

Abstract
The incidence of malignant melanoma has dramatically increased in recent years thus requiring the need for improved therapeutic strategies. In our efforts to design selective histone deactylase inhibitors (HDACI), we discovered that the aryl urea 1 is a modestly potent yet non-selective inhibitor. Structure activity relationship studies revealed that adding substituents to the nitrogen atom of the urea so as to generate compounds bearing a branched linker group results in increased potency and selectivity for HDAC6. Compound 5g shows low nanomolar inhibitory potency against HDAC6 and a selectivity of ~600-fold relative to the inhibition of HDAC1. These HDACIs were evaluated for their ability to inhibit the growth of B16 melanoma cells with the most potent and selective HDAC6I being found to decrease tumor cell growth. To the best of our knowledge, this work constitutes the first report of HDAC6 selective inhibitors that possess antiproliferative effects against melanoma cells.
Introduction
Epigenetic regulation and subsequent gene expression or silencing represents a tightly orchestrated interplay among enzymes responsible for modifying the tails of histones, around which nuclear DNA is wrapped. Among the various modifiers of the histones, the cell is capable of balancing the activity of both histone acetyltransferases (HAT) and histone deacetylases (HDAC) to attach or remove acetyl groups, respectively, from the lysine tails of these histone barrels. This particular epigenetic marker masks the positive lysine residues from interacting closely with the DNA phosphate-backbone, resulting in a more “open” chromatin state, whereas the deacetylases remove these acetyl groups, resulting in a more “closed” or compacted DNA-histone state.
There are currently no selective HDAC6I approved for clinical use in oncology. The development of such small molecules may be advantageous as a therapeutic approach, for they may possibly result in fewer side effects, which is an apparent problem associated with the pan-HDACIs.1 Recent pre-clinical efforts are being directed toward the use of HDAC6I for certain cancers, specifically in combination with known drugs.2 Literature reports suggest that HDACIs may be useful as possible therapeutics for melanoma; however, many such studies to date have focused on using pan-HDACIs, such as suberoylanilide hydroxamic acid (SAHA).3 While SAHA exhibits activity against all Zn++-dependent HDAC isozymes, it has been approved solely for the treatment of cutaneous T-cell lymphoma.4
It has previously been reported that HDAC6 forms an association with HDAC11.5 Recent efforts have begun to uncover the biological significance of HDAC11 as a participant in activating the immune response, and targeting one or both of these enzymes may be of therapeutic value.6,7 Thus HDAC6 has emerged as a possible target in the treatment of melanoma. Such an approach may well be devoid of some of the undesirable side effects of the pan-HDACI, and thus of value to explore in the context of finding safer cancer therapeutics.8
Our laboratory has previously published on the development of a variety of HDAC6 inhibitors.9, 10 A key feature of these potent and selective agents derives from the presence of the benzylic linker that is built into the canonical inhibitor structure, which consists of the usual cap-linker-zinc binding group scaffold (Figure 1). Herein we report on the use of certain urea-based branched linkers that lead to potent and selective HDAC6 inhibitors with cellular anti-melanoma activity.
Figure 1.
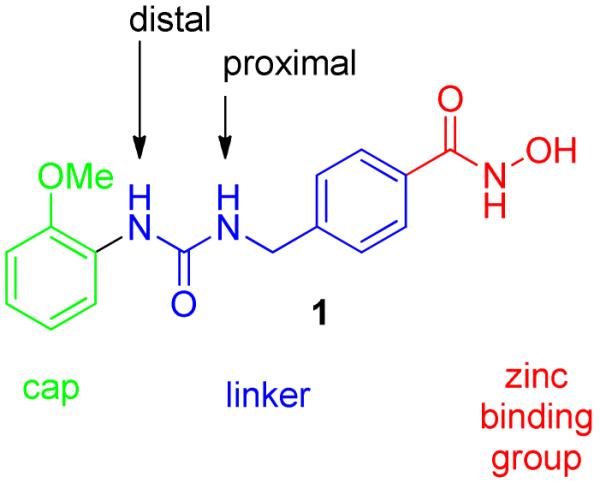
Structure of 1, a urea containing HDACI. Appendage of substitutions to the nitrogen atoms of the urea generate a branched linker motif that may interact with a side cavity at the enzyme surface.
Chemistry
Based upon our knowledge of the structural parameters essential for selective HDAC6 inhibitory activity, we chose to investigate the design of some new hydroxamate-based HDACIs that might retain the desired isozyme selectivity while at the same time optimizing for drug-like properties. Interestingly, a recent publication reported on the discovery of HDACIs without a zinc-binding group (ZBG).11 These agents possess modest activity against the Class 1 enzymes; however, our experience with HDAC6I design indicates that inclusion of the ostensibly problematic hydroxamic acid group is not a liability. Our previous efforts toward developing selective HDAC6Is were assisted by the development of a homology model.9 This model showed the entrance to the binding site was wider and shallower for HDAC6 than that of HDAC1. Further examination of this model shows a lipophilic cavity suitable for exploration in compound design. We proposed that the introduction of certain branched-elements to the linker would improve both the potency and selectivity by accessing this side cavity, thereby perhaps resulting in better interactions with the surface of HDAC6. These efforts originated by analoging around compound 1, a modestly potent inhibitor of HDAC6 that is bereft of any selectivity relative to HDAC1. With our knowledge of the HDAC6 binding site, in combination with our previous SAR data, we hypothesized that access to the HDAC6 lipophilic side-cavity could most efficiently be accomplished by appending functionality to the nitrogen atoms of the urea within the linker region of the canonical HDACI scaffold.
The synthesis of these branched ureas was readily accomplished as outlined in Scheme 1. A variety of amines were subjected to a reductive amination reaction employing methyl 4-formylbenzoate 2 to form the desired secondary amines 3a-h. Subsequent reaction of 3a-h with the appropriate isocyanates afforded the branched urea esters 4a-h. This chemistry generated a series of ureas displaying branched substitutions on the nitrogen atom proximal to the ZBG. Finally, the hydroxamic acid group was installed using hydroxylamine under basic conditions to provide the hydroxamic acids 5a-h.
Scheme 1.

Synthesis of proximal nitrogen atom substituted hydroxamic acids
Reagents and conditions: (a) R1-NH2, NaCNBH3, rt, 5% AcOH/DCM, 16 h; (b) Aryl-NCO, DCM, rt, 16 h; (c) 50% wt NH2OH, NaOH, THF/MeOH (1:1), 0 °C to rt, 30 min.
To evaluate the activity of compounds that possess a branched linker positioned at the nitrogen atom distal to the ZBG we carried out the chemistry displayed in Scheme 2. A copper-mediated Buchwald coupling reaction12 was used in order to assemble anilines 6a-b from iodobenzene, as these intermediates were not commercially available. Triphosgene chemistry was implemented to convert methyl 4-(aminomethyl)benzoate into the corresponding isocyanate in situ, which underwent reaction with secondary amines 6a-c to afford the penultimate esters 7a-c. Final conversion to the hydroxamic acids was accomplished in the usual fashion to complete the synthesis of 8a-c.
Scheme 2.
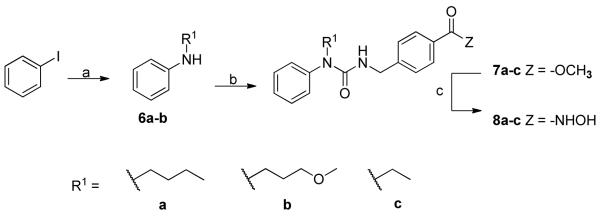
Synthesis of distal nitrogen atom substituted hydroxamic acids
Reagents and conditions: (a) R1-NH2, CuI, K3PO4, ethylene glycol, iPrOH, 80°C, 18 h; (b) i. triphosgene, sat. aq bicarbonate/DCM (1:1), 0°C, 30 min; ii. methyl 4-(aminomethyl)benzoate-HCl, Et3N, DCM, rt, 16 h; (c) 50% wt NH2OH, NaOH, THF/MeOH (1:1), 0°C to rt, 30 min.
Results and Discussion
HDAC isoform activity
Our preliminary screening efforts identified 1 to possess submicromolar HDAC inhibitory activity, however, it was not selective against representative members of the Zn++-dependent Classes 1 and 2. In an effort to improve upon these compounds, we thus chose to investigate the role of substitution on the nitrogen atoms of the urea within the linker as a possible way to impart both selectivity and potency to these compounds, and in particular for HDAC6 by accessing a unique cavity on the surface of HDAC6. A summary of the HDAC1 and HDAC6 inhibitory data obtained is presented in Table 1.
Table 1.
Initial HDAC inhibition screen of substituted urea compoundsa

