

Abstract
Translational accuracy and efficiency depends upon modification of uridines in the tRNA anticodon stem loop (ASL) by a highly conserved pseudouridine synthase TruA. TruA specifically modifies uridines at positions 38, 39 and/or 40 of tRNAs with highly divergent sequences and structures through a poorly characterized mechanism that differs from previously studied RNA modifying enzymes. The molecular basis for the site and substrate “promiscuity” was studied by determining the crystal structures of E. coli TruA in complex with two different leucyl tRNAs in conjunction with functional assays and computer simulation. The structures capture three stages of the TruA*tRNA reaction, revealing the mechanism by which TruA selects the target site. We propose that TruA utilizes the intrinsic flexibility of the ASL for site promiscuity and also to select against intrinsically stable tRNAs to avoid their overstabilization through pseudouridylation, thereby maintaining the balance between the flexibility and stability required for its biological function.
Keywords: Pseudouridine synthase, tRNA, anticodon stem loop, substrate flexibility, multisite specificity, regional specificity
Introduction
Protein synthesis involves a series of coordinated movements and conformational changes of tRNAs inside the ribosome while maintaining the anticodon-codon interaction. Many experiments suggest that sequences of tRNAs have been optimized through evolution to ensure efficient and accurate decoding during translational processes (Cochella and Green, 2005; Olejniczak et al., 2005). Extensive usage of various modified ribonucleotides identified in tRNAs, which alter the chemical and physical properties of the nucleotides, also suggests the importance of fine-tuning the conformations and dynamics of tRNAs (Agris, 2004). Among the approximately one hundred modified nucleotides identified to date, the most common is the C-C glycosidic isomer of uridine, pseudouridine (Ψ). This modification introduces one additional hydrogen bond donor at the 5-position of uridine. The critical role of Ψ in biogenesis and function of the ribosome and spliceosome has been well documented (Gutgsell et al., 2005; Yang et al., 2005).
Ψ's at positions 38-40 in the Anticodon Stem Loop (ASL) of tRNAs play an important role in maintaining translational efficiency and accuracy (Agris, 2004). Although the crystal and NMR structures of pseudouridylated and non-pseudouridylated tRNA ASL are very similar (Arnez and Steitz, 1994; Durant and Davis, 1999), Ψ's at 38-40 were shown to increase the thermal stability of the ASL, which could affect the anticodon-codon interaction or conformational changes of the tRNA during translation. X-ray and NMR studies revealed a unique hydrogen bond mediated by a water molecule between the new hydrogen bond donor of Ψ and the phosphate backbone, and UV hypochromicity suggested an enhanced base stacking of pseudouridylated RNA (Arnez and Steitz, 1994; Durant and Davis, 1999). These data suggest a molecular basis for this thermal stabilization.
Pseudouridylation of U38-40 is catalyzed by a highly conserved enzyme, TruA, which belongs to one of the five families of Ψ synthase. The crystal structure of apo-TruA from E. coli, which was solved in our laboratory as the first structure of a Ψ synthase, revealed that the enzyme is a homodimer, where each monomer consists of distinct N- and C-terminal domains juxtaposed to form an active site cleft harboring the universally conserved catalytic Asp60 (Foster et al., 2000). More recent structures of several other bacterial Ψ synthases from four other families, as well as that of H/ACA snoRNP, a eukaryote/archaea specific Ψ synthase, have shown that other Ψ synthases function as monomers. However, the overall fold and the active site are similar to a monomer of TruA, suggesting that all five families of Ψ synthase from bacteria to eukaryotes evolved from a common ancestor (del Campo et al., 2004; Hamma et al., 2005; Hoang et al., 2006; Hoang and Ferre-D'Amare, 2004; Li and Ye, 2006; Rashid et al., 2006; Sivaraman et al., 2002).
TruA is distinct from other Ψ synthases in its substrate specificity in the following ways. First, TruA modifies multiple tRNAs (for example, 17 tRNAs in E. coli) with divergent sequences in the region of modification, namely the ASL (Sprinzl et al., 1998). In contrast, TruB, which modifies U55 in nearly all tRNAs, binds to a conserved sequence of the neighboring nucleotides of U55 in the T-stem loop (Gu et al., 1998). Second, TruA can modify nucleotides that are as far as 15 Å apart using a single active site. For example, tRNAleu2 contains uridines at positions 38 and 40, and both are modified by TruA. RluD is the only other Ψ synthase in E. coli exhibiting a similar regional specificity, and its substrate is a single stem-loop within rRNA rather than multiple tRNAs as in the case of TruA. We have used a structural approach to understand this remarkable substrate specificity of TruA.
Here, we report the crystal structures of TruA in complex with tRNAleu3, and in complex with tRNAleu1 from E. coli. This is the first report of structures of a Ψ synthase bound to its intact substrate tRNA. (To date, TruB and RluA are the only other Ψ synthases which have been crystallized with a substrate, the isolated 22-nucleotide T-stem loop and the 21-nucleotide ASL of tRNA, respectively (Hoang et al., 2006;Hoang and Ferre-D'Amare, 2001; Pan et al., 2003).) Three crystal forms in our study yielded TruA•tRNA complexes in six different crystallographic environments. The six crystallographically independent TruA•tRNA complexes display the ASL in three different conformations, allowing us to model three different states along the substrate recognition pathway: the initial complex where the tRNA body docks distal to the active site, an intermediate state where the ASL bends towards the active site cleft, and the reactive conformation where a target base “flips out” and positions in the active site. Along with functional assays and computer simulations, our structures suggest molecular mechanisms by which TruA recognizes the three target sites in tRNA ASLs with divergent sequences and flips out the target nucleotides from the stacked conformation to gain the access to the base.
