

Abstract
Clostridium difficile, a proteolytic Gram-positive anaerobe, has emerged as a significant nosocomial pathogen. Stickland fermentation reactions are thought to be important for growth of C. difficile and appear to influence toxin production. In Stickland reactions, pairs of amino acids donate and accept electrons, generating ATP and reducing power in the process. Reduction of the electron acceptors proline and glycine requires the d-proline reductase (PR) and the glycine reductase (GR) enzyme complexes, respectively. Addition of proline in the medium increases the level of PR protein but decreases the level of GR. We report the identification of PrdR, a protein that activates transcription of the PR-encoding genes in the presence of proline and negatively regulates the GR-encoding genes. The results suggest that PrdR is a central metabolism regulator that controls preferential utilization of proline and glycine to produce energy via the Stickland reactions.
INTRODUCTION
Clostridium difficile, an anaerobic, Gram-positive, spore-forming bacterium, is an emerging nosocomial pathogen and the etiological agent of antibiotic-associated diarrhea and pseudomembranous colitis (1–3). In recent years, the severity and incidence of C. difficile infection have increased due to the emergence in North America and Europe of highly virulent NAP1/027 C. difficile strains (4, 5). Most pathogenic C. difficile strains produce two large cytotoxins (TcdA and TcdB) that are the primary virulence factors (6, 7).
Toxin production in C. difficile responds to environmental conditions, including the availability of specific nutrients, temperature changes, and alteration of the redox potential (8–10). In addition, the presence of a rapidly metabolizable carbon source (11, 12) or certain amino acids (13, 14) inhibits toxin gene expression. For instance, addition to the growth medium of a mixture of nine amino acids (cysteine, isoleucine, leucine, valine, methionine, threonine, tryptophan, glycine, and proline) strongly reduces toxin yield (12, 14). When cells are grown in rich medium, the toxin genes are transcribed only when the cells reach stationary phase (11, 15).
Some of the molecular mechanisms regulating C. difficile toxin gene expression have been elucidated (8, 11, 16–19). Transcription of the tcdA and tcdB genes depends on TcdR, an alternative sigma factor that directs RNA polymerase to the toxin gene promoters (20). Transcription of the tcdR gene is repressed during the rapid exponential growth phase by CodY, a global regulator of metabolism and of genes that allow adaptation to nutrient limitation (19, 21). CodY is active in cells with an excess of branched-chain amino acids (isoleucine, leucine, and valine) and GTP (22–24). When the cells reach stationary phase, the intracellular concentrations of these ligands decrease and CodY is less able to bind as a repressor, leading to derepression of tcdR transcription. How C. difficile responds to the levels of other amino acids to modulate toxin expression remains unclear.
The activity of TcdR is modulated by TcdC, an anti-sigma factor that destabilizes the TcdR–core RNA polymerase complex (25). TcdC seems to be most active in rapidly growing cells. Other sigma factors, such as SigD and SigH, are involved in toxin gene transcription by unknown mechanisms (8, 18).
Regulation of toxin production by the carbon source is mediated by CcpA, a global regulator of carbon metabolism in Gram-positive bacteria (16). CcpA is a direct repressor of the tcdA and tcdB genes. The roles of CodY and CcpA reflect a tight coupling between metabolism and virulence in Clostridium spp. and other pathogenic bacteria (26, 27).
In several nonpathogenic Clostridium species (e.g., C. sporogenes and C. sticklandii), amino acid metabolism by the Stickland reactions is a primary source of energy when bacteria are grown with amino acids as sole sources of carbon and nitrogen (28–31). Stickland reactions couple metabolism of pairs of amino acids in which one amino acid, acting as an electron donor, is oxidatively deaminated or decarboxylated and a second amino acid, acting as an electron acceptor, is reduced or reductively deaminated (Fig. 1). The most efficient electron donors are leucine, isoleucine, and alanine, and the most efficient acceptors are glycine, proline, and hydroxyproline (30, 32, 33). In C. sticklandii, the reduction of the Stickland acceptors glycine and proline is performed by two selenium-dependent reductases, glycine reductase (GR) and d-proline reductase (PR), respectively. GR catalyzes the reductive deamination of glycine to acetyl phosphate and ammonium, and PR reductively cleaves d-proline to 5-aminovalerate (34–37). Jackson et al. (38) showed that C. difficile growth requires selenium in a limiting basal medium with torula yeast extract, suggesting a key role of the Stickland reactions in C. difficile physiology (39). Based on sequence homology to C. sticklandii, genes encoding the C. difficile GR and PR subunits are clustered in two distinct genetic loci (grd and prd, respectively) on the chromosome of C. difficile strain 630 (38). The grd locus contains eight genes (Fig. 2). Two of the genes, grdA and grdB, likely encode the selenocysteine-containing subunits of GR (38). The seven genes of the putative prd operon (Fig. 2) include prdF, predicted to encode a d-proline racemase, and prdB, predicted to encode the selenium-containing subunit of PR. It is interesting to note that proP, downstream of the grd operon, is predicted to encode a proline aminopeptidase, suggesting a link between metabolism of glycine and that of proline (38). Moreover, Jackson et al. (38) found that proline and glycine specifically stimulate production of the selenoenzymes PR and GR, respectively. Interestingly, addition to the medium of glycine and proline in combination with other amino acids has been shown to decrease toxin yield (14). Therefore, a detailed understanding of the amino acid metabolism pathways and their regulation may give important insights into C. difficile pathogenesis.
Fig 1.

Overview of Stickland metabolism. Stickland reactions couple oxidation and reduction of amino acid pairs. d-Proline reductase catalyzes the reductive cleavage of the d-proline ring to yield δ-aminovaleric acid. Glycine reductase catalyzes the reductive deamination of glycine to acetyl phosphate to generate ATP via substrate-level phosphorylation through acetate kinase.
Fig 2.
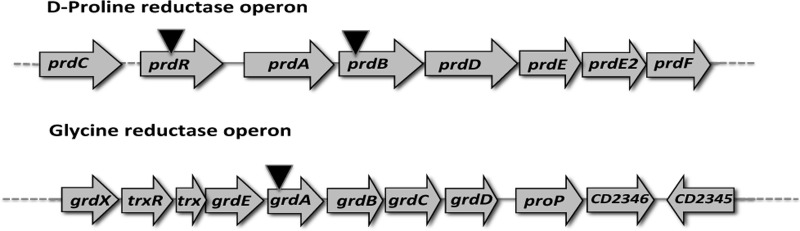
Organization of the d-proline reductase and glycine reductase gene clusters. Arrows indicate the locations of group II intron insertions in various mutant strains. The coordinates refer to the positions of the gene clusters in the genome of C. difficile strain 630.