| Compound | HDAC1 IC50 (nM) |
HDAC6 IC50 (nM) |
Fold Selective (HDAC1/HDAC6) |
|
|---|---|---|---|---|
| 1 |

|
265 ± 59 | 139 ± 27 | 2 |
| 5a |

|
2550 ± 540 | 458 ± 64 | 6 |
| 5b |

|
1910 ± 570 | 2.38 ± 0.38 | 803 |
| 5c |

|
8950 ± 770 | 468 ± 130 | 19 |
| 5d |

|
1690 ± 120 | 5.80 ± 0.50 | 292 |
| 5e |

|
5180 ± 130 | 11.7 ± 1.7 | 443 |
| 5f |

|
2250 ± 420 | 9.26 ± 0.66 | 243 |
|
5g
“Nexturastat A ” |

|
3020 ± 740 | 5.02 ± 0.060 | 600 |
| 5h |

|
3120 ± 640 | 14.0 ± 0.75 | 222 |
| 8a |

|
8120 ± 600 | 25.2 ± 2.5 | 322 |
| 8b |

|
1360 ± 450 | 60.3 ± 15.8 | 226 |
| 8c |

|
1360 ± 650 | 41.1 ± 0.40 | 330 |
| Tubastatin A b |

|
1640 ± 260 | 15.0 ± 0.01 | 1093 |
| TSA b,c |
|
5 ± 1.0 | 1.2 ± 0.30 | 4 |
| MGCD0103 |

