

Abstract
Kingella kingae is an emerging bacterial pathogen that is being recognized increasingly as an important etiology of septic arthritis, osteomyelitis, and bacteremia, especially in young children. The pathogenesis of K. kingae disease begins with bacterial adherence to respiratory epithelium, which is dependent on type IV pili and is influenced by two PilC-like proteins called PilC1 and PilC2. Production of either PilC1 or PilC2 is necessary for K. kingae piliation and bacterial adherence. In this study, we set out to further investigate the role of PilC1 and PilC2 in type IV pilus-associated phenotypes. We found that PilC1 contains a functional 9-amino-acid calcium-binding (Ca-binding) site with homology to the Pseudomonas aeruginosa PilY1 Ca-binding site and that PilC2 contains a functional 12-amino-acid Ca-binding site with homology to the human calmodulin Ca-binding site. Using targeted mutagenesis to disrupt the Ca-binding sites, we demonstrated that the PilC1 and PilC2 Ca-binding sites are dispensable for piliation. Interestingly, we showed that the PilC1 site is necessary for twitching motility and adherence to Chang epithelial cells, while the PilC2 site has only a minor influence on twitching motility and no influence on adherence. These findings establish key differences in PilC1 and PilC2 function in K. kingae and provide insights into the biology of the PilC-like family of proteins.
INTRODUCTION
Kingella kingae is a Gram-negative organism that belongs to the Neisseriaceae family and is being recognized increasingly as an important pathogen in young children. Improvements in culture-based and molecular diagnostics have identified K. kingae as a common etiology of septic arthritis, osteomyelitis, and bacteremia in the pediatric population (1–8). A recent study reported K. kingae as the leading cause of septic arthritis in children 6 to 36 months of age (1). Based on epidemiological studies, K. kingae is believed to initiate infection by colonizing the posterior pharynx, where it typically persists for weeks without producing symptoms (9–12). On occasion, the organism will breach the epithelial barrier and enter the bloodstream and then disseminate hematogenously to the joints, bones, or endocardium (6, 8). This model is supported by reports describing genotypically identical K. kingae paired isolates from the respiratory tract and blood of patients with invasive K. kingae disease (11, 13). Despite increased recognition of K. kingae as an important pathogen, little is known about the molecular mechanisms used by this organism to cause disease.
Previous work established that K. kingae produces type IV pili that are necessary for adherence to respiratory epithelial and synovial cells (14–16). Type IV pili are surface fibers that are widespread in Gram-negative bacteria and convey many phenotypes, including adherence, twitching motility, and natural competence (17, 18). These fibers are composed primarily of a single protein subunit (called PilA1 in K. kingae) and require numerous other factors for proper production. A key aspect of type IV pilus biology and the associated phenotypes is the ability of the fiber to be retracted through the outer membrane, a process catalyzed by the retraction ATPase, PilT, and associated with twitching motility (19–23). While many of the protein factors involved in type IV pilus biology have been well studied, the mechanism controlling pilus extension and pilus retraction remains poorly understood. However, current data from work on the pathogenic Neisseria species (Neisseria meningitidis and Neisseria gonorrhoeae) and Pseudomonas aeruginosa suggests that counteraction of retraction by PilC-like proteins is required for proper pilus production and twitching motility (24–26).
The PilC proteins are proposed to influence two aspects of type IV pilus biology, namely, pilus biogenesis and adherence. Like N. meningitidis and N. gonorrhoeae, K. kingae produces two PilC-like proteins called PilC1 and PilC2, which are encoded by genes at separate locations in the genome (14, 27, 28). The PilC1 and PilC2 proteins in the pathogenic Neisseria species have been identified as the adhesive component of the pilus fiber and are located at the tip of the fiber in N. gonorrhoeae (29–31). Recent observations suggest that P. aeruginosa PilY1 (the single PilC-like protein in P. aeruginosa) is also an adhesin (25, 32). In earlier work, we observed a reduction in adherence to human cells by K. kingae strain 269-492 derivatives that expressed only PilC1 or PilC2, supporting a potential role for these proteins in adherence (14). In addition, we found that elimination of both PilC1 and PilC2 in K. kingae strain 269-492 resulted in a loss of piliation, suggesting that at least one PilC protein is required for pilus production (14). Similar results were obtained with the pathogenic Neisseria species and P. aeruginosa (27, 33). Recently, P. aeruginosa PilY1 was shown to regulate pilus production and twitching motility through calcium binding (Ca binding) at a 9-amino-acid loop in the C-terminal domain (34). Using bioinformatic approaches, similar loops were identified in other PilC homologs, including N. meningitidis and N. gonorrhoeae PilC1 and PilC2 and K. kingae PilC1, but not PilC2 (34).
In contrast to the PilC1 and PilC2 proteins in N. gonorrhoeae and N. meningitidis, K. kingae PilC1 and PilC2 share only limited overall sequence homology (7% identity and 16% similarity) (14). In this regard, K. kingae represents a unique and intriguing system to investigate PilC-like protein biology. In this study, we examined Ca binding by PilC1 and PilC2 and the potential role of Ca binding in control of type IV pilus production, twitching motility, and adherence to Chang epithelial cells. We report that both PilC1 and PilC2 bind calcium, with PilC1 utilizing a 9-amino-acid PilY1-like Ca-binding site and PilC2 utilizing a 12-amino-acid human calmodulin-like Ca-binding site. While both the PilC1 and PilC2 Ca-binding sites are dispensable for pilus production, the PilC1 Ca-binding site is required for twitching motility and adherence.
MATERIALS AND METHODS
Bacterial strains.
Bacterial strains used in this study are listed in Table 1. K. kingae strains were stored at −80°C in brain heart infusion broth (BHI) with 30% glycerol. K. kingae strain KK03 is a stable high-level pilus-expressing, spreading-corroding colony type (bacterial growth spreads from the central raised colony, and colony expansion results in pitting or corroding of the agar surface) of clinical isolate 269-492. E. coli strains were stored at −80°C in Luria-Bertani (LB) broth with 15% glycerol. K. kingae strains were routinely cultured at 37°C with 5% CO2 on chocolate agar supplemented with 50 μg/ml kanamycin, 1 μg/ml erythromycin, or 2 μg/ml tetracycline, as appropriate. E. coli strains were routinely cultured at 37°C in LB broth or on LB agar supplemented with 100 μg/ml ampicillin, 50 μg/ml kanamycin, 500 μg/ml erythromycin, 25 μg/ml tetracycline, 50 μg/ml streptomycin, or 50 μg/ml chloramphenicol, as appropriate.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| E. coli strains | ||
| Dh5α | E. coli F− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) phoA supE441 thi-1 gyrA96 relA1 | 49 |
| BL21-CodonPlus(DE3)-RIPL | E. coli B F− ompT hsdS(rB− mB−) dcm+ Tetr gal λ(DE3) endA Hte [argU proLCamr] [argU ileY leuW Strep/Specr] | Agilent |
| BL21-Gold(DE3) | E. coli B F− ompT hsdS(rB− mB−) dcm+ Tetr gal λ(DE3) endA Hte | Agilent |
| K. kingae strains | ||
| KK03 | Naturally occurring spreading and corroding variant of septic arthritis clinical isolate 269-492 | 36 |
| KK03 derivatives (genotype or phenotype) | ||
| ΔpilF | KK03 with an aphA3 marked pilF deletion | This work |
