL2) exhibited an increased level of cell shape deformities, as measured by flow cytometry (Fig. 9C). This result was also confirmed by microscopy, in that MEL2 (lacking AmpH and MepA) exhibited ∼26% abnormally shaped cells whereas only ∼7% of ALM10 cells were imperfectly rod shaped. (For these purposes, an abnormally shaped cell was defined as one having an abnormal division site, branched ends, bent ends, or a curved or spiral shape, plus those that were abnormally long. Cells with slightly pointed ends, lysed cells, or those with blebs were not included.) Overproducing either AmpH or MepA in the original aberrant parent, CS315-1, did not reduce the shape defects of this strain, indicating that the strongest effects occurred in the suppressor strain. The results suggested that the presence of either AmpH or MepA could partially compensate for the loss of the other but that the loss of both seriously reversed the shape suppression pathway in strain ALM10. Overall, the results confirm that AmpH plays a role in shape maintenance and suggests that the d,d-endopeptidases in general are important contributors to cellular morphology.
Finally, the mutations present in each suppressor strain are under investigation and will be reported and characterized more fully at a later date. However, by genomic and directed DNA sequencing, we do know that the suppressor mutations are not in or near the ampH or mepA gene, nor are the mutations in or near any other known PBP gene (data not shown). Thus, the suppression mechanism does not involve changes in the promoter or coding regions of the PBP genes.
DISCUSSION
Cell sorting by FACS is a potentially powerful tool for isolating and characterizing mutants with novel morphological phenotypes. This technology enabled us to isolate the first-ever suppressor mutants from three aberrantly shaped E. coli strains lacking different sets of peptidoglycan-modifying enzymes. Preliminary characterization of these mutants led us to conclude that the endopeptidase AmpH plays a role in at least one morphological suppression pathway and that the loss of MepA undermines cell shape in this suppressor as well.
Cell sorting as an enrichment strategy.
The generation and study of bacterial morphological variation have been hampered somewhat by the technical bottleneck of manual microscopy. Even so, new and intriguing morphological mutants have been isolated by the sheer willpower of brute force visual screening (8, 9, 47), whereas others have been detected by culling through candidates collected after antibiotic selection, followed by semiautomated microscopy and analysis (48, 49). The latter technique could be adapted to search for morphological mutants, though as currently practiced only 48 isolates are visualized per individual microscope slide (49–51). In either case, whether by eye or enhanced by automation, screening by microscopy continues to be relatively arduous and entails a significant time investment. On the other hand, FACS can separate bacterial cells on the basis of light scatter or fluorescence and can therefore screen hundreds of thousands to millions of cells in less than an hour. Previously, we showed that flow cytometry could detect slight shape alterations in stained bacteria (35). Here, we employed the sorting capability of FACS to detect morphological differences among live organisms and to enrich for normally shaped cells from among a population of aberrantly shaped bacteria, illustrating the utility of this approach.
As powerful as these tools may be, the following caveats and warnings should be kept in mind when bacterial shape is analyzed by flow cytometry or when cells are sorted via FACS. First, cell sorting is an enrichment procedure and not an all-or-none selection process. Therefore, visualization by microscopy is still required to confirm the existence of appropriate mutants. Nonetheless, the technique increases the types of mutations that can be isolated and reduces enormously the numbers of candidates to be confirmed by microscopy. Second, what is being measured is light scatter, which is only an indirect measure of morphology because it is affected by cell length, width, gross shape, and possibly chemical composition. For example, harvesting cells at different points in the growth curve produced cells with different degrees of light scatter. Most of these differences were due to changes in cell length over the course of incubation, especially as the cells moved out of lag phase or into stationary phase, but alterations in light scatter also accompanied changes in optical density during the exponential phase as well. This means that experimental and reference strains must be collected under similar growth conditions, ideally on the same day from the same medium and at the same time. Notwithstanding this caveat, such sensitivity to small shape fluctuations is also an asset. Carefully employed, flow cytometry can detect morphological effects that may be difficult or impossible to identify by routine visualization, as we showed previously to be true for minor shape changes accompanying the loss of PBP5 from E. coli (35).