|
102 ± 38 | >10,00013 | -- |
IC50 values displayed are the mean of two experiments ± standard deviation obtained from curve fitting of a 10-point enzyme assay starting from 30 OM of the HDACI with 3-fold serial dilution. Values are extracted from fitting dose-response curves to the data points;
ref. 9;
Trichostatin A
The first round of analogs based on 1 maintained the same 2-methoxyphenyl cap group but contained varied substitutions on the proximal nitrogen atom (5a-d). Introducing the branching element at this position had a dramatic impact on decreasing activity at HDAC1. Interestingly, inhibition at HDAC6 was found to be dependent upon the nature of this substitution. The dimethylamino substitution as in 5a, and the 3-indolyl substitution as in 5c, both proved detrimental to HDAC6 inhibition as they were over three times less potent compared to 1. They did, however, maintain low micromolar inhibitory activity at HDAC1. As the tertiary amine in 5a would be protonated at physiological pH, it is possible that a positive charge is unfavorable for proper target binding. Likewise, the larger indole group of 5c may simply present too much steric bulk to be properly accommodated by the active site. However, the 3-hydroxypropyl derivative, 5b, and the 4-hydroxyphenylethyl derivative, 5d, displayed significant increases in the inhibition of HDAC6. These substitutions had only a marginal effect on HDAC1 activity. It is possible the hydroxyl groups of 5b and 5d are able to serve as H-bond acceptors or donors and possess favorable interactions with key amino acid residues on the HDAC6 surface, thus improving binding affinity. Confirmation of this notion would require a high quality x-ray structure of HDAC6, which is not yet available.
Concurrently, we queried the effect of masking the free hydroxyl moiety of this branched linker design to examine the influence of an H-bond acceptor. Capping the free hydroxyl with a methyl group resulted in 5e, and this modification was found to lead to a low nanomolar inhibitor with >400 fold selectivity for HDAC6. Shortening this appendage to a hydroxyethyl group as in 5f did not significantly alter HDAC6 inhibition, but did lead to slightly increased activity against HDAC1, thus lowering the selectivity for our desired isozyme. The general trend established was that an H-bond donor and a large aromatic group detracted from the desired activity profile, whereas smaller groups with H-bond acceptors favored HDAC6 activity. Interestingly, the n-butyl containing analog 5g and phenethyl analog 5h were nM potent HDAC6I. Furthermore, 5g possessed excellent selectivity over HDAC1 (600-fold). These data indicate that either a H-bond acceptor or lipophilic group introduced as the branching element offers the best gains in potency and selectivity for HDAC6.
Shifting the branching element to the distal nitrogen atom of the urea relative to the ZBG resulted in analogs 8a-c. The most potent of this series, 8a, possessed the same n-butyl substitution as found in 5g. While 8a is a nanomolar HDAC6I, it is five-times less potent and more importantly, is less selective than the proximally substituted analolg 5g. The methoxy variant 8b suffered a dramatic decrease in potency towards HDAC6. Decreasing the length of the alkyl chain as in 8c also resulted in decreased HDAC6 inhibition. For compounds possessing branched linkers originating from the urea, the most significant gains in potency and selectivity for HDAC6 comes from substitutions to the proximal nitrogen of the ZBG. Lipophilic groups and H-bond acceptors are best accommodated at this position, which we believe are most suitable to access the side cavity at the HDAC6 surface.
Evaluation of this lead against other HDACI developed by others and us reveals 5g, termed Nexturastat A, is in fact a potent and selective HDAC6I. Comparing Nexturastat A to Tubastatin A, an HDAC6I developed in our laboratory,9 reveals that the inhibition of HDAC6 has been improved while maintaining excellent selectivity relative to HDAC1. Nexturastat A also demonstrates comparable HDAC6 potency to Trichostatin A (TSA) (Table 1). Additionally, we note that the amino-benzamide ZBG has been incorporated into the design of HDACIs, and its introduction reduces Class 2 inhibition resulting in Class 1 selectivity; this is typified by MGCD0103, an HDACI that possesses antiproliferative activity and that has recently entered clinical trials.14 Compared to MGCD0103, Nexturastat A leads to a 30-fold reduction in activity at HDAC1. Since the HDAC isozymes are highly homologous, obtaining selectivity is critical to avoiding off-target effects and is crucial to the anticipated development of HDAC6I.
Several pan-selective compounds have been approved by the FDA, although only for use in cutaneous T-cell lymphoma.15 Avoiding cytotoxicity through isozyme selectivity would ultimately prove advantageous and may open doors to a variety of other therapeutic areas. Nexturastat A was thus screened against all 11 isozymes (Table 2). In the similar Class 1 and Class 4 isozymes, Nexturastat A displayed low micromolar activity compared to the low nanomolar activity against HDAC6. Moreover, it also demonstrated high levels of selective inhibition against members of the related Class 2 HDAC isozymes reaching >1000-fold selective in some cases. These data establish Nexturastat A to be a potent and isozyme selective HDAC6I. As more data emerge regarding the biological activities of the other HDAC isozymes, maintaining selectivity during inhibitor development will be paramount.
Table 2.
Inhibitory profile of Nexturastat A (5g) against HDAC1-11a
| Isoform | IC50
(μM) |
Fold selective for HDAC6 |
|---|---|---|
| HDAC1 | 3.02 ± 1.04 | 600 |
| HDAC2 | 6.92 ± 0.763 | 1380 |
| HDAC3 | 6.68 ± 1.75 | 1330 |
| HDAC4 | 9.39 ± 0.863 | 1870 |
| HDAC5 | 11.7 ± 0.141 | 2330 |
| HDAC6 | 0.00502 ± 0.00060 | -- |
| HDAC7 | 4.46 ± 0.665 | 888 |
| HDAC8 | 0.954 ± 0.0799 | 190 |
| HDAC9 | 6.72 ± 1.15 | 1340 |
| HDAC10 | 7.57 ± 0.481 | 1510 |
| HDAC11 | 5.14 ± 0.686 | 1020 |
IC50 displayed are the mean of two experiments ± standard deviation obtained from curve fitting of 10-point enzyme assay starting from 30 μM analog with 3-fold serial dilution. Values are extracted from fitting dose-response curves to the data points.
Next, we evaluated the ability of Nexturastat A to induce hyperacetylation of α-tubulin, a hallmark of HDAC6 inhibition, without elevating levels of acetylated histones. HDAC6 contains two catalytic domains. Its C-terminus domain is the functional domain for both synthetic and physiological substrates, whereas the N-terminal domain is devoid of enzymatic activity.16 Low nanomolar treatment of Nexturastat on B16 murine melanoma cells led to a dose-dependent increase of acetyl α-tubulin levels without a concomitant elevation of histone H3 acetylation (Figure 2) indicating binding to the second, enzymatically-active catalytic domain. Not until concentrations of 1 and 10 μM were used was an observable increase in histone H3 acetylation found. This is not surprising as the biochemical IC50 of aNexturastat A against the Class 1 HDACs, those responsible for histone acetylation, is in the micromolar range. There is a clear preference for activity in a cellular environment that corresponds to selective HDAC6 inhibition.