Results and Discussion
Overview of the TruA•tRNA Complex Structures
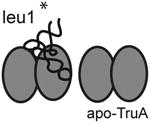
We attempted to obtain structures of E. coli TruA complexed with three E.coli tRNAs representing all of the target sites: tRNAleu1 with U at 39, tRNAleu2 with U at 38 and 40, and tRNAleu3 with U at 38. These tRNAs are type II tRNAs with a 15-nucleotide variable loop. Three crystal forms were obtained from similar buffer conditions, containing the complex of the wild type TruA (wt-TruA) and full length tRNAleu1 in crystal I, and the complex of wt-TruA and tRNAleu3 in crystal forms II and III (Table 1). No crystals were obtained with tRNAleu2. Since wt-TruA•tRNA is likely to yield product complexes, we tried to crystallize an analog of a TruA-substrate complex using a mutant enzyme with alanine substituted for the catalytic Asp60 (D60A), which binds tRNA more tightly than wt-TruA (Fig. S4). We also sought a reaction intermediate complex using tRNAleu1 with the mechanistic inhibitor, 5-fluorouridine (5f-U) at 39. No crystals were obtained in either case. The crystals that were obtained gave rise to six independent images of TruA-tRNA product complexes.
Table 1. Crystallographic Statistics.
| crystal I | crystal II | crystal III | |
|---|---|---|---|
| Asymmetric Unit Content |
|
|
|
| Data Collection | |||
| space group | P212121 | P212121 | P3221 |
| cell dimension (Å) | 96.3 × 128.6 × 159.3 | 65.0 × 149.3× 292.0 | 80.4×80.4×205.4 |
| Resolution (Å) | 20-3.5 | 50-3.9 | 20-4.0 |
| R merge (%) | 23.6 (72.2) | 21.3 (61.9) | 17.2 (53.1) |
| I/sI | 7.0 (2.4) | 4.9 (1.6) | 9.6 (3.4) |
| redundancy | 5.7 (5.8) | 2.8 (2.6) | 7.6 (6.8) |
| Refinement | |||
| No. atoms | 9935 | 14467 | 3253 |
| Solvent Content | 64.0 | 68.0 | 61.0 |
| Rwork/Rfree (%) | 24.6/27.9 | 29.8/35.0 | 25.9/35.7 |
| RMSD bond length (Å) | 0.01 | 0.01 | 0.01 |
| RMSD bond angles (°) | 1.24 | 1.29 | 1.33 |
The sequence identity between tRNAleu1 and tRNAleu3 is 65 % (the total length) and 26 % (the ASL only).
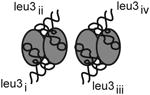
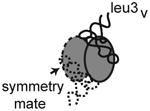
Crystal form I diffracts to 3.5 Å and the asymmetric unit contains one dimer of apo-TruA and one dimer of TruA complexed with one molecule of tRNAleu1 (Table 1). Crystal form II diffracts to 3.9 Å, and the asymmetric unit contains two dimers of TruA with each dimer bound to two molecules of tRNAleu3, thus crystal II presents four different examples of tRNAleu3 in complex with TruA. Crystal III diffracts to 4.0 Å and contains a dimer of TruA bound to two molecules of tRNAleu3 that lies on the crystallographic two fold axis. Thus the asymmetric unit contains only one image of the TruA•tRNAleu3 interaction. The five tRNAleu3s in crystals II and III are numbered (i-v) to distinguish them.
In all six crystallographic complexes, tRNA binds across the two subunits of the TruA dimer (Fig. 1A). The ASL binds in the cleft between the N- and C-terminal domains of TruA with the nucleotides at 38-40 positioned near the catalytic Asp60 (Fig. 1B). The binding mode of tRNA in our structures matches the predicted binding mode based on the structure of the apo-TruA dimer (Foster et al., 2000). In our complexes, neither TruA nor tRNA undergoes large conformational changes upon binding. All tRNAs retain the canonical L-shape with the long variable loop extruding away from TruA. The absence of interaction between the tRNA variable loop and TruA is consistent with the observation that TruA modifies both type I (short variable loop) and type II tRNAs.
Figure 1. Overview of the Structures of TruA•tRNA.

(A) Superposition of the six complexes of TruA•tRNA by aligning the TruA Cα backbones. Each monomer of TruA consists of distinct N-terminal (yellow) and C-terminal domains (red). tRNA (blue) binds to TruA across the dimerization interface. (B) The ASL binds in the cleft formed between the N- and C-terminal domains, placing the nucleotides at 38-40 (in stick model) proximal to the catalytic Asp60 (cyan). The 15-nucleotide variable loop, located between the ASL and the T-stem loop, is disordered and could not be traced completely in most tRNAs. (C) Surface representation of TruA•tRNAleu3i in two view angles shows that TruA interacts with the tRNA elbow, the D-stem backbone and the major/minor grooves of the ASL. Colors on the protein surface represent the electrostatic characteristics of the atoms; hydrophobic (white), positive (blue), negative (red), polar N (lightblue), polar O (salmon) and sulfur (yellow). Colors on tRNA represent atom identities; C (green), O (red), P (orange) and N (blue).
TruA recognizes the common shape and electrostatic properties of tRNAs. The interactions of tRNA with the protein involve the elbow where the D and T loops join together and the D-stem backbone (Fig. 1C). The exposed base and riboses on the surface of the elbow make hydrophobic contacts with a cluster of aliphatic residues in TruA. The D-stem backbone is recognized by a combination of hydrophobic and hydrophilic residues that complement the ribose and the phosphate backbone. These contacts position the ASL in the region of the active site cleft. Details of global interaction are described in Fig. S4.
The ASL appears to make contact with two loops, L1 (residues 20-33) and L8 (residues 166-173) (as named in apo-TruA (Foster et al., 2000)), which interact with the minor and the major grooves of the ASL (Fig. 1C). As in the structure of apo-TruA, these two loops exhibit the highest backbone temperature factors, which did not allow determination of the side chain conformations for all residues. However, the polar and charged characters of many residues lining the loops suggest electrostatic interaction between the ASL and these loops.
ASL-in and ASL-out Conformations of tRNA Bound to TruA
We superposed the six crystallographic complexes by aligning the Cα backbones of TruA. Unlike the elbow and the D-stem, the conformation of the ASL differs among the six tRNAs. Two primary ASL conformations were observed: one where the ASL is away from the protein, “ASL-out”, and one where it is tightly bound to the protein surface, “ASL-in” (Fig. 2A).