| ΔpilT | KK03 with an ermC marked pilT insertion | 38 |
| ΔpilC1 | KK03 with a tetM marked pilC1 deletion | This work |
| ΔpilC2 | KK03 with an aphA3 marked pilC2 deletion | This work |
| ΔpilC1 ΔpilC2 | KK03 with a tetM marked pilC1 deletion and an aphA3 marked pilC2 deletion | This work |
| ΔpilC1 PilC2mark | pilC1-bearing strain with an aphA3 insertion immediately downstream of WT pilC2 | This work |
| ΔpilC2 PilC1mark | pilC2-bearing strain with an ermC insertion upstream of WT pilC1 | This work |
| ΔpilC1 PilC2D1444A | ΔpilC1 PilC2mark strain with a pilC2 mutation resulting in expression of PilC2D1444A | This work |
| ΔpilC1 PilC2D1444K | ΔpilC1 PilC2mark strain with a pilC2 mutation resulting in expression of PilC2D1444K | This work |
| ΔpilC2 PilC1D930A | ΔpilC2 PilC1 mark strain with a pilC1 mutation resulting in expression of PilC1D930A | This work |
| ΔpilC2 PilC1D930K | ΔpilC2 PilC1 mark strain with a pilC1 mutation resulting in expression of PilC1D930K | This work |
| PilC1D930A PilC2D1444A | Strain KK03 expressing PilC1D930A and PilC2D1444A | This work |
| PilC1D930K PilC2D1444K | Strain KK03 expressing PilC1D930K and PilC2D1444K | This work |
| Plasmids | ||
| pMCSG7 | Protein expression vector for 6× His N-terminal fusions and a TEV cleavage site | 50 |
| pMCSG9 | Protein expression vector for MBP 6× His N-terminal fusions and a TEV cleavage site | 51 |
| pMCSG9/PilC1 | For expression of MBP-PilC1739–1047 | This work |
| pMCSG9/PilC1D930A | For expression of MBP-PilC1739–1047 D930A | This work |
| pMCSG9/PilC1D930K | For expression of MBP-PilC1739–1047 D930K | This work |
| pMCSG7/PilC2 | For expression of HIS-PilC2868–1502 | This work |
| pMCSG7/PilC2D1125A | For expression of HIS-PilC2868–1502 D1125A | This work |
| pMCSG7/PilC2D1125K | For expression of HIS-PilC2868–1502 D1125K | This work |
| pMCSG7/PilC2D1444A | For expression of HIS-PilC2868–1502 D1444A | This work |
| pMCSG7/PilC2D1444K | For expression of HIS-PilC2868–1502 D1444K | This work |
| pFalcon2 | Source of aphA3 kanamycin resistance gene | 52 |
| pHSXtetM4 | Source of tetM tetracycline resistance gene | 53 |
| pIDN4 | Source of ermC erythromycin resistance gene | 54 |
| pUC19/ΔpilF | pilF deletion construct | 36 |
| pUC19/ΔpilC1 | pilC1 deletion construct | 14 |
| pUC19/ΔpilC2 | pilC2 deletion construct | This work |
| pUC19/ErmpilC1 | For introduction of ermC marked pilC1 locus | This work |
| pUC19/pilC2Kan | For introduction of aphA3 marked pilC2 locus | This work |
| pUC19/PilC1D930A | For introduction of PilC1D930A | This work |
| pUC19/PilC1D930K | For introduction of PilC1D930K | This work |
| pUC19/PilC2D1444A | For introduction of PilC2D1444A | This work |
| pUC19/PilC2D1444K | For introduction of PilC2D1444K | This work |
Recombinant PilC constructs and protein purification.
All plasmids used in this study are listed in Table 1, and all primers are listed in Table S1 in the supplemental material. PilC1 residues 739 to 1047 were cloned from pUC19pilC1 with primers PilC1 738 F and PilC1 1047 R into a pMCSG9 LIC HIS MBP vector. Site-directed mutagenesis was performed to produce D930A and D930K mutations with primers PilC1D930A-F/PilC1D930A-R and PilC1D930K-F/PilC1D930K-R, respectively. All clones were confirmed with nucleotide sequencing. Resultant vectors were transformed into BL21-RIPL cells (Stratagene) and plated on LB agar plates containing 25 μg/ml tetracycline, 50 μg/ml streptomycin, 50 μg/ml chloramphenicol, and 100 μg/ml ampicillin. A single colony was used to inoculate 50 ml of LB broth containing the previously mentioned antibiotics, and cultures were incubated overnight. Cell cultures were centrifuged at 3,000 × g, and the supernatant was discarded. The resultant pellet was used to inoculate a 1.5-liter shaker flask of Terrific broth (TB) with 50 μl antifoam (Sigma-Aldrich) and the previously mentioned antibiotics. Cells were grown at 37°C until the optical density at 600 nm (OD600) reached 0.6 to 0.8. The temperature was then reduced to 18°C, and protein expression was induced with 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cells were grown overnight and harvested by centrifugation at 6,000 × g at for 15 min at 4°C, and pellets were stored at −80°C.
Thawed PilC1 pellets were suspended in lysis buffer (50 mM sodium phosphate [pH 7.6], 500 mM NaCl, 25 mM imidazole) supplemented with 0.5 mM EDTA, 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF), one tablet of a protease inhibitor cocktail (Roche), and 1 μg/ml lysozyme. After 1 h of gentle stirring on ice, the cells were sonicated on ice for 1 min and the lysate was centrifuged at 45,000 × g for 90 min at 4°C. Using an ÄKTAxpress (GE Healthcare), protein from the filtered soluble fraction was nickel purified (elution buffer consisted of 50 mM sodium phosphate [pH 7.6], 500 mM NaCl, and 500 mM imidazole) and purified on an S200 gel filtration column in the final buffer, which contained 20 mM Tris-HCl (pH 7.5), 250 mM NaCl, 2 mM dithiothreitol (DTT), and 5% glycerol. Protein was then concentrated, flash frozen in liquid nitrogen, and stored at −80°C.
PilC2 residues 868 to 1502 were cloned from genomic K. kingae DNA with primers PilC2 868 F and PilC2 end R into a pMCSG7 LIC HIS vector. Site-directed mutagenesis was performed to produce D1125A (primers PilC2D1125A-F/PilC2D1125A-R), D1125K (primers PilC2D1125K-F/PilC2D1125K-R), D1444A (primers PilC2D1444A-F/PilC2D1444A-R), and D1444K (primers PilC2D1444K-F/PilC2D1444K-R) mutations. Resultant plasmids were transformed into BL21-Gold cells (Agilent), which were incubated overnight on LB plates containing ampicillin. Subsequently, a single colony was used to inoculate 100 ml LB broth overnight containing 50 μg/ml ampicillin, and the culture was incubated overnight. Cell cultures were treated as described above except that they were induced with 0.5 mM IPTG.
Cells pellets were thawed and resuspended in buffer consisting of 10 mM Tris (pH 7.8) and 50 mM NaCl with 10 mM imidazole, DNase, and protease inhibitor tablets (Roche). Cells were sonicated, and the cell lysate was separated into soluble and insoluble fractions using high-speed centrifugation. The soluble fraction was filtered, then nickel purified (with 300 mM imidazole), buffer exchanged (no imidazole), and separated using an S200 gel filtration column on an ÄKTAxpress (GE Healthcare). If necessary, protein and storage buffers were chelated with Chelex-100 to remove bound calcium (Bio-Rad Laboratories). Purified proteins were concentrated to ∼100 μM, frozen, and stored at −80°C.
K. kingae strain construction.
Gene disruptions and directed mutations were generated in K. kingae as previously described (14, 15). Briefly, plasmid-based disruption constructs were created in E. coli, linearized, and introduced into K. kingae strain KK03 via natural transformation and plating on appropriate antibiotic-containing media. Correct localization of disruptions was confirmed by PCR or Southern blotting, and site-directed mutations were confirmed by nucleotide sequencing. The ΔpilF and ΔpilC1 deletion constructs were generated as previously described (14). As our original pilC2 disruption was an insertional transposon mutant (14), we decided to generate a targeted ΔpilC2 deletion. PCR fragments corresponding to the 5′ and 3′ regions of pilC2 were amplified individually from strain KK03 with primers pilC2Δ5′F/pilC2Δ5′R and pilC2Δ3′F/pilC2D3′R and were then ligated into EcoRI/HindIII-digested pUC19, generating pUC19pilC2::BamHI, which contains a large pilC2 internal deletion and a BamHI site. The kanamycin resistance cassette aphA3 was PCR amplified with primers aphA3FBamHI/aphA3RBamHI from pFalcon2 and ligated into BamHI-digested pUC19pilC2::BamHI, generating pUC19ΔpilC2.