Figure 2.
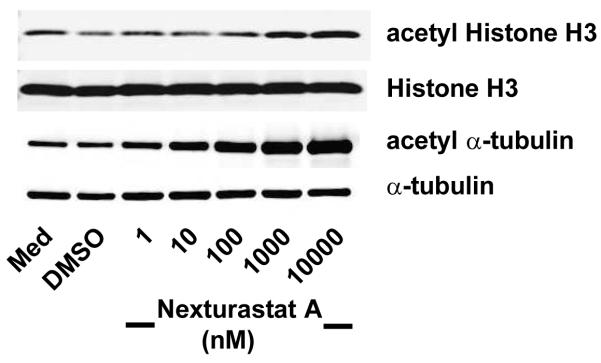
Comparison of acetylation status of tubulin and histone H3 demonstrates HDAC62substrate selectivity of Nexturastat A.
Antiproliferative activity in melanoma cells
The above compounds were then evaluated in an MTS assay to determine the ability of selective HDAC6I to exert an antiproliferative effect on B16 murine melanoma cells. Treatment with the above compounds for 48 h resulted in dose-dependent growth inhibition of the melanoma cells as summarized in Table 3. The general trend for inhibiting cell growth correlates with potency for HDAC6. However, the biochemically potent and selective 5b, performed very poorly in the cellular assay. This may possibly be attributed to its high polarity as its cLogP = 0.54 (ChemDraw 12.0), and thus the compound fails to efficiently permeate the cell membrane. Comparing the most active compounds 5d and 5f-h in this whole-cell assay reveals that the most selective HDAC6I have the greatest efficacy in inhibiting cell growth. It should also be noted that they also have higher cLogP values, possibly contributing to improved cell permeability. As the cLogP is adjusted to more optimal levels, as exemplified for Nexturastat A (cLogP = 2.20), cellular efficacy is restored, demonstrating that a proper balance of physiochemical parameters must be maintained.
Table 3.
Antiproliferative activity against B16 murine melanoma cells
| Compound | GI50 Melanoma cells (μM) |
|---|---|
| 5a | 75.3 ± 1.23 |
| 5b | >100 |
| 5c | 30.4 ± 1.32 |
| 5d | 18.4 ± 1.23 |
| 5e | 22.2 ± 1.41 |
| 5f | 19.1 ± 1.19 |
|
5g “Nexturastat A” |
14.3 ± 1.15 |
| 5h | 15.4 ± 1.20 |
| 8a | 65.8 ± 1.19 |
| 8b | >100 |
| 8c | >100 |
| Tubastatin | 40.5 ± 1.21 |
| LBH589 | 0.150 ± 0.00115 |
Compared to the pan-selective HDACI LBH589, Nexturastat A is approximately 100-fold less potent in inducing murine B16 melanoma cell death. This decreased efficacy is unlikely due to poor cell permeability, for as shown above, the treatment of B16 cells with nanomolar doses of Nexturastat A results in increased acetyl-tubulin levels (Figure 2). Additionally, both compounds possess similar cLogP values (2.64 vs 2.20 for LBH589 and Nexturastat A, respectively). Rather, the effects of non-selective HDAC inhibition with LBH589 treatment are likely contributing to its increased potency, and in particular, its Class 1 activity. It is also of interest to note that Nexturastat A has increased potency against the B16 cell line in comparison to Tubastatin A (Table 3). While a definitive explanation for this difference in cellular activity is lacking, it is possible that this is due to the improved HDAC6 activity of Nexturastat A. While HDAC6-selective inhibitors have not played a prominent role in cancer therapy to date, the present data suggest that further explorations in these directions may be valuable to pursue.
Conclusions
This study has investigated the use of a branched linker introduced into the canonical HDACI platform. Introduction of this branching element, specifically to the nitrogen atom proximal to the zinc binding group, has led to the discovery of potent inhibitors that show excellent selectivity for HDAC6 versus the full panel of HDACs. The SAR data indicate that the branched urea scaffold imparts substantial gains in the desired biochemical activity. These compounds were evaluated in a cellular context, and the best compound, Nexturastat A, was found to be capable of increasing acetylated α-tubulin levels. Moreover, Nexturastat A inhibited the growth of B16 melanoma cells, albeit with lower potency than LBH589. The work thus demonstrates that this design-platform for selective HDAC6Is, typified in Nexturastat A, leads to agents useful not only as chemical tools but also of possible value in oncology.
Experimental Section
General Information
1H and 13C spectra were obtained on a Bruker spectrometer with TMS as an internal standard. The following abbreviations for multiplicity were used: s = singlet, d = doublet, t = triplet, m = multiplet, dd = double doublet, br = broad. Reactions were monitored by TLC using precoated silica gel plates (Merck silica gel 60 F254, 250 μm thickness) and visualized under UV light. LRMS experiments were carried out using an Agilent 1946A LC-MSD with MeCN and H2O spiked with 0.1% formic acid as mobile phase. HRMS determinations were done with a Shimadzu IT-TOF instrument with MeCN and H2O spiked with 0.1% formic acid as mobile phase. Flash chromatography was accomplished using the automated CombiFlash Rf system from Teledyne ISCO and prepacked silica gel cartridges according to the recommended loading capacity. Preparatory HPLC was used in purification of all final compounds using a Shimadzu preparative liquid chromatograph with the following specifications: Column: ACE 5AQ (150 × 21.2 mm) with 5 μm particle size. Method 1–25-100% MeOH/H2O, 30 min; 100% MeOH, 5 min; 100-25% MeOH/H2O, 4 min. Method 2 – 8-100% MeOH/H2O, 30 min; 100% MeOH, 5 min; 100-8% MeOH/H2O, 4 min. Method 3 – 0% MeOH, 5 min; 0-100% MeOH/H2O, 25 min; 100% MeOH, 5 min; 100-0% MeOH/H2O, 4 min. Flow rate = 17 mL/min with monitoring at 254 and 280 nm. Both solvents were spiked with 0.05% TFA. Analytical HPLC was carried out using an Agilent 1100 series instrument with the following specifications: column: Luna 5 C18(2) 100A (150 × 4.60 mm) 5 μm particle size; gradient – 10-100% MeOH/H2O, 18 min, 100% MeOH, 3 min; 100-10% MeOH/H2O, 3 min; 10% MeOH/H2O, 5 min. Both solvents were spiked with 0.05% TFA. The purity of all tested compounds was >95%, as determined by analytical HPLC.
Methyl 4-(((3-(Dimethylamino)propyl)amino)methyl)benzoate (3a)
The synthesis of 3a is representative, general procedure A. A round-bottom flask charged with methyl 4-formyl benzoate (3-8 mg, 2 mmol) and 3-dimethylamino propylamine (0.252 mL, 2 mmol) was taken up in a solution of 5% AcOH in DCM (10 mL). After 5 minutes NaCNBH3 (1–6 mg, 2 mmol) was added in portions and the resulting mixture was allowed to stir at room temperature under an atmosphere of Ar overnight. The reaction was quenched with 1 N NaOH (10 mL) and the aqueous layer extracted with DCM (3× 10mL). The combined organic extracts were washed with brine, dried over sodium sulfate, concentrated in vacuo and purified via flash chromatography affording the product as a waxy solid (313 mg, 63%). 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 3.89 (s, 3H), 3.84 (s, 2H), 2.79 (br s, 1H), 2.68 (t, J = 6.8 Hz, 2H), 2.35 (t, J = 6.8 Hz, 2H), 2.23 (s, 6H), 1.70 (quint, J = 6.8 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 166.96, 145.27, 129.71, 128.87, 127.95, 58.04, 53.42, 51.99, 47.87, 45.36, 27.41. LRMS ESI: [M+H]+ = 251.1
Methyl 4-(((3-Hydroxypropyl)amino)methyl)benzoate (3b)
Made according to general procedure A affording a waxy solid (89 mg, 29%). 1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 8.0 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 3.91 (s, 3H), 3.90 (s, 2H), 3.78 (broad, 4H), 2.93 (t, J = 5.6 Hz, 2H), 1.78-1.73 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 166.79, 143.16, 129.77, 129.44, 128.26, 63.55, 53.14, 52.10, 48.88, 30.18. LRMS ESI: [M+H]+ = 224.2.