In this study, we investigated the transcriptional regulation of the C. difficile grd and prd gene clusters. We found that addition of proline to the growth medium increases the expression of the prd operon and decreases the expression of the grd operon. By constructing prd and grd null mutant strains, we were able to demonstrate the interdependent regulation of the two operons. That is, expression of a functional PR decreased transcription of the grd operon. Furthermore, we identified PrdR, the product of a gene located upstream of the prd operon, as the mediator of proline-dependent activation of the prd operon and proline-dependent repression of the grd operon.
MATERIALS AND METHODS
Strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. C. difficile strains were grown routinely in TY medium (11), supplemented with 250 μg/ml d-cycloserine, 40 μg/ml kanamycin, 20 μg/ml thiamphenicol, or 5 μg/ml erythromycin as needed. C. difficile strains were maintained at 37°C in an anaerobic chamber (Coy Laboratory Products) with an atmosphere of 10% H2, 5% CO2, and 85% N2. Escherichia coli strains were grown at 37°C in L medium supplemented with 20 μg/ml chloramphenicol or 100 μg/ml ampicillin, as needed.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Characteristics | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| HB101 | F− supE44 hsdS20(rB− mB−) recA13 ara-14 proA2 lacY1 galK2 rpsL20 xyl-5 mtl-1 | B. Dupuy |
| DH5α | fhuA2 Δ(argF-lacZ)U169 phoA glnV44 Φ80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | New England Biolabs, USA |
| C. difficile | ||
| JIR8094 | Ems derivative of strain 630 | 19 |
| LB-CD4 | JIR8094 prdB::ermB | This study |
| LB-CD12 | JIR8094 grdA::ermB | This study |
| LB-CD8 | JIR8094 prdR::ermB | This study |
| LB-CD13 | JIR8094 pBL58 | This study |
| LB-CD14 | LB-CD8 pBL93 | This study |
| Plasmids | ||
| pBL33 | pCE240 targeted to prdB | This study |
| pBL64 | pCR2.1-intron template part A | This study |
| pBL65 | pCR2.1-intron template part B | This study |
| pBL67 | pCE240 targeted to prdR | This study |
| pBL68 | pMC123 without HindIII and BsrGI sites | This study |
| pBL71 | pMC123 containing groupII intron targeted to prdR from pBL67 | |
| pBL91 | pBL100 targeted to grdA | This study |
| pBL93 | pBL68 containing prdR | This study |
| pBL100 | pBL68 containing un-targeted groupII intron–TargeTron vector | This study |
| pCE240 | Derivative of pJIR750ai (Sigma-Aldrich), Tmr | 42 |
| pCR2.1 | Invitrogen | |
| pMC123 | Ampr Tcr | 44 |
| pRK24 | Tra+ Mob+ Ampr Tcr | 45 |
Growth behavior analyses of C. difficile strains were performed in TY medium, with or without supplementation as indicated below, in a microtiter plate format. Briefly, mid-exponential-phase cultures adjusted to an optical density at 600 nm (OD600) of 0.1 were diluted (1:10) in 200 μl TY medium supplemented with proline (30 mM) or glycine (30 mM) in wells of a 96-well microtiter plate (Costar). Cultures were grown anaerobically at 37°C. At 10-min intervals over a 12-h period, the plates were agitated for 2 min before the OD600 was recorded using a plate reader (Bio-Tek). Data from two biological replicates, each with six technical replicates, were used to construct the growth curve for each strain.
Strain and plasmid construction.
Oligonucleotides used in this study are listed in Table 2. C. difficile strain 630 DNA, GenBank accession number AM180355 (41), was used as a template for PCR amplification. Sequencing of cloned DNA fragments was performed by the Tufts University Core Facility using an ABI 3130XL DNA sequencer.
Table 2.
Primers used in this study
| Primer name | Sequence |
|---|---|
| EBS Universal | CGAAATTAGAAACTTGCGTTCAGTAAAC |
| oLB64 | AAAAGCTTTTGCAACCCACGTCGATCGTGAATTCTGCAGCTAAGTGCGCCCAGATAGGGTG |
| oLB65 | CAGATTGTACAAATGTGGTGATAACAGATAAGTCAGCTAATTTAACTTACCTTTCTTTGT |
| oLB66 | CGCAAGTTTCTAATTTCGGTTCAGAATCGATAGAGGAAAGTGTCT |
| oLB67 | AAAAGCTTTTGCAACCCACGTCGATCGTGAAGACCCAAGTAAAGTGCGCCCAGATAGGGTG |
| oLB78 | TCCTCCAGTTTGGACTCCTGTAAC |
| oLB79 | ATTGGAACTAATGGAGCAACTG |
| oLB99 | AAAAGCTTTTGCAACCCACGTCGATCGTGAACAGTTACTGTTTGTGCGCCCAGATAGGGTG |
| oLB100 | CAGATTGTACAAATGTGGTGATAACAGATAAGTCCTGTTTCGTAACTTACCTTTCTTTGT |
| oLB101 | CGCAAGTTTCTAATTTCGGTTAACTGTCGATAGAGGAAAGTGTCT |
| oLB102 | AAAAGCTTTTGCAACCCACGTCGATCGTGAAATGTATCATTTAGTGCGCCCAGATAGGGTG |
| oLB103 | CAGATTGTACAAATGTGGTGATAACAGATAAGTCCATTTACTTAACTTACCTTTCTTTGT |
| oLB104 | CGCAAGTTTCTAATTTCGGTTTACATTCGATAGAGGAAAGTGTCT |
| oLB118 | ATAATAGGTGACCGTGATGG |
| oLB119 | TTCTTCTCTTATACCAGACATTTC |
| oLB120 | ATAGGGATTCTTCCTGTTCTTAG |
| oLB121 | TCCTATGTCTTCTTTTCTCTCTCT |
| oLB122 | CTAGCTGCTCCTATGTCTCACATC |
| oLB123 | CCAGTCTCTCCTGGATCAACTA |
| oLB131 | GTATGGATAGGTGGAGAAGTCA |
| oLB132 | CTCTTCCTCTAGTAGCTGTAATGC |
| oLB261 | CTATAGACTCTCATACAGCAGGTG |
| oLB262 | CATTATGTCCTCTTGGCTCTAAC |
| oLB265 | GGGAGAGGGTATTACACATACT |
| oLB266 | GGAGTTCCATATCTCCCAAATACC |
| oLB170 | GGTCAAGTACTAGGAGCTAAGT |
| oLB171 | CTACTTCTTCTTTAGCCTCTCCTG |
| oLB176 | CCCTGGTATCATGTCTAAAGTTG |
| oLB177 | GAGTATACTTAGCTCCTTCTCCAG |
| oLB187 | CCCGAATTCTGGATACAGGCATTCATTAATTGT |
| oLB188 | CCCAAGCTTAACACATCTATATACACAACAC |
| oLB221 | GAACTCTAATCGCAGAACCAAC |
| oLB222 | CATTCCTACTACTCCAGCTTCT |
| oLB277 | GTAGGAGAACCTATGGGAAC |
| oLB278 | TCCTTACCATTTAAGCACAC |
Null mutations in prdB and prdR were created in several steps. First, plasmids pBL33 and pBL67 were constructed by retargeting of the erm gene-containing group II intron from pCE240 (42), using for prdB the primers oLB64, oLB65, oLB66, and the EBS Universal primer and for prdR the primers oLB102, oLB103, oLB104, and the EBS Universal primer as outlined in the TargeTron user manual (Sigma-Aldrich), followed by initial cloning of the retargeted fragment in pCE240 digested with BsrGI/HindIII. Plasmid pCE240 is a derivative of pJIR750ai (Sigma-Aldrich) and is similar to pMTL007 (43). The retargeted group II intron from pBL67 was then extracted by digestion with SfoI and SphI and cloned between the SphI and SnaBI sites of pMC123 (44), resulting in pBL71. pBL33 (targeted for prdB) and pBL71 (targeted for prdR) were then introduced by transformation into E. coli strain HB101(pRK24). pRK24 is a derivative of the broad-host-range plasmid RP4, which mobilizes IncP oriT plasmids. The resulting strains were then mated with C. difficile strain JIR8094, resulting in transfer of pBL33 and pBL71 by conjugation, as previously described (45), except that transconjugants were selected on BHIS plates (45) supplemented with d-cycloserine, kanamycin, and thiamphenicol. Inactivation of prdB and prdR was selected for by screening transconjugants for erythromycin resistance, resulting in strains LB-CD4 and LB-CD8, respectively. These strains were also thiamphenicol sensitive, indicating that they no longer carried the replicating plasmid. Insertional disruption of prdB or prdR was confirmed using primer pairs oLB78/oLB79 and oLB120/oLB121, which anneal outside the region of insertion in the prdB and prdR genes, respectively.