To generate pilC alleles that encode Ca-binding site mutations, antibiotic resistance markers were inserted adjacent to the pilC gene in a pUC19 backbone, subjected to site-directed mutagenesis, sequenced, linearized, and transformed into K. kingae strain KK03. For PilC1 Ca-binding site mutations, the entire pilC1 gene and ∼1,000 bp of 5′ and 3′ flanking sequence were PCR amplified from KK03 genomic DNA with primers pilC1regionF/pilC1regionR and the ligated into EcoRI/SalI-digested pUC19, generating pUC19pilC1. The QuikChange XL-II site-directed mutagenesis kit (Agilent) and primers pilC1markF/pilC1markR were used to insert an MluI site in the gene upstream of the pilC1 promoter region, a predicted ABC-type transporter, generating pUC19/MluIpilC1. The ermC erythromycin resistance cassette was amplified from pIDN4 with primers ermCF/ermCR and was ligated into MluI-digested pUC19/MluIpilC1, generating pUC19/ErmpilC1. Restriction digestion and sequencing were used to confirm proper orientation of ermC. To confirm that the ermC marker inserted upstream of WT pilC1 did not alter the phenotypes observed in the ΔpilC2 background, a control ΔpilC2/PilC1mark (pilC2 deleted and ermC upstream of pilC1) strain was created and was confirmed to have phenotypes identical to those of the ΔpilC2 strain. Site-directed mutagenesis with primers PilC1D930A-F/PilC1D930A-R and PilC1D930K-F/PilC1D930K-R was used to generate the D930A and D930K mutations, respectively.
For the PilC2 Ca-binding site mutations, ∼2.6 kb of the 3′ region of pilC2 was amplified with primers pilC2markAF/pilC2markAR and ∼1.5 kb of sequence immediately downstream of the pilC2 open reading frame (ORF) was amplified with primers pilC2markBF/pilC2markBR individually from KK03 genomic DNA and ligated into EcoRI/HindIII-digested pUC19, generating pUC19pilC2, which contains a BamHI site inserted 20 bp downstream of the pilC2 stop codon. The kanamycin resistance cassette aphA3 was PCR amplified with primers aphA3FBamHI/aphA3RBamHI from pFalcon2 and ligated into BamHI-digested pUC19pilC2, generating pUC19pilC2Kan. To confirm that the aphA3 marker inserted immediately downstream of wild-type (WT) pilC2 did not alter the phenotypes observed in the K. kingae ΔpilC1 background, a control ΔpilC1 PilC2mark (pilC1 deleted and aphA3 inserted downstream of pilC2) strain was created and confirmed to have phenotypes identical to those of the ΔpilC1 strain. Site-directed mutagenesis with primers PilC2D1444A-F/PilC2D1444A-R and PilC2D1444K-F/PilC2D1444K-R was used to generate the D1444A and D1444K mutations, respectively.
Calcium binding assay.
A binding curve for Oregon green 488 BAPTA-5N, hexapotassium salt (Invitrogen) in 10 mM Tris (pH 7.8) and 50 mM NaCl was measured on a PHERAstar (BMGLabtech) at 488 nm. Purified PilC1 or PilC2 was chelated with Chelex-100 resin (Bio-Rad) and subsequently serially diluted 1.5- to 3-fold from ∼100 μM to ∼1 nM, and each dilution was combined with 20 μM Oregon green and 2 μM CaCl2 and measured at 488 nm on a PHERAstar (BMGLabtech) to obtain a 50% effective concentration (EC50). The EC50 was then used to calculate the respective Kd (dissociation constant) for calcium as described previously (34).
CD and thermal denaturation.
Protein samples were exchanged into chelated 10 mM KxHxPO4–50 mM NaF (pH 7.7) buffer, where x is dependent on pH, and brought to 5 μM with or without the addition of 20 μM CaCl2. A wavelength scan from 200 to 260 nm was performed on a circular-dichroism (CD) spectrometer 62 DS (Aviv) at 16°C with a 10-s averaging time. Melting temperatures were measured at 214 nm from 3°C to 95°C at 1-degree increments with a 10-s averaging time.
Pilus preparations.
Derivatives of K. kingae strain KK03 were incubated on chocolate agar for 17 to 18 h, and growth was suspended in 1.5 ml 50 mM Tris–150 mM NaCl (pH 8.0) to an OD600 of 1.0, vortexed at full speed for 1 min, and centrifuged at 21,000 × g for 2 min to pellet the bacteria. A total of 1.25 ml of the bacterium-free supernatant was subjected to 20% ammonium sulfate precipitation on ice for 2 h. Precipitated pili were collected via centrifugation at 21,000 × g for 5 min and resuspended in 1× SDS-PAGE loading buffer. Aliquots were separated on 15% SDS-PAGE gels and stained with Coomassie blue. The observed protein band was confirmed to be the major pilin subunit, PilA1, by Western blotting with antiserum GP65 as described previously (15).
Twitching-motility assays.
Twitching motility was assessed by a modified agar plate stab assay originally developed for P. aeruginosa (35). Briefly, derivatives of K. kingae strain KK03 were incubated on chocolate agar for 17 to 18 h, and growth was resuspended in 1× phosphate-buffered saline (PBS) to an OD600 of 1.0. One microliter of the bacterial suspensions was stab inoculated to the bottoms of tissue culture-treated 100-mm plates containing 10 ml chocolate agar with 1% agar and incubated at 37°C with 5% CO2 for 48 h. To reduce zone-to-zone and plate-to-plate variation, the twitching-motility plates were cooled at ambient temperature for 24 h prior to stab inoculation, and each strain set replicate was performed with the same batch of chocolate agar plates. Twitching-motility-competent strains spread from the stab inoculation site at the plate-agar interface. The agar was carefully peeled away, and the plate was air dried and stained with crystal violet to visualize the twitching-motility zones. Three diameter measurements were taken and averaged per twitching zone. All experiments were performed in triplicate, and error bars represent standard errors of the means. Statistical analysis was performed using analysis of variance (ANOVA) and Bonferroni's posttest to compare twitching zones between mutant and parental strains.
Eukaryotic cell lines.
Chang cells (Wong-Kilbourne derivative [D] of Chang conjunctiva, HeLa origin; ATCC CCL-20.2) were cultivated at 37°C with 5% CO2 in media as described previously (36).
Adherence assays.
Quantitative adherence assays were performed as described previously (14, 15). Briefly, monolayers of fixed Chang cells at a density of ∼2 × 105/well in 24-well plates were inoculated with approximately 6.5 × 106 CFU of bacteria (multiplicity of infection [MOI] of ∼30), and plates were centrifuged at 165 × g for 5 min and then incubated for 25 min at 37°C in 5% CO2. Monolayers were washed 4 times with PBS to remove nonadherent bacteria and were then treated with 1× trypsin-EDTA (Sigma) for 20 min at 37°C to release adherent bacteria. Appropriate dilutions were plated on chocolate agar, and percent adherence was calculated by dividing the number of adherent CFU by the number of inoculated CFU. All experiments were performed in triplicate. Statistical analysis was performed using ANOVA and Bonferroni's posttest to compare adherence levels between mutant and parental strains.
Transmission electron microscopy (TEM).
To assess surface piliation, K. kingae strains were resuspended in ammonium acetate, pH 7.4, and allowed to absorb onto Formvar-carbon-coated 200-mesh grids for 1 min. The grids were subsequently stained with 1% uranyl acetate for 30 s, excess liquid was wicked away, and the grids were air dried and examined on a Philips CM12 transmission electron microscope (TEI) at an accelerating voltage of 80 kV.