Methyl 4-(((2-(1H-Indol-3-yl)ethyl)amino)methyl)benzoate (3c)
Made according to general procedure A affording a waxy solid (446 mg, 72%). 1H NMR (400 MHz, CDCl3) δ 8.28 (br s, 1H), 7.98 (d, J = 8.0 Hz, 2H), 7.62 (d, J = 7.6 Hz, 1H), 7.36-7.34 (m, 3H), 7.21 (t, J = 7.6 Hz, 1H), 7.13 (t, J = 7.6 Hz, 1H), 7.00 (s, 1H), 3.92 (s, 3H), 3.87 (s, 2H), 3.02-2.98 (m, 4H), 1.68 (br s, 1H). 13C NMR (100 MHz, CDCl3) δ 167.06, 145.66, 136.34, 129.60, 128.62, 127.85, 127.31, 122.02, 121.89, 119.13, 118.74, 113.47, 111.15, 53.36, 51.97, 49.28, 25.64. LRMS ESI: [M+H]+ = 309.1.
Methyl 4-(((4-Hydroxyphenethyl)amino)methyl)benzoate (3d)
Made according to general procedure A affording a waxy solid (130 mg, 46%). 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.0 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 7.01 (d, J = 8.4 Hz, 2H), 6.71 (d, J = 8.4 Hz, 2H), 3.90 (s, 3H), 3.86 (s, 2H), 2.86 (t, J = 6.8 Hz, 2H), 2.76 (t, J = 6.8 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 167.07, 154.60, 145.02, 131.05129.78, 129.75, 128.91, 128.04, 115.55, 53.29, 52.08, 50.37, 35.02. LRMS ESI: [M+H]+ = 268.1.
Methyl 4-(((3-Methoxypropyl)amino)methyl)benzoate (3e)
Made according to general procedure A affording a colorless oil (127 mg, 54%). 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 3.86 (s, 3H), 3.80 (s, 2H), 3.41 (t, J = 6.4 Hz, 2H), 3.28 (s, 3H), 2.67 (t, J = 6.4 Hz, 2H), 1.72 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 166.88, 145.78, 129.54, 128.58, 127.76, 71.17, 58.48, 53.50, 51.85, 46.72, 29.83. LRMS ESI: [M+H]+ = 238.2.
Methyl 4-(((2-Methoxyethyl)amino)methyl)benzoate (3f)
Made according to general procedure A affording a colorless oil (29 mg, 13%). 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 3.88 (s, 3H), 3.84 (s, 2H), 3.49 (t, J = 5.2 Hz, 2H), 3.33 (s, 3H), 2.77 (t, J = 5.2 Hz, 2H), 1.88 (br, 1H). 13C NMR (100 MHz, CDCl3) δ 166.98, 145.58, 129.65, 128.74, 127.90, 71.85, 58.74, 53.47, 51.94, 48.69. LRMS ESI: [M+H]+ = 224.2.
Methyl 4-((Butylamino)methyl)benzoate (3g)
Made according to general procedure A affording colorless oil (150 mg, 68%). 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.0 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 3.91 (s, 3H), 3.84 (s, 2H), 2.62 (t, J = 7.2 Hz, 2H), 1.49 (m, 2H), 1.34 (m, 3H), 0.91 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 167.00, 145.89, 129.65, 128.70, 127.87, 53.63, 51.95, 49.14, 32.14, 20.38, 13.93. LRMS ESI: [M+H]+ = 222.1.
Methyl 4-((Phenethylamino)methyl)benzoate (3h)
Made according to general procedure A affording a colorless oil (203 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 8.01 (m, 2H), 7.35 (m, 4H), 7.22 (m, 3H), 3.93 (s, 3H), 3.88 (s, 2H), 2.89 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 166.97, 145.66, 139.81, 129.65, 128.65, 128.43, 127.82, 126.15, 53.38, 51.96, 50.46, 36.29. LRMS ESI: [M+H]+ = 270.1.
Methyl 4-((1-(3-(Dimethylamino)propyl)-3-(2-methoxyphenyl)ureido)methyl)benzoate (4a)
The synthesis of 4a is representative, general procedure B. A solution of 3a (99 mg, 0.395 mmol) in DCM (5 mL) was added the appropriate isocyante (0.053 mL, 0.395 mmol) at room temperature under and atmosphere of Ar and the resulting solution was allowed to stir overnight. The reaction was quenched with saturated bicarbonate (10 mL) and extracted with DCM (3× 10 mL). The combined organics were washed with brine (15 mL), dried over sodium sulfate, concentrated in vacuo and purified via flash chromatography affording the urea ester as a waxy solid (156 mg, 98%). 1H NMR (400 MHz, CDCl3) δ 8.64 (br s, 1H), 8.18 (d, J = 7.2 Hz, 1H), 7.99 (d, J = 8.0 Hz, 2H), 7.40 (d, J = 8.0 Hz, 2H), 6.9726.94 (m, 2H), 6.84 (d, J = 7.2 Hz, 1H), 4.64 (s, 2H), 3.91 (s, 3H), 3.81 (s, 3H), 3.42 (t, J = 6.0 Hz, 2H), 2.34 (t, J = 6.0 Hz, 2H), 2.20 (s, 6H), 1.74 (quint, J = 6.0 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 166.85, 156.64, 148.86, 143.75, 129.77, 129.43, 129.02, 127.53, 122.25, 120.94, 120.36, 109.92, 55.52, 54.60, 51.96, 49.19, 44.64, 44.18, 24.98. LRMS ESI: [M+H]+ = 400.2.
Methyl 4-((1-(3-Hydroxypropyl)-3-(2-methoxyphenyl)ureido)methyl)benzoate (4b)
Made according to general procedure B affording a waxy solid (113 mg, 74%). 1H NMR (400 MHz, CDCl3) δ 8.07-8.03 (m, 3H), 7.39 (d, J = 8.0 Hz, 2H), 7.20 (br s, 1H), 6.93-6.90 (m, 2H), 6.77-6.47 (m, 1H), 4.59 (s, 2H), 3.91 (s, 3H), 3.67-3.62 (m, 7H), 1.80-1.77 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 166.65, 156.31, 147.85, 142.24, 130.14, 129.70, 128.43, 126.77, 122.44, 121.04, 119.48, 109.88, 58.25, 55.53, 52.12, 50.41, 44.00, 30.32. LRMS ESI: [M+H]+ = 373.2.
Methyl 4-((1-(2-(1H-Indol-3-yl)ethyl)-3-(2-methoxyphenyl)ureido)methyl)benzoate (4c)
Made according to general procedure B affording a solid (200 mg, 95%). 1H NMR (400 MHz, CDCl3) δ 8.23-8.21 (m, 1H), 8.15 (br s, 1H), 7.99 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 7.6 Hz, 1H), 7.36-7.34 (m, 3H), 7.22-7.10 (m, 3H), 7.02 (s, 1H), 6.97-6.94 (m, 2H), 6.81-6.79 (m, 1H), 4.59 (s, 2H), 3.91 (s, 3H), 3.70-3.67 (m, 5H), 3.12 (t, J = 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 166.83, 155.17, 147.60, 143.28, 136.29, 129.98, 129.34, 128.79, 127.30, 127.13, 122.18, 122.08, 121.15, 119.51, 118.96, 118.46, 112.53, 111.29, 109.29, 55.56, 52.10, 50.89, 48.98. LRMS ESI: [M+H]+ = 458.2.
Methyl 4-((1-(4-Hydroxyphenethyl)-3-(2-methoxyphenyl)ureido)methyl)benzoate (4d)
Made according to general procedure B affording a solid (178 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 4.8 Hz, 1H), 8.01 (d, J = 7.6 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.11 (s, 1H), 7.02 (d, J = 8.0 Hz, 2H), 6.95-6.93 (m, 2H), 6.80-6.77 (m, 3H), 6.43 (br s, 1H), 4.53 (s, 2H), 3.91 (s, 3H), 3.73 (s, 3H), 3.56 (t, J = 6.8 Hz, 2H), 2.87 (t, J = 6.8 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 166.89, 155.25, 154.99, 147.69, 142.93, 130.04, 129.83, 129.76, 129.40, 128.50, 127.21, 122.35, 121.18, 119.10, 115.67, 109.78, 55.60, 52.15, 50.97, 50.33, 33.88. LRMS: [M+H]+ = 435.2.
Methyl 4-((1-(3-Methoxypropyl)-3-phenylureido)methyl)benzoate (4e)
Made according to general procedure B affording a colorless oil (70 mg, 73%). 1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 8.4 Hz, 2H), 7.84 (s, 1H), 7.45 (d, J = 8.4. Hz, 2H), 7.40 (d, J = 8.0 Hz, 2H), 7.30 (m, 2H), 7.02 (t, J = 7.6 Hz, 1H), 4.64 (s, 2H), 3.92 (s, 3H), 3.49 (t, J = 5.2 Hz, 2H), 3.44 (s, 3H), 3.415 (t, J = 6.4 Hz, 2H), 1.77 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 166.87, 156.36, 143.76, 139.85, 129.88, 129.18, 128.81, 127.72, 122.45, 119.17, 68.16, 58.63, 52.05, 49.41, 43.05, 27.55. LRMS ESI: [M+H]+ = 358.2.
Methyl 4-((1-(2-Methoxyethyl)-3-phenylureido)methyl)benzoate (4f)
Made according to general procedure B affording a colorless oil (40 mg, 91%). 1H NMR (400 MHz, CDCl3) δ 8.33 (br, 1H), 8.02 (d, J = 8.0 Hz, 2H), 7.35 (m, 6H), 7.02 (t, J = 7.2 Hz, 1H), 4.68 (s, 2H), 3.93 (s, 3H), 3.50 (s, 3H), 3.46 (s, 4H). 13C NMR (100 MHz, CDCl3) δ 166.88, 157.07, 143.81, 139.37, 129.92, 129.28, 128.81, 127.73, 122.34, 119.15, 72.59, 59.28, 52.07, 50.90, 48.44. LRMS ESI: [M+H]+ = 343.2.