To interrupt grdA and to make future mutant construction in C. difficile easier, we created a new set of plasmids for retargeting of the group II intron. We amplified two portions of the intron by PCR using pBL71 as the template and primers oLB102/EBS Universal and oLB103/oLB104. Amplicons were TA cloned in the pCR2.1 vector (Invitrogen), resulting in pBL64 and pBL65. A mixture (1:1) of pBL64 and pBL65 was used as the template to generate the retargeted intron fragment by mutagenic PCR.
We generated a grdA targeted intron fragment using primers oLB99, oLB100, oLB101, and EBS Universal and a pBL64-pBL65 mixed template. Following HindIII/BsrGI digestion, the PCR fragment was cloned in HindIII/BsrGI-digested pBL100 (see below). The resulting plasmid, pBL91, was introduced into E. coli strain HB101(pRK24), and the obtained strain was subsequently mated with C. difficile JIR8094 as described above. Insertional disruption was verified by PCR using primers oLB118 and oLB119.
To construct pBL100, the BsrGI and HindIII sites of pMC123 (44) were first removed in multiple steps by PCR-directed mutagenesis and blunting of digested DNA, creating pBL68. Then, pBL68 was digested by EcoRI and blunted using Klenow fragment (New England BioLabs, USA) and subsequently digested by SphI. The intron portion of pCE240 was extracted by SphI/SfoI digestion and cloned in digested pBL68, yielding pBL100.
To complement the prdR disruption, a 2,119-bp fragment containing the prdR gene and its upstream region was amplified using primers oLB187 and oLB188 and cloned between the EcoRI and HindIII sites of pBL58, generating pBL93. This plasmid was introduced into LB-CD8 as previously described (45), resulting in LB-CD14.
Southern hybridization.
C. difficile genomic DNA was digested with HindIII and subjected to agarose gel electrophoresis (0.8%). Digested DNA was then transferred to a nylon membrane (pore size = 0.45 μm) in 20× saline-sodium citrate (SSC; 1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) according to the method of Southern (46). The membrane was prehybridized for 3 h at 42°C in 50% formamide, 5× SSC, 2× Denhardt's solution, and 100 mg denatured salmon sperm DNA per ml. Overnight hybridization was carried out in the same solution at 42°C with an intron-specific, 32P-labeled PCR fragment generated using oLB277 and oLB278 as primers. The membrane was washed for 30 min in 1× SSC–0.1% SDS and for 30 min in 0.1× SSC–0.1% SDS at room temperature. Radioactive bands were visualized by PhosphorImager analysis (Molecular Dynamics).
qPCR analysis.
Cultures of C. difficile grown in TY medium were harvested at OD600s of 0.4 to 0.7 (mid-exponential phase) or 1.2 to 1.4 (stationary phase), and DNA-free RNA was prepared as previously described (19, 44). RNA was quantitated by absorbance (A260 and A260/A280 ratio) using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). Primers for quantitative reverse transcription-PCR (qPCR) were designed using the online PrimerQuest tool from Integrated DNA Technologies (http://www.idtdna.com/Scitools/Applications/Primerquest), and amplification efficiencies for each primer set were determined prior to use. To control for chromosomal DNA contamination, mock cDNA synthesis reaction mixtures containing no reverse transcriptase were used as negative controls in subsequent amplifications. cDNA samples were diluted 4-fold and used as the templates for qPCR of rpoC (primers oLB122/oLB123), prdA (primers oLB170/oLB171), prdC (primers oLB221/oLB222), prdD (primers oLB265/oLB266), prdF (primers oLB261/oLB262), grdE (primers oLB176/oLB177), and tcdA (primers oLB131/oLB132) using Roche SYBR green I PCR mix and a Roche LightCycler 480 II thermocycler. Reactions were performed in a final volume of 20 μl using 4 μl diluted cDNA and 1 μM each primer. Reactions were performed in triplicate using cDNA extracted from each of a minimum of three biological replicates, and results are presented as the means and standard deviations of the data obtained. Amplification included 45 cycles of the following steps: 10 s at 95°C, 10 s at 53°C, and 15 s at 72°C. Results were calculated using the comparative cycle threshold method (47), in which the amount of target mRNA is normalized to that of an internal control transcript (rpoC).
Proline assay.
Overnight cultures in TY medium were diluted in TY medium to an OD600 of 0.05 and incubated anaerobically at 37°C. Cells were harvested at mid-exponential growth phase (OD600 of 0.4 to 0.7) by centrifugation (10 min at 4,000 rpm) and resuspended in 3% (wt/vol) 5-sulfosalicylic acid. Silica-glass beads (0.1 mm) were added, and cells were disrupted using a Mini-BeadBeater (BioSpec Products). Silica beads and cell debris were removed by centrifugation. The proline concentration was determined by a published procedure (48), and the results were expressed relative to those for the wild-type strain.
75Se labeling experiment.