RESULTS
K. kingae PilC1 and PilC2 contain a calcium-binding site.
Based on sequence homology with P. aeruginosa PilY1 and N. meningitidis and N. gonorrhoeae PilC1 and PilC2, K. kingae encodes two PilC homologs called PilC1 and PilC2. Interestingly, while PilC1 and PilC2 in the pathogenic Neisseria species are nearly identical, the K. kingae strain 269-492 PilC1 and PilC2 proteins share only limited homology with each other and with other PilC-like protein family members (14). Most of the similarity between members of this protein class resides in the C-terminal putative pilus-interacting half of the proteins (34). Interestingly, while K. kingae is a member of the Neisseriaceae family, the PilC1 and PilC2 C-terminal domains are more similar to P. aeruginosa PilY1 than to the Neisseria PilC1 and PilC2 proteins or even to each other (Fig. 1A and C). The difference in length of PilC1 and PilC2 compared to P. aeruginosa PilY1 reflects the presence of multiple nonhomologous sequences in PilC1 and PilC2 that are absent in PilY1. Examination of the K. kingae PilC1 and PilC2 amino acid sequences revealed one predicted Ca-binding site in each protein (Fig. 1B and D). PilC1 contains a 9-residue site (922-DVNRDGVYD-930) that is similar to the Ca-binding loop in P. aeruginosa PilY1 (Fig. 1B), and PilC2 contains a 12-residue site (1433-DTNGDGKFTEAD-1444) with strong homology to the canonical EF-hand human calmodulin Ca-binding loop (Fig. 1D). An additional less conserved 12-residue calmodulin-like Ca-binding site was also identified in PilC2 (1114-DLTNPTDLTESD-1125) (Fig. 1D).
Fig 1.
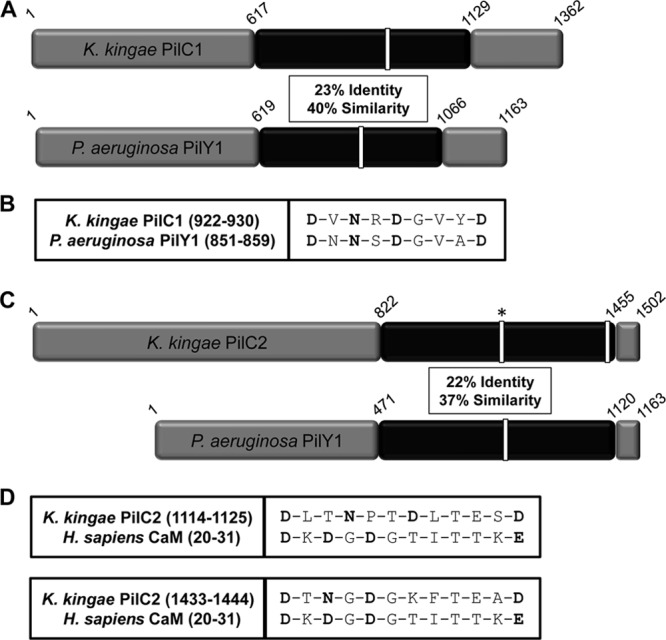
(A) Sequence comparison of K. kingae PilC1 with P. aeruginosa PilY1 highlighting the C-terminal region with the greatest amount of homology (in black). Sequence identity and similarity values are given for the region in black. White bars represent putative and confirmed calcium-binding domains in K. kingae and P. aeruginosa, respectively. (B) Alignment of the K. kingae PilC1 putative Ca-binding site with the calcium-chelating residues of P. aeruginosa PilY1, with the potential calcium-chelating residues of PilC1 shown in bold. (C) Sequence comparison of K. kingae PilC2 with P. aeruginosa PilY1 highlighting the C-terminal region with the greatest amount of homology (in black). Sequence identity and similarity values are given for the region in black. White bars represent putative and confirmed calcium-binding domains in K. kingae and P. aeruginosa, respectively. The white bar with an asterisk in the K. kingae PilC2 sequence represents a site homologous to a CaM calcium-binding site that does not bind calcium. (D) Alignment of the K. kingae PilC2 putative Ca-binding sites, with the calcium-chelating residues of human calmodulin and the potential calcium-chelating residues of PilC2 shown in bold.
To determine if the putative PilC1 Ca-binding site binds calcium, recombinant fragments of PilC1 were expressed in E. coli, purified, and tested for Ca-binding potential in vitro. A C-terminal PilC1 fragment containing amino acids 739 to 1047 was expressed as a maltose-binding protein (MBP) fusion, PilC1739–1047-MBP, and purified. As shown in Fig. 2A, PilC1739–1047-MBP exhibited a binding affinity for calcium with a Kd of 530 nM, confirming that PilC1 binds calcium. The affinity of PilC1 is comparable to the affinity of PilY1, which has a Kd of 2.6 μM (34). Orans et al. demonstrated that mutation of the terminal bidentate aspartic acid residue in the PilY1 9-amino-acid Ca-binding loop to alanine eliminated Ca binding and created a calcium-free PilY1 state, while mutation of this residue to lysine also eliminated Ca binding but created a potential calcium-bound mimic PilY1 state (34). As shown in Fig. 2A, mutation of the terminal aspartic acid residue D930 in the K. kingae PilC1 Ca-binding site to either alanine or lysine eliminated specific Ca binding. The mutant PilC1739–1047D930A-MBP and PilC1739–1047D930K-MBP exhibited wild-type CD spectra (see Fig. S1 in the supplemental material), indicating that the overall structure of these fusion proteins was not altered.
Fig 2.

K. kingae PilC1 and PilC2 contain calcium-binding sites. Calcium competition binding assays using Oregon green were performed with WT, D930A, and D930K recombinant PilC1739–1047-MBP (A); WT, D1444A, and D1444K recombinant PilC2868–1502 (B); and WT, D1125A, and D1125 recombinant PilC2868–1502 (C). Binding curves were modeled to one-site competition or linear line. Error bars represent standard errors of the means.
In order to characterize PilC2, a recombinant fragment containing amino acids 868 to 1502 was purified. As shown in Fig. 2B, purified PilC2868–1502 exhibited a binding affinity for calcium with a Kd of 5.5 μM, confirming that PilC2 binds calcium. This Kd is consistent with the previously published calmodulin values in the low micromolar range (37). Mutation of the terminal bidentate aspartic acid residue D1444 in the PilC2 Ca-binding loop to alanine or lysine eliminated Ca binding. The PilC2868–1502D1444A and PilC2868–1502D1444K proteins exhibited WT CD spectra and melting temperatures (see Fig. S2 in the supplemental material), indicating that the overall structure of these proteins was not altered. The less conserved 12-amino-acid site located at positions 1114 to 1125 was demonstrated to have no effect on Ca binding (Fig. 2C).
Taken together, these data indicate that PilC1 contains a functional Ca-binding site located in the region corresponding to amino acids 922 to 930 and that PilC2 contains a functional Ca-binding site located in the region corresponding to amino acids 1433 to 1444.
Influence of K. kingae PilC1 and PilC2 Ca-binding sites on type IV pilus production.
To investigate the influence on and role of the PilC1 and PilC2 Ca-binding sites in type IV pilus production and other type IV pilus phenotypes in K. kingae, we generated a series of mutants in the KK03 strain background, a naturally occurring spreading and corroding derivative of 269-492 that produces stable levels of type IV pili. The pilC1 and pilC2 genes are located in physically separate regions of the K. kingae genome, as highlighted in the diagrams of the mutant derivatives of KK03 in Fig. 3. To investigate the phenotypes associated with an individual pilC locus, Ca-binding site mutations were introduced into the pilC locus of interest with a nearby antibiotic resistance marker in a strain background with the other pilC gene deleted. Strains containing Ca-binding mutations in both PilC1 and PilC2 were also generated for investigation. In addition to the strains diagrammed in Fig. 3, a previously described ΔpilF type IV pilus assembly ATPase mutant that is unable to assemble type IV pili was used as a control strain (14).
Fig 3.