Figure 2. ASL-in and ASL-in Conformations.

(A) Stereo view. The superposition of the six tRNAs shows two distinct conformations of the ASL backbone (ASL-out and ASL-in) with different thermal factor profiles. The thermal factors are normalized relative to the average thermal factor of the bound protein Cα. ASL-out is more distant from the surface of TruA and exhibits significant flexibility, whereas ASL-in is bent towards TruA and shows thermal stability. A few nucleotides in ASL-out (typically nucleotides at positions 33-37) could not be traced in tRNAleu1, tRNAleu3IV and tRNAleu3V. (B) Spatial locations of target bases. Left: superposition of four “ASL-out” tRNAs with nucleotides at 38 (yellow), 39 (green) and 40 (blue) in stick model. Right: superposition of two “ASL-in” tRNAs with nucleotides 38, 39, and 40 similarly designated. In the insets, small spheres represent trajectories of the center of mass of the bases at 38-40 during the three 2 ns MD simulations. The starting structures (tRNAleu1 and tRNAleu3i) for the simulations are shown in stick model. (C) Comparison of the surface of TruA in complex with ASL-in tRNAleu3ii (left) and ASL-out tRNAleu3i (right) (L1 in blue, L8 in magenta, D60 in red, R58 in orange, R110 in green, Phe110 in cyan). Nucleotides at 38-40 are shown in stick model (only 40 is labeled). The cleft widths (double arrows) are measured between Cα's of R58 and Q170. Both proteins are in the same orientation, viewed from the bottom of the structures in (A).
ASL-out was observed in tRNAleu1 and three RNAleu3s (ii, iv and v). The ASLs in these tRNAs make only a few contacts with the protein (residues at 29-31 in L1 and with 169-170 in L8), and their phosphate backbone conformations are similar to those seen in free tRNAs (Shi and Moore, 2000). The ASL-out backbones display high thermal factors (Fig. 2A), indicating a high degree of flexibility analogous to the flexibility observed in tRNAs free in solution (Merino et al., 2005). The flexibility is also apparent in the target bases in that their spatial positions in the superposed ASL-out tRNAs are broadly distributed; for example nucleotide 38 in tRNAleu3ii occupies space that overlaps nucleotide 39 in tRNAleu3iv (Fig. 2B).
Because the different ASL-out conformations could have been induced, perhaps, by crystal contacts, we investigated the flexibility of the ASL by molecular dynamics (MD) simulations. We started a simulation from one ASL-out conformation, tRNAleu1, and asked if the trajectories of the target bases would envelope their positions in the other structures. Superposition of the trajectories obtained during the three 2 ns MD simulations that were initiated with different conditions (see Experimental Procedures) shows that each base drifts into the space occupied by the adjacent nucleobase as seen in the crystal structures (Fig. 2B inset). Thus, both crystallographic data and MD simulations support the notion that the ASL in the out conformation is highly flexible, and the target bases may have a fairly wide spatial distribution.
In contrast to ASL-out, the phosphate backbone of ASL-in observed in two tRNAleu3s (i and iii) is bent towards the enzyme and is more ordered as judged by thermal factors (Fig. 2A). The locations of the target bases are more ordered than in ASL-out (Fig. 2B). Similarly motivated MD simulations of TruA•tRNAleu3i also indicate that the bases do not intrude on each others positions, and that the conformation of the ASL is stable as shown in the crystal structure (Fig. 2B inset).
The overall conformations of TruA in ASL-out and ASL-in complexes are very similar, except in the immediate ASL binding cleft bounded by protein loops L1 and L8. In the ASL-out complexes, the conformation of L8 is similar to that in the apo-state, whereas in the ASL-in complexes, L8 moves away from L1 widening the cleft to ∼20 Å, compared to ∼12 Å in ASL-out (Fig. 2C). This allows the insertion of the ASL target strand (containing nucleotides 38-40) into the newly opened cleft. Additional electrostatic interactions are formed between the phosphate backbone and several residues in the cleft (R58 and R110) and the L8 loop (Q168, Q170 and R172). These newly formed contacts are the basis of the ordered character of the ASL-in conformation.
Base-Flip Occurs in the “ASL-in” Conformation
In five of the six tRNAs, representing both ASL-out and ASL-in conformations, bases of the TruA target nucleotides, 38, 39 and 40, remain stacked in the ASL and are 10-15 Å away from the catalytic Asp60 at the active site (Fig. 3A-B). However, in TruA•tRNAleu3i, a conformation for the ASL in which the base of G39 is flipped-out of the stem loop coexists at partial occupancy with the stacked conformation. When the crystal II structure is refined with only the stacked conformation of tRNAleu3i, strong electron density appears in the TruA active site of this complex (Fig. 3C). The density matches the shape and size of a guanine nucleotide (Fig. S2 for more discussion). G39 can be fitted to the density by a simple rotation of G39 around the 3′O(G39)---P(G40) bond with a minor rearrangement in the G40 phosphate group as observed in the ASL-in stacked conformation (Fig. 3C), whereas G40 cannot be built into the density without major distortions in the tRNA and protein structures.
Figure 3. Stacked and Flipped-out Conformations of Target Bases.

Difference maps (Fo-Fc, contoured at 2.5σ) phased with the structure of (A) TruA•tRNAleu3ii(ASL-out), and (B) TruA•tRNAleu3iii (ASL-in), where the nucleotides at 38-40 are omitted, show the target bases stacked in the ASL. Distances (dashed lines) measured between Asp60-Cγ and N1 in pyrimidines or N9 in purines indicate that the catalytic Asp60 cannot access the target bases in the stacked conformation in either ASL-out or ASL-in (C) Stereoview of the active site of TruA•tRNAleu3i with TruB (PDB: 1R3E) active site superposed. The difference map (Fo-Fc) phased with the stacked conformation (green) shows the partial occupancy of G39 in the flipped-out conformation (cyan). The simulated annealing omit map is provided in Fig. S2. Superposed TruB residues (yellow) align with homologous TruA residues. The co-crystalized reaction product, 5-fluoro-6-hydroxy Ψ (5f-6OH-Ψ) in the TruB structure aligns with the flipped-out G39. (D) Summary of the tRNA conformations.