A third important consideration is the selection of the gate (the sorting window) from which cells are collected by FACS. Our first attempts to define a sorting gate for enriching normally shaped cells resulted in the need for 15 rounds of sorting and reincubation before a population arose in which about 50% of the cells had a uniform rod-like morphology. However, revising the gate enriched suppressor mutants after as few as four iterations of sorting. This undoubtedly has the added advantage of reducing the number of extraneous mutations that may arise. Also, the sorting gate can be chosen to enrich for cells having other phenotypes. For example, although we enriched wild-type-shaped cells from among a pool of cells with abnormal morphologies, we also defined selection gates to collect cells with shapes that were even more aberrant than those in the starting population (unpublished results). The addition of fluorescent probes may further expand the sorting options.
Finally, as hard as we tried, we found that a single round of sorting was not sufficient to produce a useful enrichment. A particularly curious observation was that after a single sort, the resulting population displayed cell shapes that were worse than the original even though the cells were collected from a subpopulation that were ostensibly more like wild-type rods. This phenomenon occurred every time we selected cells (well over a dozen times), but we still have no good theory to explain it.
Endopeptidase requirement for suppressing aberrant shapes.
Using the FACS-based enrichment strategy, we isolated four mutants that suppressed abnormal cell shapes in three strains lacking different combinations of low-molecular-weight PBPs. The characterization of these suppressors will be reported elsewhere. However, we did find that two peptidoglycan endopeptidases, AmpH and MepA, are involved in the mechanism of suppressing shape abnormalities in at least one of these strains. Removing AmpH from any of the four suppressor strains produced cells with increasingly abnormal shapes, and removing both AmpH and MepA returned ALM10 to being almost as oddly shaped as its original parent, CS315-1. In addition, we could not isolate suppressor mutants if AmpH was removed from CS315-1, implying that this protein was required to generate the suppression pathway in the first place. Tellingly, the enzymatic activity of AmpH was required for suppression because modifying its active site prevented complementation of the shape defects.
AmpH is primarily a peptidoglycan d,d-endopeptidase, though it can also act as a d,d-carboxypeptidase (46). The fact that MepA is also involved strongly implies that it is the endopeptidase activity that helps suppress abnormal shapes in strain ALM10. Consistent with this enzymatic activity, removing these two enzymes from ALM10 increased the number of cross-linked muropeptides in peptidoglycan, as well as the amount of pentapeptides and anhydro-muropeptides (M. E. Laubacher and M. de Pedro, unpublished results). Previous results lead us to hypothesize that increasing amounts of pentapeptide were (somehow) responsible for creating aberrantly shaped E. coli cells (14, 52), perhaps by altering the geometry of the FtsZ ring so as to produce asymmetric septation (20). The data presented here are consistent with that interpretation because an increase in pentapeptides accompanied the reappearance of shape defects when ampH and mepA were deleted from strain ALM10. An alternate interpretation is that in this suppressor strain, the endopeptidases must cleave certain peptidoglycan cross-links to maintain a normal rod shape. In fact, in the ampH mepA mutant, the number of cross-linked muropeptides increased, as would be expected. Also, the number of cross-linked glycan chain ends increased, and the average glycan chain length became shorter (Laubacher and de Pedro, unpublished).
Huang et al. predicted that a mechanism that breaks selected bonds within the peptidoglycan can affect the geometric uniformity of the cell wall (53), which suggests one way that d,d-endopeptidases might contribute to maintenance of cell shape. Experimental support for such an endopeptidase-cell shape connection comes from finding that similar d,d-endopeptidase/d,d-carboxypeptidase enzymes are required to create the normal helical shape of H. pylori (8, 9, 54). If a similar mechanism is at work in the E. coli suppressor mutants described here, then the combined endopeptidase activities of AmpH and MepA are evidently able to compensate for the absence of PBPs 4 and 7, the other known endopeptidases. However, when PBPs 4 and 7 and AmpH are removed, then MepA by itself must not be able to support the efficient generation of suppressor mutants.
The role of PBP1A.
Strains lacking PBP1A were unable to generate suppressor mutants, but deletion of the gene encoding PBP1A from the ALM10 suppressor did not create a grossly misshapen strain. If PBP1A had been required for continued operation of the suppression pathway, then these cells should have become much more aberrant. That they did not suggests that PBP1A might be required to generate the original suppressor mutation but that the protein may not be needed to maintain cell shape once the relevant mutation is in place. This suggests that generating a new cell shape and maintaining that shape could require different functions. The current results imply that AmpH is needed to generate and maintain a new cell shape but that PBP1A is important only for generating the shape suppressor, though what the latter function might be is unclear.
Summary.