Representation of the pilC1 and pilC2 loci of K. kingae strain KK03 derivatives used in this study. The pilC1 and pilC2 genes are present at separate loci in the genome. The ΔpilC1 and ΔpilC2 mutants are marked with a tetracycline resistance cassette (Tetr) and a kanamycin resistance cassette (Kanr), respectively. Both deletion mutations were combined to generate the ΔpilC1 ΔpilC2 double mutant. ΔpilC2 PilC1D930A and ΔpilC2 PilC1D930K strains, with a ΔpilC2 PilC1mark strain (WT pilC1 with an erythromycin resistance cassette [Ermr] upstream of the promoter region) used as a control, were generated to study the role of the PilC1 Ca-binding site in the absence of pilC2. ΔpilC1 PilC2D1444A and ΔpilC1 PilC2D1444K strains, with a ΔpilC1 PilC2mark strain (WT pilC2 with the Kanr marker immediately downstream of pilC2) used as a control, were generated to study the role of the PilC2 Ca-binding site in the absence of pilC1. Mutations were combined to generate PilC1D930A PilC2D1444A and PilC1D930K PilC2D1444K strains.
To assess production of type IV pili, fibers were sheared from the surfaces of equal-OD600 suspensions of K. kingae KK03 derivatives and separated using SDS-PAGE, and the major pilin subunit was stained with Coomassie blue. In earlier work, we demonstrated that the major band at ∼14 kDa is PilA1, the major pilin subunit (15). As shown in Fig. 4, while the ΔpilF and ΔpilC1 ΔpilC2 strains were nonpiliated, all mutants that produced at least one PilC protein (whether WT or containing a mutation in the Ca-binding site) were piliated. The type IV pilus retraction machinery mutant ΔpilT was included as a control and was piliated (38). In some strains, there appeared to be slight reductions in quantity of extracellular PilA1 relative to the parental strains, including ΔpilC2 PilC1D930A, ΔpilC2 PilC1D930K, and PilC1D930A PilC2D1444A strains, indicating a minor defect in surface pilus production (Fig. 4). However, all strains examined produced surface pili, demonstrating that Ca-binding site mutant PilC1 and PilC2 are still able to promote piliation. Examination of this strain set using TEM revealed no noticeable changes in pilus number per bacterium and no differences in pilus length or morphology (data not shown). Together, these data indicate that at least one PilC protein is required to promote piliation and that the Ca-binding sites are dispensable for piliation.
Fig 4.
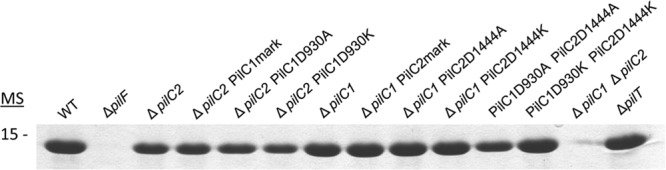
All PilC1 and PilC2 Ca-binding-site mutants produce surface pili. Pili were sheared from the surface of K. kingae strain KK03 derivatives, and the major pilin subunit, PilA1, was visualized with Coomassie blue staining following SDS-PAGE. ΔpilF and ΔpilC1 ΔpilC2 strains both express PilA1 but fail to assemble surface pili, and the ΔpilT strain is a pilus retraction mutant and is piliated. MS, molecular size (in kDa).
The PilC1 and PilC2 Ca-binding sites influence twitching motility.
We next wanted to assess the role of the PilC Ca-binding sites in twitching motility. Using a modified agar plate stab assay originally developed for use in P. aeruginosa (35), we measured the spreading zone at the plate-agar interface as a readout for twitching motility. We demonstrated the utility of this method for assessing twitching motility by showing that a strain lacking PilT, the type IV pilus retraction ATPase, still produced surface pili but was unable to form a spreading zone (38). As shown in Fig. 5, the ΔpilC2 and control ΔpilC2 PilC1mark strains produced statistically significantly larger twitching zones than did the parent strain (P < 0.05), while the ΔpilC1 and ΔpilC1 PilC2mark strains produced twitching zones that were slightly smaller than but were not statistically significantly different from those of the parent strain. These data indicate that PilC1 and PilC2 differentially control twitching motility and that PilC2 serves to dampen twitching motility when PilC1 is present. Interestingly, the PilC1D930A and PilC1D930K mutations exhibited significantly reduced twitching motility compared to the parent and wild-type strains (P < 0.05), similar to the nontwitching ΔpilF and ΔpilC1 ΔpilC2 strains. In contrast, the ΔpilC1 PilC2D1444A strain demonstrated no defect in twitching motility compared to the parental ΔpilC1 strain and control ΔpilC1 PilC2mark strain, while the ΔpilC1 PilC2D1444K strain had a modest but statistically significant (P < 0.05) reduction in twitching zone. Reduced twitching zones were also evident for the PilC1D930A PilC2D1444A and PilC1D930K PilC2D1444K double mutants compared to those of the parental and wild-type strains. Despite the smaller twitching zones in these three mutants, the zones were significantly larger than the zones produced by the twitching-deficient controls (P < 0.05), indicating reduced and not absent twitching motility. Compared to the wild-type strain, the mutants fall into four distinct statistically significant categories: null (ΔpilF, ΔpilC1 ΔpilC2, ΔpilC2 PilC1D930A, and ΔpilC2 PilC1D930K strains), WT (WT, ΔpilC1, ΔpilC1 PilC2mark, and ΔpilC1 PilC2D1444A strains), <WT (ΔpilC1 PilC2D1444K, PilC1D930A PilC2D1444A, and PilC1D930K PilC2D1444K strains), and >WT (ΔpilC2 and ΔpilC2 PilC1mark strains). Together, these data demonstrate that the PilC1 Ca-binding site is essential for twitching motility, while the PilC2 Ca-binding site has only a minor influence on twitching motility.
Fig 5.
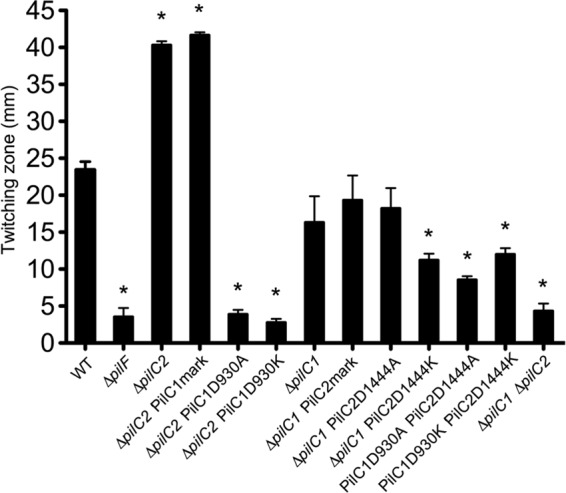
The PilC1 and PilC2 Ca-binding sites influence twitching motility. K. kingae strain KK03 and derivatives were assessed for twitching motility using a modified agar plate stab assay. Twitching-zone diameters were measured in triplicate, and averages were calculated from three independent experiments. Error bars represent standard errors of the means. Statistical analysis was performed using ANOVA with Bonferroni's posttest to compare twitching-motility zones between the mutants and the parental strains as noted in the text. Asterisks indicate statistically significant (P < 0.05) differences between the mutants and the relevant parental strain.
The PilC1 Ca-binding site but not the PilC2 Ca-binding site is necessary for adherence.