The “G39-flipped-out” conformation of TruA•tRNAleu3i is a relevant model for the reactive conformation for a U39 modification. The catalytic Asp60 is oriented towards the bound base as required by its role of catalytic nucleophile in the proposed chemical mechanism (Gu et al., 1999; Hamilton et al., 2005). In addition, superposition of the structure of the TruB-RNA complex (Hoang and Ferre-D'Amare, 2001; Pan et al., 2003) with the TruA•tRNAleu3i complex reveals that the flipped out G39 aligns well with the target base of the co-crystallized reaction product in the TruB structure (Fig. 3C).
Finding G39 in the flipped out conformation suggests that TruA flips out any nucleotide in a target position regardless of the base identity and incorporates it into the active site that is large and mainly hydrophobic. The size and hydrophobicity of the active site is conserved among all Ψ synthases whose structures have been solved. Although the large size of the hydrophobic active site in Ψ synthases does not allow discrimination between nucleotides based on size and shape, it may be important for the chemical process of pseudouridylation, which involves a 180° rotation of the base to shift the atom position that bridges to the ribose ring.
In the other five complexes of TruA•tRNA, there is no electron density for a nucleotide ‘flipped out’ into the active site. In the ASL-out conformation, the target strand is∼5-10 Å farther from the active site than in tRNAleu3i, and flipping out any of the target bases would expose them to the bulk solvent, which is energetically unfavorable. In tRNAleu3iii, the ASL is closer to the protein (ASL-in), but G36 makes crystal contacts that interfere with the positional adjustment of nucleotides 36-38 that appears to be required for base-flipping. In tRNAleu3i, the electron density for the nucleotides at 36-38 in the G39-flipped-out conformation of the ASL is poorly defined, indicating significant dynamics. However, it is clear that those nucleotides have to be in a different conformation than the G39-stacked conformation in order to connect with the flipped-out G39, and the packing environment allows such conformational rearrangements.
Three Conformations of the ASL
From our six crystal structures of tRNAs, we identified three primary conformations of the ASL: ASL-out, ASL-in/base-stacked and ASL-in/base-flipped-out (Fig. 3D). Although these are almost certainly product complexes, NMR and X-ray studies have shown that both pseudouridylated and non-pseudouridylated ASLs can adopt similar conformations (Arnez and Steitz, 1994; Durant and Davis, 1999). The ASL-out conformation was observed with two different tRNAs, tRNAleu1 and -leu3, and in four different crystal packing environments. The ASL-in conformation was observed in two crystal packing environments. The multiplicity of these observations supports their mechanistic relevance, and presents a picture of conformational plasticity of the ASL, which may be an important trait of the substrate that allows TruA to modify target bases at three different positions. The conformational plasticity of the ASL can explain the tendency of the complex to crystallize in several different forms in similar buffer conditions, and the limited resolution of all three crystal forms. We propose that the three conformations represent three different states of TruA•tRNA along the substrate recognition pathway: the initial docking complex, an intermediate complex prior to the flipping out of the target base, and the chemically competent reactive conformation, respectively.
Conserved Arg58 Facilitates Base-Flipping
The mechanism for base-flipping by TruA was investigated by Targeted Molecular Dynamics (TMD) simulation, a computational method that is used to identify the transition pathway between two known conformations. In this case, the two alternate conformations of TruA•tRNAleu3i, G39-stacked and G39-flipped-out, were chosen as the initial and the final structures, respectively. To focus our study on the base-flipping pathway of a uridine in a known TruA substrate, tRNAleu3/U39 (presented in Fig. 4C), G39 was replaced by uridine, the G39 base-pairing partner was changed from C31 to A31, and U38 was changed to A38. During the simulation, an external force was gradually applied to bring the U39 base in the stacked conformation closer to the flipped-out conformation. The rest of the RNA and the protein were free to accommodate the conformational change of U39. The simulation was repeated three times, each time with a different initial distribution of sodium ions and initial velocities of individual atoms.
Figure 4. The Mechanism for Base-Flipping of U38, U39 and U40 from TMD Simulations.

(A) TMD snapshots of TruA•tRNA when (a) U39 is stacked in the ASL, (b-c) U39 is partially flipped-out, and (d) U39 is fully flipped-out and positioned in the active site. (B) the (A-d) structure viewed from a different angle to show the conformation of the ASL with the extruded U39. (C) Pseudouridylation efficiencies of tRNAleu3, leu3/U39 and leu3/U40 by wt-TruA and R58A. Nucleotides mutated from tRNAleu3 in tRNAleu3/U39 and leu3/U40 are colored blue. Wobble base-pair is indicated with **. (D) Binding affinities of R58A to tRNAleu3, leu3/U39 or leu3/U40, and are compared with that of wt-TruA to tRNAleu3/U39. Quantitations of gel shift assays are plotted with ± standard deviation denoted by the error bars. Values of R2 for fitting are greater than 0.9 in all cases. The gel images are in Fig. S3. (E) The sequence alignment of TruA from bacteria and eukaryotes shows that Arg58 (green arrow) is strictly conserved in the TruA family. Below are the sequence alignments of selected enzymes from all five families of Ψ synthase, showing residues corresponding to residues 57-60 in E. coli TruA.
In all three simulations, Arg58 interacts extensively with U39 during the course of base-flipping. First, while U39 remains stacked in the stem loop, Arg58 forms a hydrogen bond with the O2′ hydroxyl group of the U39 sugar and O2 of the U39 base (Fig. 4A-a). Next, the U39 base flips out from the minor groove of the ASL, and becomes flanked between the two guanidinium groups of Arg58 and Arg110, that form a π–cation sandwich(Fig. 4A-b). As U39 rotates further, it gradually loses the interaction with the two guanidinium groups and moves towards the aliphatic chain of Arg58, to form a hydrophobic contact (Fig. 4A-c). Subsequently, the U39 base moves further along the side chain of Arg58 and approaches the catalytic Asp60 in the active site (Fig. 4A-d).