Methyl 4-((1-Butyl-3-phenylureido)methyl)benzoate (4g)
Made according to general procedure B affording colorless oil (59 mg, 98%). 1H NMR (400 MHz, CDCl3) δ8.04 (d, J = 8.0 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 7.27 (m, 4H), 7.03 (t, J = 7.2 Hz, 1H), 6.32 (s, 1H), 4.65 (s, 2H), 3.93 (s, 3H), 3.36 (t, J = 7.2 Hz, 2H), 1.64 (m, 2H), 1.37 (m, 2H), 0.96 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 166.74, 155.30, 143.11, 138.83, 130.14, 129.50, 128.85, 127.04, 123.16, 119.90, 52.12, 50.49, 47.74, 30.52, 20.18, 13.81. LRMS ESI: [M+H]+ = 341.1
Methyl 4-((1-Phenethyl-3-phenylureido)methyl)benzoate (4h)
Made according to general procedure B affording a colorless oil (113 mg, 92%). 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.4 Hz, 2H), 7.36 (m, 4H), 7.30 (d, J = 7.2 Hz, 1H), 7.21 (m, 4H), 7.08 (d, J = 8.0 Hz, 2H), 6.99 (t, J = 7.2 Hz, 1H), 6.00 (s, 1H), 4.58 (s, 2H), 3.91 (s, 3H), 3.59 (t, J = 6.8 Hz, 2H), 2.90 (t, J = 6.8 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 166.71, 155.61, 142.99, 138.86, 138.72, 130.06, 129.43, 129.00, 128.86, 128.65, 127.28, 126.92, 122.95, 119.83, 52.08, 50.38, 49.94, 34.74. LRMS ESI: [M+H]+ = 389.2.
4-((1-(3-(Dimethylamino)propyl)-3-(2-methoxyphenyl)ureido)methyl)-N-hydroxybenzamide (5a)
The synthesis of 5a is representative, general procedure C. Solid NaOH (125 mg, 3.12 mmol) was dissolved in an aq. solution (50% wt, 1 mL) at 0 °C. Then a solution of 4a (156 mg, 0.390 mmol) in THF/MeOH (1:1, 6 mL total) was added dropwise where the biphasic solution became homogenous upon compete addition. The resulting solution was allowed to stir 30 min at room temperature. The reaction was quenched with AcOH (0.223 mL, 3.90 mmol) and concentrated in vacuo, and the crude product was purified via HPLC method 2 and neutralized with bicarbonate wash affording the title compound (20 mg, 13%). 1H NMR (400 MHz, DMSO-d6) δ 11.22 (br s, 1H), 9.02 (br s, 1H), 7.80-7.76 (m, 3H), 7.39 (d, J = 8.4 Hz, 2H), 7.00-6.94 (m, 2H), 6.88-6.84 (m, 2H), 4.62 (s, 2H), 3.72 (s, 3H), 3.43-3.40 (m, 2H), 2.82 (br s, 2H), 2.56 (s, 6H), 1.91-1.86 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 155.35, 149.62, 141.56, 131.69, 128.60, 127.24, 127.15, 127.02, 123.14, 121.40, 120.23, 110.78, 55.62, 54.33, 49.32, 44.24, 42.76, 23.45. HRMS ESI: calc. for C21H28N4O4 [M+H]+ m/z = 401.2183; found 401.2164.
N-Hydroxy-4-((1-(3-hydroxypropyl)-3-(2-methoxyphenyl)ureido)methyl)benzamide (5b)
Made according to general procedure C and purified via method 3 affording the title compound (95 mg, 84 %). 1H NMR (400 MHz, DMSO-d6) δ 11.19 (br s, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.76-7.71 (m, 3H), 7.37 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 4.0 Hz, 2H), 6.88-06.85 (m, 1H), 4. 58 (s, 2H), 3.74 (s, 3H), 3.48 (t, J = 5.6 Hz, 2H), 3.40 (t, J = 6.8 Hz, 2H), 1.7321.69 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 164.10, 155.18, 149.08, 141.99, 131.56, 128.91, 127.18, 127.10, 126.93, 122.61, 120.49, 120.29, 110.76, 57.59, 55.69, 49.28, 43.97, 30.61. HRMS ESI: calc. for C19H23N3O5 [M+H]+ m/z = 374.1710; found 374.1693.
4-((1-(2-(1H-Indol-3-yl)ethyl)-3-(2-methoxyphenyl)ureido)methyl)-N-hydroxybenzamide (5c)
Made according to general procedure C and purified via method 1 affording the title compound (62 mg, 57%). 1H NMR (400 MHz, DMSO-d6) δ 11.21 (br s, 1H), 10.86 (s, 1H), 10.12 (br s, 1H), 7.86 (t, J = 7.6 Hz, 1H), 7.75 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 8.0 Hz, 1H), 7.42-7.40 (m, 3H), 7.34 (d, J = 8.0 Hz, 1H), 7.18 (s, 1H), 7.07 (t, J = 7.2 Hz, 1H), 7.01-6.96 (m, 3H), 6.90-6.86 (m, 1H), 4.64 (s, 2H), 3.71 (s, 3H), 3.62 (t, J = 7.6 Hz, 2H), 3.00 (t, J = 7.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 163.97, 158.37, 154.72, 148.88, 141.90, 136.21, 131.65, 128.70, 127.12, 122.98, 122.65, 120.97, 120.35, 118.31, 118.18, 111.41, 111.11, 110.66, 55.66, 49.77, 48.47, 23.89. HRMS ESI: calc. for C26H26N4O4 [M+H]+ m/z = 459.2027; found 459.2030.
N-Hydroxy-4-((1-(4-hydroxyphenethyl)-3-(2-methoxyphenyl)ureido)methyl)benzamide (5d)
Made according to general procedure C and purified via method 2 affording the title compound (63 mg, 63%). 1H NMR (400 MHz, DMSO-d6) δ 11.20 (br s, 1H), 9.20 (br s, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.74 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.4 Hz, 2H), 7.37 (s, 1H), 7.04 (d, J = 8.4 Hz, 2H), 6.98 (d, J = 8.0 Hz, 2H), 6.89-6.85 (m, 1H), 6.69 (d, J = 8.4 Hz, 2H), 4.57 (s, 2H), 3.75 (s, 3H), 3.49 (t, J = 7.6 Hz, 2H), 2.75 (t, J = 7.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 164.00, 155.76, 154.62, 148.97, 141.88, 131.63, 129.63, 128.83, 127.12, 122.72, 120.44, 120.33, 115.22, 110.69, 55.69, 49.68, 49.49, 33.21. HRMS ESI: calc. for C24H25N3O5 [M+H]+ m/z = 436.1867; found 436.1858.
N-Hydroxy-4-((1-(3-methoxypropyl)-3-phenylureido)methyl)benzamide (5e)
Made according to general procedure C and purified via method 2 affording the title compound (48 mg, 76%). 1H NMR (400 MHz, DMSO-d6) δ 11.19 (br, 1H), 8.36 (s, 1H), 7.72 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 7.6 Hz, 2H), 7.32 (d, J = 8.4 Hz, 2H), 7.23 (m, 2H), 6.94 (t, J = 7.2 Hz, 1H), 4.61 (s, 2H), 3.34 (m, 4H), 3.21 (s, 3H), 1.74 (m, 2H). 13C NMR (100 MHz, DMSO2d6) δ164.04, 155.30, 142.25, 140.42, 131.49, 128.31, 127.06, 121.87, 119.88, 69.16, 57.88, 49.08, 43.56, 27.81. HRMS ESI: calc. for C19H23N3O4 [M+H]+ m/z = 358.1761; found 358.1785.
N-Hydroxy-4-((1-(2-methoxyethyl)-3-phenylureido)methyl)benzamide (5f)
Made according to general procedure C and purified method 2 affording the title compound (20 mg, 50%). 1H NMR (400 MHz, DMSO-d6) δ 11.17 (s, 1H), 9.01 (br, 1H), 8.44 (s, 1H), 7.72 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 7.6 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 7.23 (t, J = 7.6 Hz, 2H)H, 6.94 (t, J = 7.2 Hz, 1H), 4.64 (s, 2H), 3.49 (s, 4H), 3.28 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 164.06, 155.46, 142.17, 140.30, 131.43, 128.32, 126.97, 121.83, 119.64, 70.88, 58.31, 49.82, 46.35. HRMS ESI: calc. for C18H21N3O4 [M+H]+ m/z = 344.1605; found 344.1601.
4-((1-Butyl-3-phenylureido)methyl)-N-hydroxybenzamide (5g)
Made according to general procedure C and purified by method 2 affording the title compound (40 mg, 68%). 1H NMR (400 MHz, DMSO-d6) δ 11.17 (br, 1H), 8.36 (s,1H), 7.72 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 7.6 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 7.22 (t, J = 7.6 Hz, 2H), 6.94 (t, J = 7.2 Hz, 1H), 4.62 (s, 2H), 3.30 (m, 1H), 1.48 (m, 2H), 1.27 (m, 2H), 0.86 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 164.01, 155.22, 142.30, 140.45, 131.42, 128.19, 127.00, 126.96, 121.80, 120.04, 49.01, 46.11, 29.64, 19.43, 13.77. HRMS ESI: calc. for C19H23N3O3 [M+H]+ m/z = 342.1812; found 342.1802.
N-Hydroxy-4-((1-phenethyl-3-phenylureido)methyl)benzamide (5h)