We next chose to investigate the influence of PilC1 and PilC2 Ca binding on adherence to human epithelial cells. Using a previously established adherence assay (14, 15), we determined the relative levels of adherence to Chang epithelial cells by the series of Ca-binding site mutants. Of note, we previously observed reduced adherence of the 269-492ΔpilC1 and 269-492pilC2::aphA3 mutants compared to strain 269-492 (14). In contrast, in this study, the ΔpilC1 derivative of KK03 had only a slight defect in adherence, and the ΔpilC2 derivative of KK03 had no defect in adherence, compared to the KK03 parent (Fig. 6). Examination of the adherence to Chang cells by the PilC Ca-binding mutants revealed stark differences between PilC1 and PilC2 (Fig. 6). The ΔpilC2 PilC1D930A and ΔpilC2 PilC1D930K strains exhibited marked deficiencies in adherence compared to the ΔpilC2 and ΔpilC2 PilC1mark control strains (P < 0.05) and adhered at levels similar to those of the nonpiliated ΔpilF and ΔpilC1 ΔpilC2 controls. However, the ΔpilC1 PilC2D1444A and ΔpilC1 PilC2D1444K strains displayed normal adherence, comparable to adherence by the parental ΔpilC1 and ΔpilC1 PilC2mark control strains. The PilC1D930A PilC2D1444A and PilC1D930K PilC2D1444K double mutants also adhered at levels similar to those of the ΔpilC1 strains that express either WT or mutant PilC2, indicating that PilC2 is able to promote adherence in the presence of Ca-binding mutant PilC1, regardless of PilC2 Ca-binding state. These results demonstrate that the PilC1 Ca-binding site is necessary and the PilC2 Ca-binding site is dispensable for adherence to Chang cells.
Fig 6.
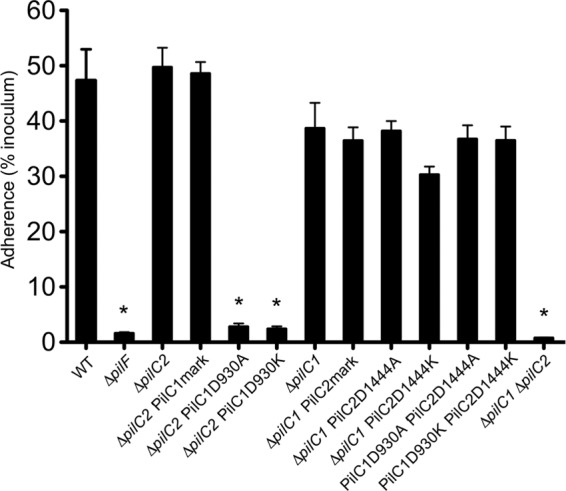
The PilC1 but not PilC2 Ca-binding site is essential for adherence. K. kingae strain KK03 and derivatives were investigated for adherence to Chang human epithelial cells using a quantitative adherence assay. Experiments were performed in triplicate, and averages were calculated from three independent experiments. Error bars represent standard errors of the means. Statistical analysis was performed using ANOVA and Bonferroni's posttest to compare adherence levels between the mutants and the parental strains as noted in the text. Asterisks indicate statistically significant (P < 0.05) differences between the mutants and the relevant parental strain.
DISCUSSION
K. kingae produces type IV pili that mediate adherence to human cells (14, 15). The PilC-like proteins play key roles in type IV pilus biology in K. kingae and numerous other Gram-negative bacteria of medical importance (17, 18). In this study, we found that PilC1 and PilC2 bind calcium in vitro and that significant differences in type IV pilus phenotypes exist in the PilC1 and PilC2 Ca-binding-site mutants. Both wild-type and Ca-binding mutant PilC1 and PilC2 were able to promote pilus production when expressed individually. However, Ca-binding mutant PilC1 eliminated twitching motility and adherence, while Ca-binding mutant PilC2 had little or no effect on twitching motility and no effect on adherence. Strains that expressed both PilC1 and PilC2 Ca-binding mutants demonstrated phenotypes identical to those of the PilC2 single mutant. These data demonstrate important differences in PilC1 and PilC2 control of type IV pilus phenotypes in K. kingae.
In the pathogenic Neisseria species, PilC1 and PilC2 are highly homologous to each other. N. gonorrhoeae PilC1 and PilC2 function interchangeably in promoting pilus biogenesis, natural competence, and adherence (39, 40). N. meningitidis PilC1 and PilC2 both promote pilus production and natural competence but differentially promote adherence to certain cell types (28, 30, 41, 42). Sequence analysis suggests that differences in PilC1- and PilC2-mediated adherence are based in the N-terminal portion of the proteins (43). Given the highly homologous nature of the PilC proteins in the pathogenic Neisseria species, especially in the C-terminal portion of the protein and in the 9-amino-acid putative Ca-binding loop, it is likely that Ca binding is an important factor for Neisseria type IV pilus biology. In contrast, the related Neisseriaceae family member K. kingae expresses two PilCs that share only limited sequence homology and are differentially controlled by Ca binding. We speculate that differences in host niches may be a driving factor influencing the mechanism of PilC function. K. kingae inhabits the posterior pharynx as a commensal but tends to cause disease primarily in the joints and bones, two calcium-rich sites. Of note, human plasma has a free calcium concentration of 1.03 to 1.30 mM, and human joint fluid has a calcium concentration of 4 mM or higher (44). These high-calcium environments may promote the Ca-dependent functions of PilC1, with no specific effect on PilC2. While calcium is the predominant divalent cation associated with the 9-amino-acid and 12-amino-acid consensus binding motifs, magnesium, zinc, and manganese may bind to these motifs as well (32, 45–48). In future studies, we will examine whether these additional divalent cations influence PilC1 or PilC2 function.
The first report that calcium plays a key role in regulating type IV pilus production and twitching motility described the study of PilY1, a PilC-like protein in P. aeruginosa (34). Orans et al. crystallized the C-terminal domain of PilY1 and identified a 9-amino-acid Ca-binding site. These investigators showed that mutation of the bidentate aspartic acid residue in the Ca-binding loop to alanine (D859A) resulted in significantly reduced pilus production and twitching motility, mimicking the effect of a ΔpilY1 mutation. Mutation of the same residue to lysine resulted in increased pilus production and reduced twitching motility, mimicking a ΔpilT pilus retraction mutation. Interestingly, K. kingae PilC1 contains a similar 9-amino-acid site, and mutation of the predicted bidentate residue to alanine or lysine eliminated twitching motility but did not have a major impact on surface pilus production, thus contrasting with PilY1. Hence, while control of twitching motility in PilY1 is due to the influence of the Ca-binding site on pilus production, the K. kingae PilC1 Ca-binding site directly impacts twitching motility and adherence, not surface pilus production. Interestingly, expression of the alanine or lysine mutant proteins in K. kingae resulted in identical phenotypes, again contrasting with PilY1. Orans et al. modeled the PilY1 D859K mutation and found that the amino group of the lysine side chain potentially mimics a calcium ion, creating a pseudo-calcium-bound PilY1 state. One potential reason for the differences observed between the K. kingae PilC1D930K mutant and the PilY1D859K mutant may be differences in the structures of the regions surrounding the Ca-binding loop in the two proteins, as the primary amino acid sequence of the loop is largely conserved (Fig. 1B).
To dissect the roles of PilC1 and the Ca-binding site in K. kingae type IV pilus-mediated twitching motility and adherence, it is important to examine the relationship between the two phenotypes. We have found that disruption of the K. kingae type IV pilus retraction machinery, pilTU, results in increased surface piliation, significantly reduced adherence, and loss of twitching motility (38). Interestingly, adherence by the ΔpilT mutant is reduced to approximately 50% of the wild type (38) rather than eliminated, as observed with the ΔpilC2 PilC1D930A and ΔpilC2 PilC1D930K mutants (Fig. 6). Taken together, these data suggest that the adherence defect of these two mutants is not completely a result of the defect in twitching motility but is a consequence of altered PilC1 adherence-promoting activity. In accord with these findings, Johnson et al. found that in vitro Ca binding by P. aeruginosa PilY1 influences RGD-mediated PilY1 binding to integrins, supporting a specific role for the 9-amino-acid Ca-binding site in adherence in this protein class (32).