After U39 is positioned at the active site, Arg58 becomes flexible and no longer forms a specific interaction with the ASL. The space vacated by the extrusion of U39 is occupied by A38, but with a significant alteration in dynamics, resulting in enhanced local motion in the anticodon loop (Fig. 4B). This is consistent with our crystal structure where the nucleotides at 36-38 were disordered in the G39-flipped-out conformation of tRNAleu3i. Our simulations suggest a dual role for Arg58 in base-flipping: (i) facilitating the initial stage of base-flipping by stabilizing the base that is partially extruded from the RNA stem loop, and (ii) guiding the U39 base towards the active site via hydrophobic interactions with the long aliphatic side chain. The former mechanism is reminiscent of the one employed by the archaeal endonuclease for stabilization of the unstacked base by two Arg's (Xue et al., 2006).
A Mutational Test
The role of Arg58 in TruA activity was further explored by characterizing a mutant enzyme where Arg58 is substituted by Ala (R58A). Pseudouridylation activities were assayed by measuring 3H release from C5 of the target uridine in the final step of the reaction. As substrates, we used tRNAleu3, with U at position 38, and constructed two tRNAs, tRNAleu3/U39 and tRNAleu3/U40, where U's were placed at 39 and 40, respectively, so that the three target positions could be assayed in similar tRNA backgrounds (Fig. 4C). Wt-TruA pseudouridylates U's at all three positions with efficiencies (kcat/KM) differing by less than ten-fold, while R58A is inactive towards all three U's (kcat/KM < 1 ×10-7 s-1 μM-1, the detection limit). The wild type and mutant enzymes have similar thermal stabilities based on identical tryptophan fluorescence curves over the range of melting temperatures, indicating that the R58A mutation does not drastically perturb the enzyme structure.
The ability of R58A to bind tRNA was evaluated using gel shift assays (Fig. 4D). R58A binds to tRNAleu3, tRNAleu3/U39 and tRNAleu3/U40, with Kd's of 0.62, 0.35 and 1.12 μM, respectively. Wt-TruA binds tRNAleu3/U39 with a Kd of 1.05 μM. The similar Kd values of R58A and wt-TruA indicate that the lack of detectable enzyme activity by R58A toward U38, U39 and U40 is not due to a lack of affinity for the substrate.
Arg58 is strictly conserved in the TruA family; however, the importance of this residue was not recognized previously because it is not conserved in all Ψ synthase families; it is conserved only in the family of TruA, RluA and RsuA (Fig. 4E). Other active site residues, including the essential nucleophile Asp60, are conserved and similarly poised for reaction in all families of Ψ synthase whose structures are known. Thus, the lack of conservation of Arg58 suggests that it does not function in the chemical process of pseudouridylation. The functional data presented here together with sequence homology data indicate that the role of Arg58 occurs after initial binding, but prior to the chemical step. Our simulations predict that the role is assisting base-flipping, and the biochemistry is consistent with this role. Recent structure of RluA in complex with the isolated ASL also shows that Arg in equivalent position plays the key role in base flipping of the target nucleotide (Hoang et al., 2006).
Flexibility of the ASL Correlates with Modification Efficiency
The conformational snapshots of the ASL observed in our structures led us to hypothesize that the flexibility of the ASL might affect modification efficiency. To test this hypothesis we engineered mutants that would affect the flexibility of the ASL of tRNAleu3, where the target U is in the loop, and tRNAleu3/U40, where the target U is in the stem. To increase flexibility we decreased the G:C content or changed the base-pair partner of the target U from A to a non-Watson-Crick base pair. To decrease flexibility, we introduced a Watson-Crick base-pair partner for the target. We evaluated these substrates using the 3H release assay.
When flexibility of the ASL was increased by mutating the two G:C base pairs in the stem of the ASL of tRNAleu3 into A:U pairs, the kcat/KM increased 2-fold (leu3 vs. leu3A in Fig. 5A). When flexibility was decreased by base-pairing the target U38 of tRNAleu3 with A32 instead of with U32, the kcat/KM decreased 10-fold (leu3 vs. leu3B in Fig. 5A). The decreased efficiency of tRNAleu3B is rescued nearly to the level of tRNAleu3 when stem flexibility was increased by replacing one G:C pair by an A:U pair (leu3B vs. leu3C in Fig. 5A). When the target U was in the stem in tRNAleu3B/U40, a 250-fold decrease in the kcat/KM was observed upon changing the U40 base-pairing partner from the “wobble” partner, G30, to the Watson-Crick partner, A30 (leu3/U40 vs. leu3/U40A in Fig. 5B). In each case, greater flexibility of the ASL correlates with greater efficiency of pseudouridylation, and reduced flexibility correlates with reduced efficiency.
Figure 5. Modification Efficiencies of Variants of tRNAs.

(A) tRNAleu3. The left hand diagram of the ASL shows the target base in red; on the right, the introduced variations are colored blue. Initial velocity plots are color coded to correspond to the variant on the left. Modification efficiency is expressed as kcat/KM, s-1μM-1. (B) tRNAleu3/U40 and introduced variant, color coding as in (A). (C) tRNAala1 variants and tRNAala1/tRNAleu3 chimeras. The top row shows tRNAala1 wild type (blue cartoon) and tRNAleu3 (magenta cartoon). The adjacent diagrams show details of the ASLs and variable loops (VAL). The red dotted line in the tRNAala1 ASL represents the additional base-pairs predicted from the mfold program. The diagram of tRNAala1 shows introduced variations with modification efficiencies (kcat/KM, s-1μM-1) adjacent to each variant. The bottom row shows tRNAala1/tRNAleu3 chimeras with modification efficiencies (kcat/KM, s-1μM-1) below.