C. difficile strains were cultivated in TY medium with or without sodium selenite (50 nM, 4 μCi of 75Se, University of Missouri Research Reactor), and cell extracts were obtained as previously described (38). The protein concentration was determined by the Bradford assay (49) using bovine serum albumin as a standard. Selenoproteins in cell extracts were identified after electrophoresis in 15% polyacrylamide gels containing SDS and visualization of radioisotope bands by PhosphorImager analysis (Molecular Dynamics), after overlaying a Coomassie blue-stained gel to determine the approximate molecular weights of the radioactive protein bands.
RESULTS
Stickland-type metabolism of proline is required for optimal growth of C. difficile.
In many Clostridium spp., including C. difficile, Stickland reactions are thought to be important for growth, at least under certain conditions (38, 50). In order to confirm the role of Stickland reactions in growth of C. difficile, we introduced insertion mutations into the prdB and grdA genes (Fig. 2). Insertional disruptions were verified by PCR (see Materials and Methods), and a Southern blot confirmed that each mutant strain had a single intron insertion (Fig. 3A). The resulting strains, LB-CD4 and LB-CD12, respectively, were tested for the ability to produce the selenoenzymes PR and GR by labeling of the proteins with 75Se. Figure 3B shows that three major radioactive bands corresponding to the PrdB subunit of PR and the GrdA and GrdB subunits of GR were detected in extracts of wild-type cells. No band corresponding to the PrdB subunit of PR was detected in strain LB-CD4 (prdB mutant), and no bands corresponding to the GrdA and GrdB subunits of GR were detected in the grdA null mutant. The lack of the GrdB band in the grdA null mutant suggests that the grdA mutation is polar (grdB is downstream of grdA [Fig. 2]). These results indicate that LB-CD4 and LB-CD12 are defective in producing the cysteine-containing subunits of the selenoenzyme complexes PR and GR, respectively.
Fig 3.
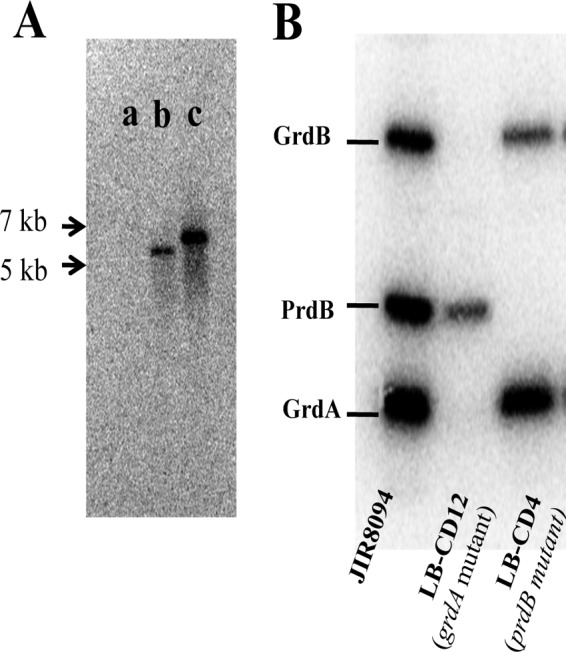
Mutant characterization. (A) Southern blot analysis of HindIII-digested genomic DNA from wild-type (JIR8094) (a), prdB mutant (LB-CD4) (b), and grdA mutant (LB-CD12) (c) strains with an intron probe. (B) Radiolabeling (75Se) of C. difficile wild-type and mutant strains. Wild-type (JIR8094), grdA mutant (LB-CD12), and prdB mutant (LB-CD4) strains were grown in TY medium with 75Se (4 μCi) for 24 h, harvested, and lysed by sonication. The extracts were subjected to SDS-15% polyacrylamide gel electrophoresis.
We then assessed the growth behavior of the mutant strains in TY medium supplemented with 30 mM proline (Fig. 4A) or 30 mM glycine (Fig. 4B). The growth rate of the mutant strains was similar to that of the wild-type (JIR8094) strain in TY medium alone (Table 3) and in TY medium supplemented with glycine (Fig. 4B and Table 3). In contrast, addition of l-proline to the medium increased the growth rate of the wild-type and the grdA mutant strains but had no effect on the doubling time or the growth behavior of the prdB mutant (LB-CD4) (Fig. 4A and Table 3). It should be noted, however, that at low cell densities, the growth rates of all strains in all media were similar, suggesting that the bacteria do not begin to utilize proline until some other constituents of TY medium have been consumed. In addition, the growth of the mutant strains in defined medium (14) supplemented with proline was appraised. Surprisingly, mutants were viable and did not exhibit a severe growth defect compared to the wild type (data not shown), suggesting that other metabolic pathways bypass the lack of a functional PR or GR. Therefore, the proline reductase pathway is needed for optimal growth of C. difficile in proline-supplemented medium, but the glycine reduction pathway does not play a significant role in the growth of C. difficile under the conditions tested.
Fig 4.

Growth of C. difficile wild-type and mutant strains supplemented with l-proline or glycine. C. difficile wild-type (JIR8094 [○]), LB-CD4 (prdB::ermB [◇]), and LB-CD12 (grdA::ermB [△]) strains were grown in TY medium alone or supplemented with 30 mM l-proline (A) or 30 mM glycine (B). The C. difficile wild type (JIR8094 [●]) was grown in TY medium alone. Cultures were grown anaerobically in a microtiter plate format at 37°C, and OD600 measurements were taken every 10 min using a plate reader (Bio-Tek).
Table 3.
Doubling times of wild-type and mutant strains in TY medium with proline and glycinea
| Strain | Genotype | Doubling time (min) in: |
||
|---|---|---|---|---|
| TY medium alone | TY medium + proline | TY medium + glycine | ||
| JIR8094 | Wild type | 55.41 ± 2.00 | 41.51 ± 0.18 | 56.51 ± 2.72 |
| LB-CD4 | prdB::erm | 57.43 ± 2.43 | 55.81 ± 0.68 | 54.43 ± 2.57 |
| LB-CD12 | grdA::erm | 57.44 ± 3.00 | 39.11 ± 0.57 | 58.67 ± 1.57 |
| LB-CD8 | prdR::erm | 63.83 ± 6.99 | 70.41 ± 4.13 | 64.15 ± 4.63 |
Strains were grown in TY medium alone or supplemented with 30 mM l-proline or 30 mM glycine. Doubling time was calculated during a 60-min period starting at 120 min of growth; values are means ± standard deviations.
Proline activates transcription of the prd operon and inhibits transcription of the grd operon.
It has been shown previously for C. difficile that addition of glycine to the growth medium increases the level of GR and that addition of proline to the growth medium both increases PR levels and decreases GR levels (38). To see if these effects could be explained by regulation at the transcriptional level, we used qPCR to compare the relative amounts of prdA, prdD, prdF, and grdE mRNAs in the mutant and wild-type strains. As shown in Fig. 5A, the level of prdA mRNA increased about 10-fold when wild-type cells were grown in the presence of 30 mM l-proline. Similarly, prdD and prdF transcripts were about 10-fold more abundant in the presence of exogenous l-proline (Fig. 5C and D). The levels of grdE mRNA increased about 3-fold in wild-type cells grown in the presence of 30 mM glycine (Fig. 5B). Interestingly, we found that grdE mRNA in the wild-type strain was reduced about 80-fold when proline was added to the medium (Fig. 5B), consistent with the reduced amount of GR protein found in cells grown with proline (18). These data demonstrate that the prd and grd gene clusters are induced at the transcriptional level in response to proline and glycine, respectively, and that proline plays a role in the transcription of the grd genes.