In contrast to the situation with PilC1, the K. kingae PilC2 Ca-binding site is dispensable for twitching motility and adherence. We originally identified two potential Ca-binding sites in PilC2 with homology to the 12-amino-acid Ca-binding loop of human calmodulin and no sites with homology to the PilY1-like loop. In vitro analysis demonstrated that only one of these sites was functional. However, our data clearly show that PilC2 is able to promote pilus production, twitching motility, and adherence when the confirmed Ca-binding site is mutated. These results suggest that K. kingae PilC1 and PilC2 utilize different mechanisms in relation to Ca binding to promote twitching motility and adherence.
At this time, we can only speculate on the potential mechanisms by which Ca binding by PilC1 and PilC2 influences type IV pilus phenotypes. In the case of PilC1, we suggest that Ca binding or Ca responsiveness may be essential for proper protein structure. In this model, the Ca-binding-site mutant PilC1 has an altered protein conformation that retains the ability to promote pilus assembly but is not able to promote twitching motility and adherence. In contrast, PilC2 appears to function completely independently of Ca binding at the site identified in this report (Fig. 1B). We envision two potential scenarios to explain our findings: (i) PilC2 contains an additional cryptic Ca-binding site in the N-terminal 857 amino acids that were not included in the PilC2 fragment used for the in vitro Ca-binding studies, or (ii) PilC2 utilizes a novel mechanism that promotes twitching motility and adherence regardless of the functionality of the Ca-binding site. In the study by Johnson et al. that identified RGD-mediated PilY1 binding to integrins, analysis revealed a second Ca-binding site in PilY1 (32). While sequence analysis of PilC2 did not reveal any additional potential Ca-binding sites, we cannot rule out this possibility.
The results presented in this report establish key differences in the role of Ca binding in the function of the K. kingae PilC1 and PilC2 proteins. However, the precise mechanisms used by these proteins to control type IV pilus phenotypes remain unclear and will require future investigation. A greater understanding of K. kingae PilC1 and PilC2 function may provide insights into species-specific control mechanisms of type IV pilus phenotypes and suggest novel strategies for prevention or treatment of K. kingae disease.
Supplementary Material
Footnotes
Published ahead of print 14 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02186-12.
REFERENCES
- 1. Chometon S, Benito Y, Chaker M, Boisset S, Ploton C, Berard J, Vandenesch F, Freydiere AM. 2007. Specific real-time polymerase chain reaction places Kingella kingae as the most common cause of osteoarticular infections in young children. Pediatr. Infect. Dis. J. 26:377–381 [DOI] [PubMed] [Google Scholar]
- 2. Gené A, Garcia-Garcia JJ, Sala P, Sierra M, Huguet R. 2004. Enhanced culture detection of Kingella kingae, a pathogen of increasing clinical importance in pediatrics. Pediatr. Infect. Dis. J. 23:886–888 [DOI] [PubMed] [Google Scholar]
- 3. Lehours P, Freydiere AM, Richer O, Burucoa C, Boisset S, Lanotte P, Prere MF, Ferroni A, Lafuente C, Vandenesch F, Megraud F, Menard A. 2011. The rtxA toxin gene of Kingella kingae: a pertinent target for molecular diagnosis of osteoarticular infections. J. Clin. Microbiol. 49:1245–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moumile K, Merckx J, Glorion C, Berche P, Ferroni A. 2003. Osteoarticular infections caused by Kingella kingae in children: contribution of polymerase chain reaction to the microbiologic diagnosis. Pediatr. Infect. Dis. J. 22:837–839 [DOI] [PubMed] [Google Scholar]
- 5. Verdier I, Gayet-Ageron A, Ploton C, Taylor P, Benito Y, Freydiere AM, Chotel F, Berard J, Vanhems P, Vandenesch F. 2005. Contribution of a broad range polymerase chain reaction to the diagnosis of osteoarticular infections caused by Kingella kingae: description of twenty-four recent pediatric diagnoses. Pediatr. Infect. Dis. J. 24:692–696 [DOI] [PubMed] [Google Scholar]
- 6. Yagupsky P. 2004. Kingella kingae: from medical rarity to an emerging paediatric pathogen. Lancet Infect. Dis. 4:358–367 [DOI] [PubMed] [Google Scholar]
- 7. Yagupsky P, Dagan R, Howard CW, Einhorn M, Kassis I, Simu A. 1992. High prevalence of Kingella kingae in joint fluid from children with septic arthritis revealed by the BACTEC blood culture system. J. Clin. Microbiol. 30:1278–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yagupsky P, Porsch E, St Geme JW., III 2011. Kingella kingae: an emerging pathogen in young children. Pediatrics 127:557–565 [DOI] [PubMed] [Google Scholar]
- 9. Yagupsky P, Dagan R, Prajgrod F, Merires M. 1995. Respiratory carriage of Kingella kingae among healthy children. Pediatr. Infect. Dis. J. 14:673–678 [DOI] [PubMed] [Google Scholar]
- 10. Yagupsky P, Peled N, Katz O. 2002. Epidemiological features of invasive Kingella kingae infections and respiratory carriage of the organism. J. Clin. Microbiol. 40:4180–4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yagupsky P, Porat N, Pinco E. 2009. Pharyngeal colonization by Kingella kingae in children with invasive disease. Pediatr. Infect. Dis. J. 28:155–157 [DOI] [PubMed] [Google Scholar]
- 12. Yagupsky P, Weiss-Salz I, Fluss R, Freedman L, Peled N, Trefler R, Porat N, Dagan R. 2009. Dissemination of Kingella kingae in the community and long-term persistence of invasive clones. Pediatr. Infect. Dis. J. 28:707–710 [DOI] [PubMed] [Google Scholar]
- 13. Basmaci R, Ilharreborde B, Bidet P, Doit C, Lorrot M, Mazda K, Bingen E, Bonacorsi S. 2012. Isolation of Kingella kingae in the oropharynx during K. kingae arthritis in children. Clin. Microbiol. Infect. 18:E134–E136 doi:10.1111/j.1469-0691.2012.3799.x [DOI] [PubMed] [Google Scholar]
- 14. Kehl-Fie TE, Miller SE, St Geme JW., III 2008. Kingella kingae expresses type IV pili that mediate adherence to respiratory epithelial and synovial cells. J. Bacteriol. 190:7157–7163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kehl-Fie TE, Porsch EA, Miller SE, St Geme JW., III 2009. Expression of Kingella kingae type IV pili is regulated by sigma54, PilS, and PilR. J. Bacteriol. 191:4976–4986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kehl-Fie TE, Porsch EA, Yagupsky P, Grass EA, Obert C, Benjamin DK, Jr, St Geme JW., III 2010. Examination of type IV pilus expression and pilus-associated phenotypes in Kingella kingae clinical isolates. Infect. Immun. 78:1692–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pelicic V. 2008. Type IV pili: e pluribus unum? Mol. Microbiol. 68:827–837 [DOI] [PubMed] [Google Scholar]
- 18. Burrows LL. 2005. Weapons of mass retraction. Mol. Microbiol. 57:878–888 [DOI] [PubMed] [Google Scholar]
- 19. Wolfgang M, Lauer P, Park HS, Brossay L, Hebert J, Koomey M. 1998. PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Mol. Microbiol. 29:321–330 [DOI] [PubMed] [Google Scholar]
- 20. Anantha RP, Stone KD, Donnenberg MS. 1998. Role of BfpF, a member of the PilT family of putative nucleotide-binding proteins, in type IV pilus biogenesis and in interactions between enteropathogenic Escherichia coli and host cells. Infect. Immun. 66:122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chiang P, Sampaleanu LM, Ayers M, Pahuta M, Howell PL, Burrows LL. 2008. Functional role of conserved residues in the characteristic secretion NTPase motifs of the Pseudomonas aeruginosa type IV pilus motor proteins PilB, PilT and PilU. Microbiology 154:114–126 [DOI] [PubMed] [Google Scholar]