A similar relationship between substrate flexibility and enzyme activity has been observed in several other tRNA modifying enzymes, such as TruB, TruD and archeosineguanine transglycosylase. In each case, enzymatic activity increases when the structural integrity and stability of the tRNA substrate is compromised by either natural or engineered mutations (Gu et al., 1998; Kaya and Ofengand, 2003; Watanabe et al., 2001). TruA and these enzymes, whose accepted function is to stabilize tRNA structures through their respective modification of nucleotides, could have evolved to recognize intrinsically unstable tRNAs with higher efficiencies and discriminate against intrinsically stable tRNAs to avoid their overstabilization, thereby maintaining the balance between the flexibility and stability required for its biological function.
Efficient Modification Depends Not Only on the ASL (flexibility), but Also on Other Parts of the tRNA
In E. coli, three tRNAs (tRNAala1, tRNAval1 and tRNAval2) are not pseudouridylated by TruA despite having U at position 38. We prepared tRNAala1 and measured its ability to serve as a substrate for TruA in the 3H release assay. Wt-TruA activity against this substrate is below the detection limit (kcat/KM < 1 ×10-7 s-1 μM-1). To test whether tRNAala1 binds to TruA, we measured the binding affinities of tRNAala1 to wt-TruA and D60A by gel-shift assay, and compared them with those of tRNAleu3. The Kd's of tRNAala1 to wt-TruA and D60A are 2.7 and 1.7 μM, respectively, which are 1.6- and 7-fold higher than those of tRNAleu3, 1.7 and 0.27 μM, respectively (Fig. S5), indicating that the initial complex can be formed with this substrate.
In order to probe the reasons for the resistance to modification after initial binding, we constructed mutations in the ASL of tRNAala1 where we could test each position with a non-Watson-Crick partner. The TruA kcat/KM remained below the detection limit for position 38 paired with a U (ala1A in Fig. 5C), while positions 39 and 40 (ala1/U39 and ala1/U40 in Fig. 5C) each displayed kcat/KM values of ∼1×10-5 s-1 μM-1, more than 100-fold improvement over tRNAala1. This suggests that both positional and flexibility factors are contributing factors. But these activities remain10,000-fold lower than with tRNAleu3 as a substrate.
To further explore the basis of resistance to modification, we constructed chimeric tRNAs (Fig. 5C). A substrate with the tRNAleu3 ASL grafted to the tRNAala1 body yielded a kcat/KM of 7.6×10-4, only ∼200-fold lower than when tRNAleu3 is the substrate, while the tRNAala1 ASL grafted to the tRNAleu3 body yielded a modification efficiency nearly 1,000-fold lower than that for tRNAleu3. The result with tRNAala1//leu3(ASL) indicates that the ASL makes an important contribution to modification efficiency; however the significant modification efficiency of tRNAleu3//ala1(ASL) indicates that the ASL is not the only factor. To test whether the variable loop (VAL) affects the activity, the 15-nucleotide VAL of tRNAleu3 was added to tRNAala1//leu3(ASL) in place of the 5-nucleotide VAL. The kcat/KM value further increases to 3 ×10-2 s-1 μM-1 (ala1//leu3(ASL) vs. ala1//leu3(ASL+VAL) in Fig. 5C), which is only 5.6-fold lower than for tRNAleu3. However, substitution of the VAL without changing the ASL does not lead to a detectable improvement in the modification efficiency (ala1 vs. ala1//leu3(VAL) in Fig. 5C).
The secondary structure of the tRNAala1 ASL predicted based on the sequence shows an unusual preference of a 7-base-paired stem structure to a normal 5-base-paired stem structure in spite of the penalty for the 3-nucleotide loop (Fig. 5C) (Zuker, 2003). The same computational protocol predicts the tRNAleu3 ASL to have the normal 5-base-paired stem. It is possible that the tRNAala1 ASL is more resistant to the conformational change required for modification as compared to the tRNAleu3 ASL, and the sequences and structures of the variable loop and other parts of the body indirectly influence the conformational adaptability of the ASL.
A Model for Substrate Recognition by TruA
i. Mechanism for Sequence-Independent Targeting the ASL
We propose that TruA first recognizes the elbow and the D-stem through their shape and electrostatic properties that are invariant among tRNAs, and that these interactions establish the orientation of tRNA relative to TruA and precisely position the ASL near the active site cleft (Fig. 6). In our ASL-out structures, the dynamics of the ASL and the interacting protein loops (L1 and L8) contrast with the thermal stability of the elbow and D-stem, indicating that the interactions at the ASL are likely to be transient and the stability of the initial docking complex is maintained largely through interactions at the elbow and D-stem. The previous observation that an isolated ASL does not serve as a substrate for TruA is consistent with this proposal (Huang et al., 1998).
Figure 6. The Proposed Model for the Target Site Recognition by TruA.

In the initial docking complex (ASL-out), tRNA binds to TruA through the stable interactions at the elbow and the D-stem while maintaining flexibility of the ASL. In the second step, the ASL bends towards TruA and forms intermediate states (ASL-in/base-stacked). Due to the dynamic nature of the ASL in the initial docking complex, three distinct intermediates can form, each placing a different target nucleotide adjacent to Arg58. Three distinct reactive conformations are formed when the nucleotide interacting with Arg58 flips out and becomes positioned at the active site near the catalytic Asp60.
Such a mechanism for targeting the ASL is reminiscent of the “ruler” mechanism utilized by other enzymes such as the eukaryal splicing endonuclease (Xue et al., 2006) and the ribonuclease, Dicer (MacRae et al., 2006), where a rigid protein scaffold provides a measure for the distance from a reference point to a target point on an RNA. The ruler mechanism would explain how TruA can modify U40 in both mature and pre-tRNAtyrs, even though the precursor ASL includes a 12-nucleotide intron (Ciampi et al., 1977). Because the intron is inserted between 37 and 38, the location of U40 relative to the core of tRNA remains the same, allowing U40 to be correctly positioned for flipping into the TruA active site in both precursor and mature tRNAs.
ii. Mechanism for Regional Specificity
We propose that the key to regional specificity lies in the plasticity of the ASL in the initial docking complex. In the ASL-in complex, the strong interaction between the RNA and the protein would make it difficult to move a base ∼8-15 Å as required for the one- or two-nucleotide shift, whereas in the initial docking complex the dynamics of the ASL backbone and the overlapping spatial positions of nucleotides at 38-40 would allow formation of a specific ASL-in conformation for each target position. The ASL-in conformation in our crystal structures mimics the intermediate state where U39 approaches the active site (ASL-in/39-stacked), leaving positions corresponding to U38 or U40 stacked close to their initial position. Thus, our model suggests that any base at 38-40 can be sampled by having the ASL oscillating between ASL-out and three different ASL-in conformations, either within a single binding event or through multiple binding events (Fig. 6). The fact that G39 was flipped out in our structure indicates that sampling is position specific, rather than base specific. Specificity at each position is achieved by the chemistry of uridine—the only base capable of being modified by the Ψ synthase active site.