Made according to general procedure C and purified by method 2 affording the title compound (71 mg, 63%). 1H NMR (400 MHz, DMSO-d6) δ 11.15 (br, 1H), 7.72 (d, J = 8.0 Hz, 2H), 7.45 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 8.0 Hz, 2H), 7.24 (m, 7H), 6.95 (t, J = 7.2 Hz, 1H), 4.60 (s, 2H), 3.54 (t, J = 7.6 Hz, 2H), 2.82 (t, J = 7.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 164.04, 155.12, 142.12, 140.35, 139.04, 131.50, 128.77, 128.31, 128.20, 127.09, 127.05, 126.16, 121.91, 120.13, 49.20, 48.04, 33.91. HRMS ESI: calc. for C23H23N3O3 [M+H]+ m/z = 390.1812; found 390.1793.
N-Butylaniline (6a)
Was synthesized in an analogous manner previously reported.12 Briefly, CuI (19 mg, 0.1 mmol) and freshly ground K3PO4 (849 mg, 4 mmol) were placed in a sealed tube followed by sequential addition of isopropanol (2 mL), ethylene glycol (0.222 mL, 4.0 mL), phenyl-iodide (0.224 mL, 2.0 mmol) and n-butylamine (0.237 mL, 2.4 mmol). The tube was then sealed and stirring commenced at 80 °C for 18 h. After cooling to room temperature the reaction was diluted with water:ethyl ether (1:1, 10 mL). The aqueous layer was extracted with ether (3× 5mL), washed with brine (15 mL), dried over sodium sulfate and concentrated in vacuo. Purification via flash chromatography afforded the title compound as a yellow oil (235 mg, 79%). 1H NMR (400 MHz, CDCl3) δ 7.19 (m, 2H), 6.70 (t, J = 7.2 Hz, 1H), 6.61 (d, J = 8.4 Hz, 2H), 3.60 (br, 1H), 3.12 (t, J = 7.2 Hz, 2H), 1.62 (m, 2H), 1.44 (m, 2H), 0.98 (t, J = 7.2 Hz, 3H). Spectra matches that reported in the literature.17
N-(3-Methoxypropyl)aniline (6b)
Made following the same procedure for 6a affording a light yellow oil (282 mg, 85%). 1H NMR (400 MHz, CDCl3) δ 7.18 (m, 2H), 6.69 (t, J = 7.2 Hz, 1H), 6.61 (d, J = 8.4 Hz, 2H), 3.92 (br, 1H), 3.52 (t J = 6.0 Hz, 2H), 3.60 (s, 3H), 3.23 (t, J = 6.8 Hz, 2H), 1.90 (m, 2H). Spectra matches that reported in the literature.18
Methyl 4-((3-Butyl-3-phenylureido)methyl)benzoate (7a)
Methyl 4-(aminomethyl)benzoate hydrochloride (101 mg, 0.5 mmol) was taken up in a biphasic solution of DCM:sat. bicarbonate (1:1, 4 mL) and added triphosgene (49 mg, 0.17 mmol) at 0 °C. After 30 min, the aqueous layer was extracted with DCM (3× 5mL), washed with brine (15 mL) and concentrated in vacuo. The crude isocyante was taken up in DCM (2 mL) and added 6a (75 mg, 0.5 mmol) and Et3N (0.209 mL, 1.5 mmol) and resulting solution allowed to stir overnight at room temperature. The reaction was quenched with with sat. bicarbonate (5 mL) and extracted with DCM (3× 5mL). The combined organics were washed with brine (15 mL), dried over sodium sulfate, concentrated in vacuo. The crude material was purified via flash chromatography affording the title compound as an off-white waxy solid (93 mg, 55%). 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.0 Hz, 2H), 7.42 (t, J = 7.6 Hz, 2H), 7.32 (m, 1H), 7.25 (m, 4H), 4.45 (t, J = 5.6 Hz, 1H), 4.41 (d, J = 6.0 Hz, 2H), 3.89 (s, 3H), 3.70 (t, J = 7.6 Hz, 2H), 1.48 (m, 2H), 1.31 (m, 2H), 0.89 (t, 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 166.89, 156.87, 145.14, 141.61, 130.08, 129.78, 128.85, 128.73, 127.81, 126.96, 52.01, 49.21, 44.25, 30.68, 19.92, 13.82. LRMS ESI: [M+H]+ = 341.1.
Methyl 4-((3-(3-Methoxypropyl)-3-phenylureido)methyl)benzoate (7b)
Made according to that of 7a except using 6b as the secondary amine affording the title compound as an off-white waxy solid (65 mg, 36%). 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8.0 Hz, 2H), 7.43 (t, J = 7.6 Hz, 2H), 7.33 (m, 1H), 7.27 (m, 4H), 4.69 (br, 1H), 4.42 (6.0 Hz, 2H), 3.90 (s, 3H), 3.80 (t, J = 7.2 Hz, 2H), 3.43 (t, J = 6.4 Hz, 2H), 3.27 (s, 3H), 1.83 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 166.88, 156.95, 145.05, 141.63, 130.10, 129.80, 128.90, 127.79, 127.00, 70.23, 58.54, 52.03, 46.86, 44.29, 28.82. LRMS ESI: [M+H]+ = 357.1.
Methyl 4-((3-Ethyl-3-phenylureido)methyl)benzoate (7c)
Made according to that of 7a except using commercially available N-ethylaniline as the secondary amine affording the title compound as an off2white waxy solid (197 mg, 63%). 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 8.4 Hz, 2H), 7.24 (t, J = 7.6 Hz, 2H), 7.32 (t, J = 7.2 Hz, 1H), 7.26 (m, 4H), 4.57 (br, 1H), 4.41 (d, J = 5.6 Hz, 1H), 3.88 (s, 3H), 3.76 (dd, J = 14, 7.2 Hz, 2H), 1.12 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 166.82, 156.64, 145.09, 141.22, 130.03, 129.72, 128.79, 127.82, 126.92, 51.95, 44.17, 44.09, 13.82. LRMS ESI: [M+H]+ = 313.1.
4-((3-Butyl-3-phenylureido)methyl)-N-hydroxybenzamide (8a)
Made according to general procedure C and purified by method 2 affording the title compound as an off2white solid (74 mg, 80%). 1H NMR (400 MHz, DMSO-d6) δ 11.13 (br, 1H), 8.72 (br, 1H), 7.66 (d, J =8.0 Hz, 2H), 7.43 (t, J = 7.6 Hz, 2H), 7.27 (m, 5H), 6.23 (t, J = 5.6 Hz, 1H), 4.20 (d, J = 5.6 Hz, 2H), 3.57 (t, J = 6.8 Hz, 2H), 1.35 (m, 2H), 1.23 (m, 2H), 0.82 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 164.17, 156.48, 144.50, 142.12, 130.129.55, 128.22, 126.67, 126.61, 48.44, 43.38, 30.21, 19.35, 13.72. HRMS ESI: calc. for C19H23 N3O3 [M+H]+ m/z = 342.1812; found 342.1825.
N-Hydroxy-4-((3-(3-methoxypropyl)-3-phenylureido)methyl)benzamide (8a)
Made according to general procedure C and purified by method 2 affording an off2white solid (59 mg, 91%). 1H NMR (400 MHz, DMSO-d6) δ 11.14 (br, 1H), 7.66 (d, J = 8.0 Hz, 2H), 7.43 (t, J = 7.6 Hz, 2H), 7.27 (m, 5H), 6.27 (t, J = 6.0 Hz, 1H), 4.21 (d, J = 5.8 Hz, 2H), 3.62 (t, J = 7.2 Hz, 2H), 3.28 (t, J = 6.4 Hz, 2H), 3.14 (s, 3H), 1.63 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 164.13, 156.51, 144.44, 142.19, 130.89, 129.57, 128.18, 126.70, 126.64, 69.49, 57.80, 46.35, 43.39, 28.32. HRMS ESI: calc. for C19H23N3O4 [M+H]+ m/z = 358.1761; found 358.1749.
4-((3-Ethyl-3-phenylureido)methyl)-N-hydroxybenzamide (8c)
Made according to general procedure C and purified by method 2 affording an off2white solid (91 mg, 96%). 1H NMR (400 MHz, DMSO-d6) δ 11.13 (br, 1H), 7.67 (d, J = 8.0 Hz, 2H), 7.43 (t, J = 7.6 Hz, 2H), 7.26 (m, 5H), 6.30 (t, J = 6.0 Hz, 1H), 4.21 (d, J = 5.6 Hz, 2H), 3.61 (dd, J = 14, 7.2 Hz, 2H), 0.99 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 164.21, 156.34, 144.54, 142.00, 130.92, 129.57, 128.32, 126.75, 126.67, 43.70, 43.39, 13.76. HRMS ESI: calc. for C17H19N3O3 [M+H]+ m/z = 314.1499; found 314.1489.
HDAC inhibition assays
HDAC inhibition assays were performed by Reaction Biology Corp. (Malvern, PA) using isolated human, recombinant full2length HDAC1 and -6 from a baculovirus expression system in Sf9 cells. An acetylated fluorogenic peptide,RHKKAc, derived from residues 379-382 of p53 was used as substrate. The reaction buffer was made up of 50 mM Tris-HCl pH 8.0, 127 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mg/mL BSA, and a final concentration of 1% DMSO. Compounds were delivered in DMSO and delivered to enzyme mixture with preincubation of 5-10 min followed by substrate addition and incubation for 2 h at 30 °C. Trichostatin A and developer were added to quench the reaction and generate fluorescence, respectively. Dose-response curves were generated starting at 30 μM compound with three-fold serial dilutions to generate a 10-dose plot. IC50 values were then generated from the resulting plots, and the values expressed are the average of duplicate trials ± standard deviation.