By experiment and in theory, anything that promotes, inhibits, speeds up, delays, or reorients FtsZ polymerization can regulate cell length or alter gross cell shape (1, 14, 20, 22–24). In addition, cell size or width can be changed by subtle mutations in the machinery responsible for elongating bacterial cells (47, 48). It is quite likely that bacterial morphology will be affected or regulated by currently unknown, less conspicuous mechanisms and routes. However, identifying these new mechanisms will be complicated by the very nature of the phenotype, which demands a heavy emphasis on visual screening. Here, we show that flow cytometry and FACS sorting are underutilized but powerful tools for measuring, separating, and collecting individual bacterial cells based on morphological differences. By pursuing a similar approach, N. Salama and colleagues (Fred Hutchinson Cancer Research Center) have also sorted bacterial cells on the basis of forward- and side-scattered light to enrich for H. pylori cell shape mutants having different cell curvatures (N. Salama, personal communication). These observations confirm that the strategy will be useful for asking morphological questions in multiple organisms. In short, the ability of FACS to detect, quantify, and enrich mutants that affect cell shape promises to become a valuable tool in identifying new types of morphological pathways and effectors.
Supplementary Material
ACKNOWLEDGMENTS
We thank Andrea Harris, director of the UAMS Flow Cytometry Laboratory, and Alan Gies, director of the UAMS DNA Sequencing Facility.
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM061019 and by the Arkansas Biosciences Institute, the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 14 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01450-12.
REFERENCES
- 1. Young KD. 2010. Bacterial shape: two-dimensional questions and possibilities. Annu. Rev. Microbiol. 64:223–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. den Blaauwen T, de Pedro MA, Nguyen-Disteche M, Ayala JA. 2008. Morphogenesis of rod-shaped sacculi. FEMS Microbiol. Rev. 32:321–344 [DOI] [PubMed] [Google Scholar]
- 3. Young KD. 2007. Bacterial morphology: why have different shapes? Curr. Opin. Microbiol. 10:596–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Young KD. 2006. The selective value of bacterial shape. Microbiol. Mol. Biol. Rev. 70:660–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Justice SS, Hunstad DA, Cegelski L, Hultgren SJ. 2008. Morphological plasticity as a bacterial survival strategy. Nat. Rev. Microbiol. 6:162–168 [DOI] [PubMed] [Google Scholar]
- 6. Rosen DA, Hooton TM, Stamm WE, Humphrey PA, Hultgren SJ. 2007. Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med. 4:e329 doi:10.1371/journal.pmed.0040329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Horvath DJ, Jr, Li B, Casper T, Partida-Sanchez S, Hunstad DA, Hultgren SJ, Justice SS. 2011. Morphological plasticity promotes resistance to phagocyte killing of uropathogenic Escherichia coli. Microbes Infect. 13:426–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sycuro LK, Pincus Z, Gutierrez KD, Biboy J, Stern CA, Vollmer W, Salama NR. 2010. Peptidoglycan crosslinking relaxation promotes Helicobacter pylori's helical shape and stomach colonization. Cell 141:822–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sycuro LK, Wyckoff TJ, Biboy J, Born P, Pincus Z, Vollmer W, Salama NR. 2012. Multiple peptidoglycan modification networks modulate Helicobacter pylori's cell shape, motility, and colonization potential. PLoS Pathog. 8:e1002603 doi:10.1371/journal.ppat.1002603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frirdich E, Biboy J, Adams C, Lee J, Ellermeier J, Gielda LD, Dirita VJ, Girardin SE, Vollmer W, Gaynor EC. 2012. Peptidoglycan-modifying enzyme Pgp1 is required for helical cell shape and pathogenicity traits in Campylobacter jejuni. PLoS Pathog. 8:e1002602 doi:10.1371/journal.ppat.1002602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rowan NJ, Kirf D, Tomkins P. 2009. Studies on the susceptibility of different culture morphotypes of Listeria monocytogenes to uptake and survival in human polymorphonuclear leukocytes. FEMS Immunol. Med. Microbiol. 57:183–192 [DOI] [PubMed] [Google Scholar]