Fig 5.

Expression of the prd and grd genes of wild-type and prdR mutant strains in the presence of l-proline or glycine. C. difficile wild-type (JIR8094), LB-CD8 (prdR::ermB), and LB-CD14 (prdR::ermB, prdR+) strains were grown to mid-exponential phase in TY medium alone or supplemented with 30 mM l-proline or 30 mM glycine. RNA was harvested, cDNA was synthesized, and qPCR was performed using gene-specific primers for prdA (A), grdE (B), prdF (C), prdD (D), and prdC (E). Results were normalized to an internal control gene (rpoC) and are presented as the ratio of each transcript level to that of wild-type cells grown in TY medium. The means and standard deviations of at least three biological replicates, each assayed in triplicate, are shown, with the exception of LB-CD8 samples in panels C, D, and E, for which data were obtained from a single biological replicate performed in triplicate.
PrdR is required for proline-dependent activation of the prd pathway.
Jackson et al. (38) reported the presence, immediately upstream of the C. difficile prd operon, of a 1,761-bp open reading frame, named prdR, that encodes a putative activator of sigma-54-dependent transcription. A previous study revealed that prdR homologs are found upstream of the proline reductase gene cluster in most of the species that possess such genes (51). BLAST analysis indicated that prdR is also present upstream of a prd gene cluster in all sequenced C. difficile genomes (data not shown). To test whether PrdR plays a role in the proline-dependent regulation of the Stickland reaction genes, we created an insertion mutation in prdR, using TargeTron technology (see Materials and Methods). Disruption of the prdR gene was verified by PCR (see Materials and Methods). Using qPCR, we measured the relative amounts of prdA, prdD, prdF, and grdE mRNAs in the wild-type and prdR mutant (LB-CD8) strains. Even in the absence of exogenous proline, the amount of prdA transcript in the prdR mutant was reduced about 2-fold compared to that in the wild type (Fig. 5A). Moreover, when the cells were grown in the presence of proline, prdA mRNA levels were ∼35-fold lower in the prdR mutant than in wild-type cells (Fig. 5A). Similarly, prdD and prdF transcripts were less abundant in the prdR mutant than in the wild-type strain grown in TY medium containing l-proline (Fig. 5C and D) or in TY medium alone. Interestingly, grdE levels were increased about 3-fold in the prdR mutant compared to those in the wild type and were not affected by the presence of proline in the medium (Fig. 5B), suggesting that PrdR participates in proline-dependent negative regulation of the grd operon.
Upstream of prdR, we found two genes, CD3246 and prdC. In C. sticklandii, prdC encodes a selenoprotein required for electron transfer by the PR complex (51). Despite the lack of a selenium signature in C. difficile PrdC, we hypothesized that PrdC might be part of the PR complex and thus might be regulated by PrdR. To test this hypothesis, we measured by qPCR the amount of prdC mRNA in the wild type and prdR mutant. We observed that prdC levels increased about 10-fold when wild-type cells were grown in the presence of l-proline (Fig. 5E). In the prdR mutant, prdC levels were 2-fold lower than in the wild type grown in TY medium alone and 30-fold less abundant than in the wild type grown in TY medium supplemented with l-proline (Fig. 5E), suggesting that PrdR is responsible for the proline-dependent activation of prdC expression.
To confirm that proline-dependent regulation was due to the disruption of prdR, we complemented strain LB-CD8 (prdR mutant) in trans with a plasmid carrying the prdR coding sequence and 260 bp of upstream sequence. The resulting strain, LB-CD14, was tested for its ability to restore prdA mRNA to wild-type levels. The empty vector, pBL58, was introduced into the wild-type strain to control for plasmid presence. The strain containing pBL58 (LB-CD13) and the wild-type strain exhibited comparable levels of prdA mRNA (data not shown). As shown in Fig. 5A, prdA levels in LB-CD14 were similar to or slightly higher than those in the wild-type strain grown under the same conditions, i.e., with or without added l-proline. Hence, we conclude that prdR is required for proline-dependent activation of the prd operon.
Given the importance of PrdR in the expression of the Stickland reaction genes and the role of PR in growth (see above), we assessed the effect of a prdR mutation on growth in TY medium with and without supplementation with l-proline or glycine. Results shown in Fig. 3 and Table 3 indicate that the prdR mutant exhibited a slightly lower growth rate than the wild type under all conditions tested. This result shows that PrdR is needed for maximal growth of C. difficile.
Effects of disruption of prdB and grdA on expression of Stickland metabolism genes.
Because the presence of the Stickland acceptors proline and glycine affects expression of the Stickland metabolism genes, we hypothesized that the PR and GR enzymes control their own expression by modulating the intracellular pools of proline. To test this hypothesis, we first measured the intracellular concentration of free proline in strains LB-CD4, LB-CD8, and LB-CD12. As expected, free proline in LB-CD4 (prdB) was about 5-fold more abundant than in the wild-type control. No changes were observed in strain LB-CD12 (grdA) compared to the wild type, and intermediate levels were observed in strain LB-CD8 (prdR) (Fig. 6). These data show that intracellular proline concentrations depend on the activity of PR but not on the activity of GR.
Fig 6.

Analysis of proline content in C. difficile cells. Extracts from C. difficile wild-type (JIR8094), LB-CD4 (prdA::ermB), LB-CD12 (grdA::ermB), and LB-CD8 (prdR::ermB) strains harvested after growth to mid-exponential phase in TY medium were assayed for free proline using a published procedure (48), and the results were expressed relative to those for the wild-type strain. Results represent the means and standard deviations of three biological replicates.
Therefore, we tested if expression of Stickland metabolism genes was altered in the absence of PR. The level of prdA mRNA in strain LB-CD4 (prdB) was much higher than in wild-type cells (Fig. 7A), consistent with the idea that blocking metabolism of proline causes it to accumulate to a level that induces prdA transcription. Furthermore, whereas the addition of proline to wild-type cells led to a dramatic increase in prdA transcription, there was no significant proline-dependent increase in prdA expression in prdB mutant cells (Fig. 7A), implying that prdA transcription had reached its maximum level in strain LB-CD4 without added proline.
Fig 7.