- 22. Merz AJ, So M, Sheetz MP. 2000. Pilus retraction powers bacterial twitching motility. Nature 407:98–102 [DOI] [PubMed] [Google Scholar]
- 23. Whitchurch CB, Hobbs M, Livingston SP, Krishnapillai V, Mattick JS. 1991. Characterisation of a Pseudomonas aeruginosa twitching motility gene and evidence for a specialised protein export system widespread in eubacteria. Gene 101:33–44 [DOI] [PubMed] [Google Scholar]
- 24. Wolfgang M, Park HS, Hayes SF, van Putten JP, Koomey M. 1998. Suppression of an absolute defect in type IV pilus biogenesis by loss-of-function mutations in pilT, a twitching motility gene in Neisseria gonorrhoeae. Proc. Natl. Acad. Sci. U. S. A. 95:14973–14978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heiniger RW, Winther-Larsen HC, Pickles RJ, Koomey M, Wolfgang MC. 2010. Infection of human mucosal tissue by Pseudomonas aeruginosa requires sequential and mutually dependent virulence factors and a novel pilus-associated adhesin. Cell. Microbiol. 12:1158–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morand PC, Bille E, Morelle S, Eugene E, Beretti JL, Wolfgang M, Meyer TF, Koomey M, Nassif X. 2004. Type IV pilus retraction in pathogenic Neisseria is regulated by the PilC proteins. EMBO J. 23:2009–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jonsson AB, Nyberg G, Normark S. 1991. Phase variation of gonococcal pili by frameshift mutation in pilC, a novel gene for pilus assembly. EMBO J. 10:477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nassif X, Beretti JL, Lowy J, Stenberg P, O'Gaora P, Pfeifer J, Normark S, So M. 1994. Roles of pilin and PilC in adhesion of Neisseria meningitidis to human epithelial and endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 91:3769–3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rudel T, Scheurerpflug I, Meyer TF. 1995. Neisseria PilC protein identified as type-4 pilus tip-located adhesin. Nature 373:357–359 [DOI] [PubMed] [Google Scholar]
- 30. Rahman M, Kallstrom H, Normark S, Jonsson AB. 1997. PilC of pathogenic Neisseria is associated with the bacterial cell surface. Mol. Microbiol. 25:11–25 [DOI] [PubMed] [Google Scholar]
- 31. Scheuerpflug I, Rudel T, Ryll R, Pandit J, Meyer TF. 1999. Roles of PilC and PilE proteins in pilus-mediated adherence of Neisseria gonorrhoeae and Neisseria meningitidis to human erythrocytes and endothelial and epithelial cells. Infect. Immun. 67:834–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson MD, Garrett CK, Bond JE, Coggan KA, Wolfgang MC, Redinbo MR. 2011. Pseudomonas aeruginosa PilY1 binds integrin in an RGD- and calcium-dependent manner. PLoS One 6:e29629 doi:10.1371/journal.pone.0029629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alm RA, Hallinan JP, Watson AA, Mattick JS. 1996. Fimbrial biogenesis genes of Pseudomonas aeruginosa: pilW and pilX increase the similarity of type 4 fimbriae to the GSP protein-secretion systems and pilY1 encodes a gonococcal PilC homologue. Mol. Microbiol. 22:161–173 [DOI] [PubMed] [Google Scholar]
- 34. Orans J, Johnson MD, Coggan KA, Sperlazza JR, Heiniger RW, Wolfgang MC, Redinbo MR. 2010. Crystal structure analysis reveals Pseudomonas PilY1 as an essential calcium-dependent regulator of bacterial surface motility. Proc. Natl. Acad. Sci. U. S. A. 107:1065–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alm RA, Mattick JS. 1995. Identification of a gene, pilV, required for type 4 fimbrial biogenesis in Pseudomonas aeruginosa, whose product possesses a pre-pilin-like leader sequence. Mol. Microbiol. 16:485–496 [DOI] [PubMed] [Google Scholar]
- 36. Kehl-Fie TE, St Geme JW., III 2007. Identification and characterization of an RTX toxin in the emerging pathogen Kingella kingae. J. Bacteriol. 189:430–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Linse S, Helmersson A, Forsen S. 1991. Calcium binding to calmodulin and its globular domains. J. Biol. Chem. 266:8050–8054 [PubMed] [Google Scholar]
- 38. Porsch EA, Kehl-Fie TE, Geme JW., III 2012. Modulation of Kingella kingae adherence to human epithelial cells by type IV pili, capsule, and a novel trimeric autotransporter. mBio 3:e00372–12 doi:10.1128/mBio.00372-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rudel T, van Putten JP, Gibbs CP, Haas R, Meyer TF. 1992. Interaction of two variable proteins (PilE and PilC) required for pilus-mediated adherence of Neisseria gonorrhoeae to human epithelial cells. Mol. Microbiol. 6:3439–3450 [DOI] [PubMed] [Google Scholar]
- 40. Rudel T, Facius D, Barten R, Scheuerpflug I, Nonnenmacher E, Meyer TF. 1995. Role of pili and the phase-variable PilC protein in natural competence for transformation of Neisseria gonorrhoeae. Proc. Natl. Acad. Sci. U. S. A. 92:7986–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ryll RR, Rudel T, Scheuerpflug I, Barten R, Meyer TF. 1997. PilC of Neisseria meningitidis is involved in class II pilus formation and restores pilus assembly, natural transformation competence and adherence to epithelial cells in PilC-deficient gonococci. Mol. Microbiol. 23:879–892 [DOI] [PubMed] [Google Scholar]
- 42. Morand PC, Drab M, Rajalingam K, Nassif X, Meyer TF. 2009. Neisseria meningitidis differentially controls host cell motility through PilC1 and PilC2 components of type IV pili. PLoS One 4:e6834 doi:10.1371/journal.pone.0006834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morand PC, Tattevin P, Eugene E, Beretti JL, Nassif X. 2001. The adhesive property of the type IV pilus-associated component PilC1 of pathogenic Neisseria is supported by the conformational structure of the N-terminal part of the molecule. Mol. Microbiol. 40:846–856 [DOI] [PubMed] [Google Scholar]
- 44. Maroudas A. 1979. Physiochemical properties of articular cartilage, p 215–290 In Freeman MAR.(ed), Adult articular cartilage. Pitman Medical, Kent, United Kingdom [Google Scholar]
- 45. Bogden JD, Singh NP, Joselow MM. 1974. Cadmium, lead, and zinc concentrations in whole-blood samples of children. Environ. Sci. Technol. 8:740–742 [Google Scholar]
- 46. Kawasaki H, Kurosu Y, Kasai H, Isobe T, Okuyama T. 1986. Limited digestion of calmodulin with trypsin in the presence or absence of various metal ions. J. Biochem. 99:1409–1416 [DOI] [PubMed] [Google Scholar]
- 47. Milne DB, Sims RL, Ralston NV. 1990. Manganese content of the cellular components of blood. Clin. Chem. 36:450–452 [PubMed] [Google Scholar]
- 48. Warren JT, Guo Q, Tang WJ. 2007. A 1.3-A structure of zinc-bound N-terminal domain of calmodulin elucidates potential early ion-binding step. J. Mol. Biol. 374:517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 50. Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. 2002. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 25:8–15 [DOI] [PubMed] [Google Scholar]
- 51. Donnelly MI, Zhou M, Millard CS, Clancy S, Stols L, Eschenfeldt WH, Collart FR, Joachimiak A. 2006. An expression vector tailored for large-scale, high-throughput purification of recombinant proteins. Protein Expr. Purif. 47:446–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hendrixson DR, Akerley BJ, DiRita VJ. 2001. Transposon mutagenesis of Campylobacter jejuni identifies a bipartite energy taxis system required for motility. Mol. Microbiol. 40:214–224 [DOI] [PubMed] [Google Scholar]
- 53. Seifert HS. 1997. Insertionally inactivated and inducible recA alleles for use in Neisseria. Gene 188:215–220 [DOI] [PubMed] [Google Scholar]
- 54. Hamilton HL, Schwartz KJ, Dillard JP. 2001. Insertion-duplication mutagenesis of Neisseria: use in characterization of DNA transfer genes in the gonococcal genetic island. J. Bacteriol. 183:4718–4726 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.