- 12. Margolin W. 2009. Sculpting the bacterial cell. Curr. Biol. 19:R812–R822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Boer PAJ. 2010. Advances in understanding E. coli cell fission. Curr. Opin. Microbiol. 13:730–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Young KD. 2003. Bacterial shape. Mol. Microbiol. 49:571–580 [DOI] [PubMed] [Google Scholar]
- 15. Varma A, Young KD. 2004. FtsZ collaborates with penicillin binding proteins to generate bacterial cell shape in Escherichia coli. J. Bacteriol. 186:6768–6774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carballido-Lopez R. 2006. Orchestrating bacterial cell morphogenesis. Mol. Microbiol. 60:815–819 [DOI] [PubMed] [Google Scholar]
- 17. Takacs CN, Poggio S, Charbon G, Pucheault M, Vollmer W, Jacobs-Wagner C. 2010. MreB drives de novo rod morphogenesis in Caulobacter crescentus via remodeling of the cell wall. J. Bacteriol. 192:1671–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones LJ, Carballido-Lopez R, Errington J. 2001. Control of cell shape in bacteria: helical, actin-like filaments in Bacillus subtilis. Cell 104:913–922 [DOI] [PubMed] [Google Scholar]
- 19. Kruse T, Moller-Jensen J, Lobner-Olesen A, Gerdes K. 2003. Dysfunctional MreB inhibits chromosome segregation in Escherichia coli. EMBO J. 22:5283–5292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Potluri L-P, de Pedro MA, Young KD. 2012. Escherichia coli low-molecular-weight penicillin-binding proteins help orient septal FtsZ, and their absence leads to asymmetric cell division and branching. Mol. Microbiol. 84:203–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kawai Y, Asai K, Errington J. 2009. Partial functional redundancy of MreB isoforms, MreB, Mbl and MreBH, in cell morphogenesis of Bacillus subtilis. Mol. Microbiol. 73:719–731 [DOI] [PubMed] [Google Scholar]
- 22. Wang JD, Levin PA. 2009. Metabolism, cell growth and the bacterial cell cycle. Nat. Rev. Microbiol. 7:822–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weart RB, Lee AH, Chien AC, Haeusser DP, Hill NS, Levin PA. 2007. A metabolic sensor governing cell size in bacteria. Cell 130:335–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chien AC, Hill NS, Levin PA. 2012. Cell size control in bacteria. Curr. Biol. 22:R340–R349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu M, Kleckner N. 1994. Molecular cloning and characterization of the pgm gene encoding phosphoglucomutase of Escherichia coli. J. Bacteriol. 176:5847–5851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hill NS, Kadoya R, Chattoraj DK, Levin PA. 2012. Cell size and the initiation of DNA replication in bacteria. PLoS Genet. 8:e1002549 doi:10.1371/journal.pgen.1002549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davey HM, Kell DB. 1996. Flow cytometry and cell sorting of heterogeneous microbial populations: the importance of single-cell analyses. Microbiol. Rev. 60:641–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bergquist PL, Hardiman EM, Ferrari BC, Winsley T. 2009. Applications of flow cytometry in environmental microbiology and biotechnology. Extremophiles. 13:389–401 [DOI] [PubMed] [Google Scholar]
- 29. Ferrari BC, Oregaard G, Sorensen SJ. 2004. Recovery of GFP-labeled bacteria for culturing and molecular analysis after cell sorting using a benchtop flow cytometer. Microb. Ecol. 48:239–245 [DOI] [PubMed] [Google Scholar]
- 30. Cheng Q, Ruebling-Jass K, Zhang J, Chen Q, Croker KM. 2012. Use FACS sorting in metabolic engineering of Escherichia coli for increased peptide production. Methods Mol. Biol. 834:177–196 [DOI] [PubMed] [Google Scholar]
- 31. Eun YJ, Utada AS, Copeland MF, Takeuchi S, Weibel DB. 2011. Encapsulating bacteria in agarose microparticles using microfluidics for high-throughput cell analysis and isolation. ACS Chem. Biol. 6:260–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Walberg M, Gaustad P, Steen HB. 1996. Rapid flow cytometric assessment of mecillinam and ampicillin bacterial susceptibility. J. Antimicrob. Chemother. 37:1063–1075 [DOI] [PubMed] [Google Scholar]
- 33. Jang KI, Kim MG, Ha SD, Kim KS, Lee KH, Chung DH, Kim CH, Kim KY. 2007. Morphology and adhesion of Campylobacter jejuni to chicken skin under varying conditions. J. Microbiol. Biotechnol. 17:202–206 [PubMed] [Google Scholar]
- 34. Løbner-Olesen A, Skarstad K, Hansen FG, von Meyenburg K, Boye E. 1989. The DnaA protein determines the initiation mass of Escherichia coli K-12. Cell 57:881–889 [DOI] [PubMed] [Google Scholar]