Expression of the prdA and grdE genes of wild-type and mutant strains in the presence of l-proline or glycine. C. difficile wild-type (JIR8094), LB-CD4 (prdB::ermB), and LB-CD12 (grdA::ermB) strains were grown to mid-exponential phase in TY medium alone or supplemented with 30 mM l-proline or 30 mM glycine. RNA was harvested, cDNA was synthesized, and qPCR was performed using gene-specific primers for prdA (A) and grdE (B). Results were normalized to an internal control gene (rpoC) and are presented as the ratio of each transcript level to that of wild-type cells grown in BHIS. The means and standard deviations of at least three biological replicates, each assayed in triplicate, are shown.
Surprisingly, grdE transcripts in strain LB-CD4 were more abundant (∼10-fold) than in wild-type cells (Fig. 7B) and ∼3-fold more abundant than in the prdR mutant (Fig. 5B and 7B). Because PrdR is present in LB-CD4 and the high intracellular concentration of proline in LB-CD4 does not repress grdE expression, we conclude that PrdR does not directly mediate the inhibition of the grd operon by proline. Instead, our results suggest that the activity of PR plays a more direct role than does PrdR in negatively controlling expression of the glycine reductase pathway.
In a similar manner, we tested whether GR plays a role in regulating expression of the prd and grd genes. No changes in prdA mRNA levels were seen in strain LB-CD12 (grdA) relative to those in wild-type cells when the cells were grown in the presence of proline (Fig. 7A). However, we noticed a small increase (∼3-fold) in the amount of prdA transcript in the grdA mutant grown with or without glycine, indicating that GR plays a minor role in the repression of PR. In contrast, grdE levels in LB-CD12 were ∼10-fold higher than in wild-type cells and ∼12-fold more abundant in glycine-supplemented medium (Fig. 7B). Furthermore, induction of grdE expression in the presence of glycine was notably higher in the grdA mutant than in the wild-type strain (Fig. 7B). These results indicate that the grd gene cluster is activated by glycine and repressed directly or indirectly by GR or by another protein or metabolite product of the grd gene cluster.
Effects of disruption of prdB and prdR on expression of a C. difficile toxin gene.
The presence of certain amino acids, including proline, inhibits toxin production (13, 14). To test whether the effect of proline on toxin production is mediated by the Stickland reactions, we measured the relative amounts of tcdA transcript in wild-type, LB-CD4, and LB-CD8 strains in the early stationary phase of growth. We observed that tcdA mRNA levels decreased about 2.5-fold (P value < 0.05) when wild-type cells were grown in the presence of exogenous proline (Fig. 8). Similarly, tcdA levels in LB-CD4 grown with or without proline were ∼2.5-fold lower than in the wild-type cells (Fig. 8). On the other hand, we observed similar (or slightly higher) levels of tcdA transcripts in strain LB-CD8 compared to those in the wild type in cells grown without proline supplementation. However, the relative amount of tcdA mRNA in LB-CD8 was ∼4-fold higher than in the wild type grown in the presence of exogenous proline, suggesting that PrdR represses tcdA expression. Thus, this result indicates that proline negatively affects toxin expression at the transcriptional level and shows that PrdR mediates this repression either directly or indirectly.
Fig 8.

Expression of the tcdA gene of wild-type and mutant strains in the presence of l-proline. C. difficile wild-type (JIR8094), LB-CD4 (prdB::ermB), and LB-CD8 (prdR::ermB) strains were grown for 7 h to stationary phase in TY medium alone or supplemented with 30 mM l-proline. RNA was harvested, cDNA was synthesized, and qPCR was performed using gene-specific primers for tcdA. Results were normalized to an internal control gene (rpoC) and are presented as the ratio of each transcript level to that of wild-type cells grown in TY medium. The means and standard deviations of biological replicates are shown.
DISCUSSION
Stickland metabolism has been shown to be an important source of energy for nontoxigenic Clostridium spp. in media containing amino acids as sole carbon and nitrogen sources. For C. difficile, both glycine and l-proline are required for optimal growth in defined media (50, 52, 53). In addition, Jackson et al. (38) suggested that because of the selenium requirement of C. difficile for growth in limiting basal medium, the Stickland selenium-containing reductases (PR and GR) play a key role in the physiology of the bacteria. In the current work, we show that even in rich complex medium where bacteria can find and utilize a multitude of carbon and nitrogen sources, the proline-dependent expression of the PR is used for optimal growth of C. difficile (Fig. 4). However, the lack of GR does not affect growth in rich medium. Hence, the proline pathway of Stickland metabolism might be an important source of energy in rich medium but the glycine pathway is not.
It has been shown previously for C. difficile that (i) proline stimulates production of PR and inhibits production of GR (38) and (ii) glycine stimulates production of GR. In addition, an earlier report (54) indicated that supplementation of the medium with l-proline decreases GR activity in C. sporogenes and suggested that the inhibition occurs at the level of GR synthesis. We show here that addition of proline to the medium increases the expression of the prd genes and decreases the expression of grdE (Fig. 5A and B). Furthermore, we identified PrdR, an apparent sigma-54-dependent activator encoded by a gene located upstream of the prd operon (Fig. 2), as the mediator of proline-dependent activation of the prd gene cluster and prdC and proline-dependent repression of the grd operon (Fig. 5). We also found that glycine activates expression of grdE and that a functional PR is required for proline-dependent repression of grdE. (The prdB mutation in fact resulted in increased expression of grdE [Fig. 5B and Fig. 7].) Given that the prdB mutant accumulates proline and that proline seems to activate PrdR, we surmise that the proline-dependent repression of grdE expression is not directly mediated by PrdR.
If it is not activation of PrdR that explains repression of grdE in proline-containing medium, what could be responsible? First, the enzymatic activity of PR could produce an inhibitor of grdE expression or consume an activator of grdE. Second, the prdB mutation is likely to have a polar effect on expression of downstream genes, including prdF, which encodes proline racemase. If d-proline, the substrate of PR, rather than l-proline regulates grdE expression, the absence of prdF expression in a prdB mutant would limit inhibition of grdE expression. Alternatively, 5-aminovalerate, the end product of d-proline reduction (Fig. 1), might be a signal for grdE repression. However, addition of either 5-aminovalerate or d-proline to prdB mutant cells failed to restore repression of grdE (data not shown), suggesting that neither d-proline nor 5-aminovalerate, assuming it is imported, is the mediator of grdE repression.
Recent work showed that CodY, a global regulator in Gram-positive bacteria that modulates gene expression in response to the availability of branched-chain amino acids (isoleucine, leucine, and valine) and GTP (23), represses toxin expression in C. difficile by binding to the promoter region of tcdR, which encodes a sigma factor needed for transcription of the toxin genes (19). CodY proteins also control virulence gene expression in other Gram-positive pathogens, such as Staphylococcus aureus (40, 55, 56), Streptococcus pneumoniae (57), and Listeria monocytogenes (58). Interestingly, isoleucine and leucine are among the most efficient Stickland donors (59). Therefore, it is tantalizing to think that Stickland metabolism might play an indirect role in toxin gene expression by reducing CodY activity by draining the pools of isoleucine, leucine, and valine. On the other hand, the effect of proline on toxin production (12, 14) might be at least partly independent of CodY. Consistent with this idea, our results show that toxin expression is reduced in the prdB null mutant, which accumulates proline, and is slightly increased in the prdR mutant compared to the wild type (Fig. 8). Whether PrdR acts directly on toxin gene expression remains to be tested.