Tubulin and histone acetylation western blot assay
B16 melanoma cells were plated at 105 cells/well in 12 well plates and allowed to adhere overnight. A 50 mM stock of compound was then added by serial dilutions in complete medium to the indicated concentrations. Cells were incubated for 24 h under humidified conditions (37°C, 5% CO2). Wells were then washed with cold PBS, and cells were lysed in a buffer containing 10 mM Tris2HCl pH 8.0, 10% SDS, 4 mM urea, 100 mM DTT, and 1× protease inhibitor (Roche). Cells were lysed for 30 min on ice, then sonicated for 8 min (8 cycles of 30 s on/30 s rest). Cells were then boild for 10 min with 6× gel loading buffer and resolved on 4-15% gradient gels and subsequently transferred onto nitrocellulose membranes. Membranes were blocked with 5% milk in PBS-T and detection of specific antigens using antibodies against acetyl-H3 and H3 (Cell Signaling), and acetyl-α-tubulin and α-tubulin (Sigma). Bands were detected by scanning blots with LI2COR Odyssey imaging system using both 700 and 800 channels.
B16 melanoma cell growth inhibition assay
B16 murine melanoma cells were plated at 5×103/well in 96 well flat bottom plates. The following day, media was changed to that containing various concentrations of HDACi or matched DMSO vehicle concentrations diluted in complete medium done in triplicate. Cells were incubated for 48 hours at 37°C and 5% CO2. Density of viable, metabolically active cells was quantified using a standard MTS assay (CellTiter 96® AQueous One, Promega, Madison, WI) as per manufacturer’s instructions. Briefly, 20μL of reagent were added per well and incubated at 37°C for 3 hours. Absorbances at 490nM were measured spectrophotometrically with background subtraction at 690 nM. All values were then normalized and expressed as a percentage of medium control (100%).
ACKNOWLEDGMENT
This work was supported by grants provided by the NIH (CA134807 to EMS and APK) and the International Rett Syndrome Foundation (IRSF).
Abbreviations used
- HDAC
histone/protein decetylase
- HDACI
histone deacetylase inhibitor
- TSA
trichostatin A
- ZBG
zinc-binding group
References
- 1.Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, Lombard D, Mizeracki A, Matthias G, Alt FW, Khochbin S, Matthias P. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Molecular and Cellular Biology. 2008;28(5):1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, Cirstea D, Rodig S, Eda H, Scullen T, Canavese M, Bradner J, Anderson KC, Jones SS, Raje N. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119(11):2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peltonen K, Kiviharju TM, Jarvinen PM, Ra R, Laiho M. Melanoma cell lines are susceptible to histone deacetylase inhibitor TSA provoked cell cycle arrest and apoptosis. Pigment Cell Research. 2005;18(3):196–202. doi: 10.1111/j.1600-0749.2005.00225.x. [DOI] [PubMed] [Google Scholar]; Facchetti F, Previdi S, Ballarini M, Minucci S, Perego P, La Porta CAM. Modulation of pro- and anti-apoptotic factors in human melanoma cells exposed to histone deacetylase inhibitors. Apoptosis. 2004;9(5):573–582. doi: 10.1023/B:APPT.0000038036.31271.50. [DOI] [PubMed] [Google Scholar]
- 4.Wagner JM, Hackanson B, Lubbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clinical Epigenetics. 2010;1(3-4):117–136. doi: 10.1007/s13148-010-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. Journal of Biological Chemistry. 2002;277(28):25748–25755. doi: 10.1074/jbc.M111871200. [DOI] [PubMed] [Google Scholar]
- 6.Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, Nguyen D, Wright KL, Atadja PW, Bhalla K, Pinilla-Ibarz J, Seto E, Sotomayor EM. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nature Immunology. 2009;10(1):92–100. doi: 10.1038/ni.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Cheng F, Woan K, Sahakian E, Merino O, Rock-Klotz J, Vicente-Suarez I, Pinilla-Ibarz J, Wright KL, Seto E, Bhalla K, Villagra A, Sotomayor EM. Histone Deacetylase Inhibitor LAQ824 Augments Inflammatory Responses in Macrophages through Transcriptional Regulation of IL-10. The Journal of Immunology. 2011;186(7):3986–3996. doi: 10.4049/jimmunol.1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, Marks PA. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(28):9633–9638. doi: 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational Design and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor, Tubastatin A. Journal of the American Chemical Society. 2010;132(31):10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalin JH, Butler KV, Akimova T, Hancock WW, Kozikowski AP. Second-Generation Histone Deacetylase 6 Inhibitors Enhance the Immunosuppressive Effects of Foxp3+ T-Regulatory Cells. Journal of Medicinal Chemistry. 2012;55(2):639–651. doi: 10.1021/jm200773h. [DOI] [PubMed] [Google Scholar]
- 11.Vickers CJ, Olsen CA, Leman LJ, Ghadiri MR. Discovery of HDAC Inhibitors That Lack an Active Site Zn2+-Binding Functional Group. ACS Medicinal Chemistry Letters. 2012;3(6):505–508. doi: 10.1021/ml300081u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwong FY, Klapars A, Buchwald SL. Copper-catalyzed coupling of alkylamines and aryl iodides: An efficient system even in an air atmosphere. Organic Letters. 2002;4(4):581–584. doi: 10.1021/ol0171867. [DOI] [PubMed] [Google Scholar]
- 13.Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget M-C, Kalita A, Liu J, Lu A-H, Zhou NZ, Robert M-F, Gillespie J, Wang JJ, Ste-Croix H, Rahil J, Lefebvre S, Moradei O, Delorme D, MacLeod AR, Besterman JM, Li Z. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Molecular Cancer Therapeutics. 2008;7(4):759–768. doi: 10.1158/1535-7163.MCT-07-2026. [DOI] [PubMed] [Google Scholar]
- 14.Zhou N, Moradei O, Raeppel S, Leit S, Frechette S, Gaudette F, Paquin I, Bernstein N, Bouchain G, Vaisburg A, Jin Z, Gillespie J, Wang J, Fournel M, Yan PT, Trachy-Bourget MC, Kalita A, Lu A, Rahil J, MacLeod AR, Li Z, Besterman JM, Delorme D. Discovery of N-(2-aminophenyl)4- 4-pyridin-3-ylpyrimidin-2-ylamino)methyl benzamide (MGCD0103), an orally active histone deacetylase inhibitor. Journal of Medicinal Chemistry. 2008;51(14):4072–4075. doi: 10.1021/jm800251w. [DOI] [PubMed] [Google Scholar]
- 15.Kelly WK, Marks PA. Drug Insight: histone deacetylase inhibitors - development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nature Clinical Practice Oncology. 2005;2(3):150–157. doi: 10.1038/ncponc0106. [DOI] [PubMed] [Google Scholar]
- 16.Zou H, Wu YQ, Navre M, Sang BC. Characterization of the two catalytic domains in histone deacetylase 6. Biochemical and Biophysical Research Communications. 2006;341(1):45–50. doi: 10.1016/j.bbrc.2005.12.144. [DOI] [PubMed] [Google Scholar]
- 17.Okano K, Tokuyama H, Fukuyama T. Synthesis of secondary arylamines through copper-mediated intermolecular aryl amination. Organic Letters. 2003;5(26):4987–4990. doi: 10.1021/ol035942y. [DOI] [PubMed] [Google Scholar]
- 18.Guo DL, Huang H, Xu JY, Jiang HL, Liu H. Efficient Iron-Catalyzed N-Arylation of Aryl Halides with Amines. Organic Letters. 2008;10(20):4513–4516. doi: 10.1021/ol801784a. [DOI] [PubMed] [Google Scholar]