- 35. Meberg BM, Paulson AL, Priyadarshini R, Young KD. 2004. Endopeptidase penicillin-binding proteins 4 and 7 play auxiliary roles in determining uniform morphology of Escherichia coli. J. Bacteriol. 186:8326–8336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schnaitman CA, McDonald GA. 1984. Regulation of outer membrane protein synthesis in Escherichia coli K-12: deletion of ompC affects expression of the OmpF protein. J. Bacteriol. 159:555–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD. 1999. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J. Bacteriol. 181:3981–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meberg BM, Sailer FC, Nelson DE, Young KD. 2001. Reconstruction of Escherichia coli mrcA (PBP 1a) mutants lacking multiple combinations of penicillin binding proteins. J. Bacteriol. 183:6148–6149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heidrich C, Ursinus A, Berger J, Schwarz H, Höltje JV. 2002. Effects of multiple deletions of murein hydrolases on viability, septum cleavage, and sensitivity to large toxic molecules in Escherichia coli. J. Bacteriol. 184:6093–6099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 41. Potluri L, Karczmarek A, Verheul J, Piette A, Wilkin JM, Werth N, Banzhaf M, Vollmer W, Young KD, Nguyen-Disteche M, den Blaauwen T. 2010. Septal and lateral wall localization of PBP5, the major d,d-carboxypeptidase of Escherichia coli, requires substrate recognition and membrane attachment. Mol. Microbiol. 77:300–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herzenberg LA, Parks D, Sahaf B, Perez O, Roederer M. 2002. The history and future of the fluorescence activated cell sorter and flow cytometry: a view from Stanford. Clin. Chem. 48:1819–1827 [PubMed] [Google Scholar]
- 43. Gant VA, Warnes G, Phillips I, Savidge GF. 1993. The application of flow cytometry to the study of bacterial responses to antibiotics. J. Med. Microbiol. 39:147–154 [DOI] [PubMed] [Google Scholar]
- 44. Banzhaf M, van den Berg van Saparoea B, Terrak M, Fraipont C, Egan A, Philippe J, Zapun A, Breukink E, Nguyen-Distèche M, den Blaauwen T, Vollmer W. 2012. Cooperativity of peptidoglycan synthases active in bacterial cell elongation. Mol. Microbiol. 85:179–194 [DOI] [PubMed] [Google Scholar]
- 45. Henderson TA, Young KD, Denome SA, Elf PK. 1997. AmpC and AmpH, proteins related to the class C β-lactamases, bind penicillin and contribute to the normal morphology of Escherichia coli. J. Bacteriol. 179:6112–6121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gonzalez-Leiza SM, de Pedro MA, Ayala JA. 2011. AmpH, a bifunctional dd-endopeptidase and dd-carboxypeptidase of Escherichia coli. J. Bacteriol. 193:6887–6894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Alyahya SA, Alexander R, Costa T, Henriques AO, Emonet T, Jacobs-Wagner C. 2009. RodZ, a component of the bacterial core morphogenic apparatus. Proc. Natl. Acad. Sci. U. S. A. 106:1239–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dye NA, Pincus Z, Fisher IC, Shapiro L, Theriot JA. 2011. Mutations in the nucleotide binding pocket of MreB can alter cell curvature and polar morphology in Caulobacter. Mol. Microbiol. 81:368–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Christen B, Fero MJ, Hillson NJ, Bowman G, Hong SH, Shapiro L, McAdams HH. 2010. High-throughput identification of protein localization dependency networks. Proc. Natl. Acad. Sci. U. S. A. 107:4681–4686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Werner JN, Chen EY, Guberman JM, Zippilli AR, Irgon JJ, Gitai Z. 2009. Quantitative genome-scale analysis of protein localization in an asymmetric bacterium. Proc. Natl. Acad. Sci. U. S. A. 106:7858–7863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Werner JN, Gitai Z. 2010. High-throughput screening of bacterial protein localization. Methods Enzymol. 471:185–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Varma A, de Pedro MA, Young KD. 2007. FtsZ directs a second mode of peptidoglycan synthesis in Escherichia coli. J. Bacteriol. 189:5692–5704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huang KC, Mukhopadhyay R, Wen B, Gitai Z, Wingreen NS. 2008. Cell shape and cell-wall organization in Gram-negative bacteria. Proc. Natl. Acad. Sci. U. S. A. 105:19282–19287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bonis M, Ecobichon C, Guadagnini S, Prevost MC, Boneca IG. 2010. A M23B family metallopeptidase of Helicobacter pylori required for cell shape, pole formation and virulence. Mol. Microbiol. 78:809–819 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.