In conclusion, our results and those of others (38, 54) show that the Stickland metabolism genes of C. difficile are regulated in response to the availability of proline and glycine and that these genes are needed for optimal growth in defined and complex media. With the mutants in hand, we will be able to assess the role of Stickland metabolism in animal models of C. difficile infection.
ACKNOWLEDGMENTS
We thank J. Sorg, S. McBride, A. Roux, and B. Belitsky for helpful suggestions and discussions during the course of this work and for criticism of the manuscript, and we thank J. P. van Pijkeren and the Britton laboratory at Michigan State University for sharing their group II intron algorithm.
Research reported in this article was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01 GM042219.
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 7 December 2012
REFERENCES
- 1. Lyerly DM, Krivan HC, Wilkins TD. 1988. Clostridium difficile: its disease and toxins. Clin. Microbiol. Rev. 1:1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnson S, Clabots CR, Linn FV, Olson MM, Peterson LR, Gerding DN. 1990. Nosocomial Clostridium difficile colonisation and disease. Lancet 336:97–100 [DOI] [PubMed] [Google Scholar]
- 3. Pépin J, Valiquette L, Alary ME, Villemure P, Pelletier A, Forget K, Pepin K, Chouinard D. 2004. Clostridium difficile-associated diarrhea in a region of Quebec from 1991 to 2003: a changing pattern of disease severity. CMAJ 171:466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kelly CP, LaMont JT. 2008. Clostridium difficile—more difficult than ever. N. Engl. J. Med. 359:1932–1940 [DOI] [PubMed] [Google Scholar]
- 5. Warny M, Pepin J, Fang A, Killgore G, Thompson A, Brazier J, Frost E, McDonald LC. 2005. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet 366:1079–1084 [DOI] [PubMed] [Google Scholar]
- 6. Lyras D, O'Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI. 2009. Toxin B is essential for virulence of Clostridium difficile. Nature 458:1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713 [DOI] [PubMed] [Google Scholar]
- 8. Karlsson S, Dupuy B, Mukherjee K, Norin E, Burman LG, Akerlund T. 2003. Expression of Clostridium difficile toxins A and B and their sigma factor TcdD is controlled by temperature. Infect. Immun. 71:1784–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamakawa K, Karasawa T, Ikoma S, Nakamura S. 1996. Enhancement of Clostridium difficile toxin production in biotin-limited conditions. J. Med. Microbiol. 44:111–114 [DOI] [PubMed] [Google Scholar]
- 10. Onderdonk AB, Lowe BR, Bartlett JG. 1979. Effect of environmental stress on Clostridium difficile toxin levels during continuous cultivation. Appl. Environ. Microbiol. 38:637–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dupuy B, Sonenshein AL. 1998. Regulated transcription of Clostridium difficile toxin genes. Mol. Microbiol. 27:107–120 [DOI] [PubMed] [Google Scholar]
- 12. Karlsson S, Lindberg A, Norin E, Burman LG, Akerlund T. 2000. Toxins, butyric acid, and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infect. Immun. 68:5881–5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karasawa T, Maegawa T, Nojiri T, Yamakawa K, Nakamura S. 1997. Effect of arginine on toxin production by Clostridium difficile in defined medium. Microbiol. Immunol. 41:581–585 [DOI] [PubMed] [Google Scholar]
- 14. Karlsson S, Burman LG, Akerlund T. 1999. Suppression of toxin production in Clostridium difficile VPI 10463 by amino acids. Microbiology 145(Part 7):1683–1693 [DOI] [PubMed] [Google Scholar]
- 15. Hundsberger T, Braun V, Weidmann M, Leukel P, Sauerborn M, von Eichel-Streiber C. 1997. Transcription analysis of the genes tcdA-E of the pathogenicity locus of Clostridium difficile. Eur. J. Biochem. 244:735–742 [DOI] [PubMed] [Google Scholar]
- 16. Antunes A, Martin-Verstraete I, Dupuy B. 2011. CcpA-mediated repression of Clostridium difficile toxin gene expression. Mol. Microbiol. 79:882–899 [DOI] [PubMed] [Google Scholar]
- 17. Underwood S, Guan S, Vijayasubhash V, Baines SD, Graham L, Lewis RJ, Wilcox MH, Stephenson K. 2009. Characterization of the sporulation initiation pathway of Clostridium difficile and its role in toxin production. J. Bacteriol. 191:7296–7305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saujet L, Monot M, Dupuy B, Soutourina O, Martin-Verstraete I. 2011. The key sigma factor of transition phase, SigH, controls sporulation, metabolism, and virulence factor expression in Clostridium difficile. J. Bacteriol. 193:3186–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dineen SS, Villapakkam AC, Nordman JT, Sonenshein AL. 2007. Repression of Clostridium difficile toxin gene expression by CodY. Mol. Microbiol. 66:206–219 [DOI] [PubMed] [Google Scholar]
- 20. Mani N, Dupuy B. 2001. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc. Natl. Acad. Sci. U. S. A. 98:5844–5849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dineen SS, McBride SM, Sonenshein AL. 2010. Integration of metabolism and virulence by Clostridium difficile CodY. J. Bacteriol. 192:5350–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ratnayake-Lecamwasam M, Serror P, Wong KW, Sonenshein AL. 2001. Bacillus subtilis CodY represses early-stationary-phase genes by sensing GTP levels. Genes Dev. 15:1093–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sonenshein AL. 2007. Control of key metabolic intersections in Bacillus subtilis. Nat. Rev. Microbiol. 5:917–927 [DOI] [PubMed] [Google Scholar]
- 24. Brinsmade SR, Kleijn RJ, Sauer U, Sonenshein AL. 2010. Regulation of CodY activity through modulation of intracellular branched-chain amino acid pools. J. Bacteriol. 192:6357–6368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matamouros S, England P, Dupuy B. 2007. Clostridium difficile toxin expression is inhibited by the novel regulator TcdC. Mol. Microbiol. 64:1274–1288 [DOI] [PubMed] [Google Scholar]
- 26. Sonenshein AL. 2005. CodY, a global regulator of stationary phase and virulence in Gram-positive bacteria. Curr. Opin. Microbiol. 8:203–207 [DOI] [PubMed] [Google Scholar]
- 27. Görke B, Stulke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6:613–624 [DOI] [PubMed] [Google Scholar]
- 28. Nisman B, Raynaud M, Cohen GN. 1948. Extension of the Stickland reaction to several bacterial species. Arch. Biochem. 16:473. [PubMed] [Google Scholar]