There seems to be no predetermined order for modification of multiple U's in a single tRNA. For example, for tRNAleu1, the modification efficiency is significantly lower when U is at position 40 versus at 38 or 39 (unpublished data), whereas for tRNAala1, position 38 is the most inefficiently modified site (Fig. 5C). Thus, each site has a probability of modification (and hence a rate constant) that may depend on the neighboring sequence of the tRNA. Likewise, the site which becomes modified first is stochastically determined and may perturb the equilibrium for other neighboring sites in the tRNA.
iii. Mechanism for Base-Flipping
Many enzymes facilitate base-flipping by reducing the energetic barrier along the pathway and stabilizing the integrity of the structure of the nucleic acid in the flipped-out state. Our simulations suggest that TruA employs only the former mechanism by utilizing Arg58 to stabilize the partially flipped base through a stacking interaction. This is in contrast to other enzymes using arginine as a stabilizing agent. The locally disordered base-flipped-out ASL in the TruA complex contrasts with the substrate in RluA where stabilization is effected by insertion of an arginine into the space vacated by the flipped-out nucleotide (Hoang et al., 2006), and the RNA uridine methyltransferase RumA complex, where the RNA refolds so that other bases occupy the vacated site (Lee et al., 2005). TruA may have evolved to avoid over-stabilization of the flipped-out state since it needs to retain the flexibility required for sampling three different sites.
Contrast with TruB•T-stem Loop
TruB specifically targets U55 in the T-stem loop of almost all tRNAs. Structural alignment of TruB•T-stem loop and TruA•tRNA by overlapping the Cα's from their conserved core scaffolds shows that both the ASL and the T-stem loop bind to the same cleft but from opposite orientations, leading to different interactions outside the stem loop. TruA, as a dimeric enzyme, utilizes its dimerization partner subunit (subunit B in Fig. 7) to interact with the tRNA elbow. TruB, which is functional as a monomer, uses a unique C-terminal domain for the recognition of the long acceptor stem loop of the tRNA. The recognition mechanism of the target site also differs in that TruA binds the ASL through the initial docking of the tRNA elbow and the D-stem (the ruler mechanism), whereas TruB directly docks the T-stem loop into the active site, inducing the thumb domain to clasp onto nucleotides near the target site and His43 to insert into the vacated space of the base-flipped-out stem loop. The extensive interactions between TruB and the neighboring region of the target site are in contrast to small contact between TruA and the ASL near the target site, and have been proposed to be responsible for site-specific identification of U55 through recognition of the secondary structure and sequence in the neighboring nucleotides (Hoang and Ferre-D'Amare, 2001; Pan et al., 2003).
Figure 7. Comparison of RNA Recognition Mechanisms of TruA and TruB.

Structural alignment of TruA and TruB using their Cα backbones (Guda et al., 2004). TruA (subunit A: orange and subunit B: yellow) and TruB (PDB:1R3E, core domain: slate, C-terminal domain: green, and thumb domain: cyan). The magnified view (lower left) shows the conservation of active site residues, including the catalytic Asp60 (bold italic). On the right, TruA and TruB are shown separately in the same orientation. The tRNA (magenta) bound to TruA is from the crystal structure (present work). The tRNA (magenta) bound to TruB is modeled based on the co-crystallized T-stem loop (purple) and other RNA fragments (gray) that mimic the acceptor stem loop.
TruA and TruB appear to have evolved independently from their common ancestor to accommodate their divergence in substrate specificity. The single site specificity of TruB utilizes accessory domains along with conformational changes (induced fit) to actively interrogate the substrate sequence near the target site; whereas the regional substrate specificity of TruA utilizes dimerization to provide a scaffold for recognition of the conserved elbow structure of tRNA, while retaining flexibility in the ASL for multisite selection.
Conclusions
The ability to modify three target bases located in differing surrounding sequences and secondary structures and distant by up to 15 Å distinguishes TruA from other Ψ synthases. The structural, computational and functional studies reported here provide the basis for a substrate recognition model for this remarkable regional selectivity. By binding to the conserved parts of tRNAs (elbow and D-stem backbone), TruA recognizes multiple tRNAs independent of sequence variations. Anchored at these two regions, TruA positions the ASL near the active site without constraining its flexibility, thereby increasing the effective concentration of each target position, 38, 39 and 40, in the vicinity of the active site. The thermal motions of the ASL allow the nucleotides at each of the three sites to be dynamically accessible for modification. The active site determines which bases get acted upon, namely, only uridines since they have the correct chemistry.
Experimental Procedures
E. coli TruA and the D60A mutant were cloned from our previous constructs (Huang et al., 1998) into pET28b vectors, over expressed in BL21*(DE3) cells and purified by Ni2+-affinity and gel filtration chromatography. tRNAleu1, -leu2 and -leu3 were transcribed in vitro by T7 RNA polymerase (Sherlin et al., 2001). For generation of tRNA with the homogeneous ends, a plasmid construct that encodes two ribozymes at 5′ and 3′ ends (5′-HH/3′-HDV) was used as the template (Walker et al., 2003). tRNAs were gel purified, concentrated to 20 mg/ml and mixed with TruA or D60A (10-15 mg/ml) at a molar ratio of 1:1.5-2 for the crystal drop. Crystals were obtained from drops containing TruA and tRNAleu1 (crystal I) or TruA and tRNAleu3 (crystal II and III), but not from drops containing tRNAleu2. Crystals I grew by the hanging-drop vapor diffusion method at room temperature using the sample mixture and well buffer (20% PEG 3350, 0.2 M K3Citrate) mixed at a 1:1 volume ratio. Crystal II and III were obtained by the same method using the sample mixture of TruA and tRNAleu3 and well buffer containing 20% PEG 3350, 0.2 M K3Citrate, pH 7.5, or 20% PEG 3350, 0.2 M K3Citrate, pH 6.5, 5 mM spermine.