- 29. Stickland LH. 1934. Studies in the metabolism of the strict anaerobes (genus Clostridium): the chemical reactions by which Cl. sporogenes obtains its energy. Biochem. J. 28:1746–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stickland LH. 1935. Studies in the metabolism of the strict anaerobes (genus Clostridium): the oxidation of alanine by Cl. sporogenes. IV. The reduction of glycine by Cl. sporogenes. Biochem. J. 29:889–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stickland LH. 1935. Studies in the metabolism of the strict anaerobes (genus Clostridium): the reduction of proline by Cl. sporogenes. Biochem. J. 29:288–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stadtman TC. 1956. Studies on the enzymic reduction of amino acids: a proline reductase of an amino acid-fermenting Clostridium, strain HF. Biochem. J. 62:614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stadtman TC, Elliott P. 1957. Studies on the enzymic reduction of amino acids. II. Purification and properties of D-proline reductase and a proline racemase from Clostridium sticklandii. J. Biol. Chem. 228:983–997 [PubMed] [Google Scholar]
- 34. Cone JE, Del Rio RM, Davis JN, Stadtman TC. 1976. Chemical characterization of the selenoprotein component of clostridial glycine reductase: identification of selenocysteine as the organoselenium moiety. Proc. Natl. Acad. Sci. U. S. A. 73:2659–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cone JE, del Rio RM, Stadtman TC. 1977. Clostridial glycine reductase complex. Purification and characterization of the selenoprotein component. J. Biol. Chem. 252:5337–5344 [PubMed] [Google Scholar]
- 36. Kabisch UC, Grantzdorffer A, Schierhorn A, Rucknagel KP, Andreesen JR, Pich A. 1999. Identification of D-proline reductase from Clostridium sticklandii as a selenoenzyme and indications for a catalytically active pyruvoyl group derived from a cysteine residue by cleavage of a proprotein. J. Biol. Chem. 274:8445–8454 [DOI] [PubMed] [Google Scholar]
- 37. Seto B, Stadtman TC. 1976. Purification and properties of proline reductase from Clostridium sticklandii. J. Biol. Chem. 251:2435–2439 [PubMed] [Google Scholar]
- 38. Jackson S, Calos M, Myers A, Self WT. 2006. Analysis of proline reduction in the nosocomial pathogen Clostridium difficile. J. Bacteriol. 188:8487–8495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jackson-Rosario S, Cowart D, Myers A, Tarrien R, Levine RL, Scott RA, Self WT. 2009. Auranofin disrupts selenium metabolism in Clostridium difficile by forming a stable Au-Se adduct. J. Biol. Inorg. Chem. 14:507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Montgomery CP, Boyle-Vavra S, Roux A, Ebine K, Sonenshein AL, Daum RS. 2012. CodY deletion enhances in vivo virulence of community-associated methicillin-resistant Staphylococcus aureus clone USA300. Infect. Immun. 80:2382–2389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeno-Tarraga AM, Wang H, Holden MT, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, Sharp S, Simmonds M, Stevens K, Unwin L, Whithead S, Dupuy B, Dougan G, Barrell B, Parkhill J. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 38:779–786 [DOI] [PubMed] [Google Scholar]
- 42. Ho TD, Ellermeier CD. 2011. PrsW is required for colonization, resistance to antimicrobial peptides, and expression of extracytoplasmic function sigma factors in Clostridium difficile. Infect. Immun. 79:3229–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. 2007. The ClosTron: a universal gene knock-out system for the genus Clostridium. J. Microbiol. Methods 70:452–464 [DOI] [PubMed] [Google Scholar]
- 44. McBride SM, Sonenshein AL. 2011. The dlt operon confers resistance to cationic antimicrobial peptides in Clostridium difficile. Microbiology 157:1457–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bouillaut L, McBride SM, Sorg JA. 2011. Genetic manipulation of Clostridium difficile. Curr. Protoc. Microbiol. 20:9A.2.1–9A.2.17 doi:10.1002/9780471729259.mc09a02s20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Southern EM. 1975. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 98:503–517 [DOI] [PubMed] [Google Scholar]
- 47. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 48. Whatmore AM, Chudek JA, Reed RH. 1990. The effects of osmotic upshock on the intracellular solute pools of Bacillus subtilis. J. Gen. Microbiol. 136:2527–2535 [DOI] [PubMed] [Google Scholar]
- 49. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 50. Osgood DP, Wood NP, Sperry JF. 1993. Nutritional aspects of cytotoxin production by Clostridium difficile. Appl. Environ. Microbiol. 59:3985–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fonknechten N, Chaussonnerie S, Tricot S, Lajus A, Andreesen JR, Perchat N, Pelletier E, Gouyvenoux M, Barbe V, Salanoubat M, Le Paslier D, Weissenbach J, Cohen GN, Kreimeyer A. 2010. Clostridium sticklandii, a specialist in amino acid degradation: revisiting its metabolism through its genome sequence. BMC Genomics 11:555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haslam SC, Ketley JM, Mitchell TJ, Stephen J, Burdon DW, Candy DC. 1986. Growth of Clostridium difficile and production of toxins A and B in complex and defined media. J. Med. Microbiol. 21:293–297 [DOI] [PubMed] [Google Scholar]
- 53. Karasawa T, Ikoma S, Yamakawa K, Nakamura S. 1995. A defined growth medium for Clostridium difficile. Microbiology 141(Part 2):371–375 [DOI] [PubMed] [Google Scholar]
- 54. Venugopalan V. 1980. Influence of growth conditions on glycine reductase of Clostridium sporogenes. J. Bacteriol. 141:386–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Majerczyk CD, Sadykov MR, Luong TT, Lee C, Somerville GA, Sonenshein AL. 2008. Staphylococcus aureus CodY negatively regulates virulence gene expression. J. Bacteriol. 190:2257–2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Majerczyk CD, Dunman PM, Luong TT, Lee CY, Sadykov MR, Somerville GA, Bodi K, Sonenshein AL. 2010. Direct targets of CodY in Staphylococcus aureus. J. Bacteriol. 192:2861–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hendriksen WT, Bootsma HJ, Estevao S, Hoogenboezem T, de Jong A, de Groot R, Kuipers OP, Hermans PW. 2008. CodY of Streptococcus pneumoniae: link between nutritional gene regulation and colonization. J. Bacteriol. 190:590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bennett HJ, Pearce DM, Glenn S, Taylor CM, Kuhn M, Sonenshein AL, Andrew PW, Roberts IS. 2007. Characterization of relA and codY mutants of Listeria monocytogenes: identification of the CodY regulon and its role in virulence. Mol. Microbiol. 63:1453–1467 [DOI] [PubMed] [Google Scholar]
- 59. Baker HA. 1961. Fermentation of nitrogenous organic compounds, p 151–188 In Gunsalus IC, Stanier RY. (ed), The bacteria, vol 2 Academic Press, New York, NY [Google Scholar]