Data Collection and Refinement
Crystals were frozen using a cryoprotectant (mother liquor with 20 % glycerol). Diffraction data were collected at the Advance Light Source (ALS, Lawrence Berkeley Lab, CA) beamline 8.3.1. Data were processed and scaled using DENZO and SCALEPACK (Otwinowski and Minor, 1997). All structures were solved by molecular replacement with PHASER (Storoni et al., 2004) using the structure of the dimeric apo TruA (1D0J) and a tRNAphe with the ASL omitted (1EHZ). The crystal structures were refined by manual rebuilding in O (Jones et al., 2001) alternated with the simulated-annealing, conjugate gradient minimization and restrained individual B factor refinement against a maximum likelihood target in CNS (Brunger et al., 1998). Anisotropic motions of sub-domains were modeled by TLS refinement using REFMAC (Winn et al., 2001) on crystal structures I and III (see Supplementary Material). Molecular graphics figures were generated using PyMOL (Delano, 2005).
Kinetic Assay and Gel Shift Assay for wt-TruA, and the D60A and R58A mutants
The R58A mutant was engineered using the QuickChange protocol (Stratagene). The final construct was confirmed by DNA sequencing. The mutant was prepared in the same manner as wt-TruA. tRNAs for kinetic assays were labeled with [5-3H]uridine during the in vitro transcription using the mixture of 0.1 mM [5-3H]UTP (20 Ci/mmol) and 0.4 mM UTP. tRNAs for gel shift assay were 3′-end labeled with [5′32P]pCp by using T4 RNA ligase. In both cases, unincorporated nucleotides were removed using the NucAway kit (Ambion).
The 3H release assay was performed as described (Huang et al., 1998) with minor changes as described in Supplementary Information. All experiments were performed in duplicate or triplicate. The kinetic data were analyzed using the software Kaleidograph.
For the gel shift assay, reaction mixtures of 20 nM 32P-labeled tRNAs and the specified concentration of wt-TruA, D60A or R58A were incubated in the binding buffer (40 mM Tris-HCl (pH 8.0), 50 mM NH4Cl, 20 mM DTT, 0.1 mg/ml bovine serum albumin, 5 mM spermidine and 15% glycerol) at room temperature for 10 minutes. Aliquots of 10 μl were analyzed on 8% acrylamide gels in 1×TAE buffer at 4°C. The gels were dried and exposed to phosphorimager (Amersham). The image was scanned and quantified using ImageQuant™. All assays were performed in duplicate or triplicate.
MD &TMD Simulation
For the MD simulations of ASL-out and ASL-in complexes, crystal structures of TruA• tRNAleu1(ASL-out) and TruA• tRNAleu3i(ASL-in) were individually solvated in a ‘box’ of water (TIP3P model (Jorgensen et al., 1983)). For computational efficiency, spherical boundary condition was used with the radius of 30 Å centered at nucleotide 40 of each tRNA. Other constraints for the boundary condition were adopted from the reference (Huang et al., 2003). Sodium ions were added at random positions to make the spherical system electronically neutral. Initial velocities were randomly assigned from the Maxwell distribution for 25°C. An MD simulation was performed for 0.5 ns for equilibration and continued for 2 ns for production run. In both ASL-in and –out simulations, the RMSD of the protein Cα relative to that of the crystal structure fluctuates between 0.6-0.9 Å during the production dynamics, indicating that both simulations are stable.
For TMD simulations, the stacked and flipped-out conformations of tRNAleu3i bound to TruA were used as the initial and the target structures. Three nucleotides in tRNAleu3 were manually mutated into those in tRNAleu3/U39. The initial and the target structures were separately solvated and MD simulations for equilibration were performed as above. During the TMD production run, a harmonic force (50 kcal/mol/Å) was applied to the U39 base so that the RMSD relative to the final flipped-out U39 decreases by 1 Å every 200 ps. For all computational procedures, the program CHARMM version 31 (Harvard University) (Brooks et al., 1983) was used with the CHARMM27 force field parameters.
Supplementary Material
Acknowledgments
We thank members of the Stroud laboratory for helpful advice, especially Janet Finer-Moore and Patricia Greene for insightful discussion throughout the project. The plasmid template for tRNA synthesis was a gift from Dr. Conn, G. L. (UMIST, UK). This work was supported by United States Public Health Service, National Institutes of Health Grant GM51232 (PI Janet Finer-Moore) and by a Larry Hillblom Foundation fellowship (to S. H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agris PF. Decoding the genome: a modified view. Nucleic Acids Res. 2004;32:223–238. doi: 10.1093/nar/gkh185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnez JG, Steitz TA. Crystal structure of unmodified tRNAGln complexed with glutaminyl-tRNA synthetase and ATP suggests a possible role for pseudouridines in stabilization of RNA structure. Biochemistry. 1994;33:7560. doi: 10.1021/bi00190a008. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromoleclar energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges N, Pannu NS, et al. Crystallography and NMR system (CNS): a new software system for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Ciampi MS, Arena F, Cortese R. Biosynthesis of pseudouridine in the in vitro transcribed tRNATYR precursor. FEBS Lett. 1977;77:75–82. doi: 10.1016/0014-5793(77)80196-8. [DOI] [PubMed] [Google Scholar]
- Cochella L, Green R. An active role for tRNA in decoding beyond codon:anticodon pairing. science. 2005;308:1178–1180. doi: 10.1126/science.1111408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Campo M, Ofengand J, Malhotra A. Crystal structure of the catalytic domain of RluD, the only rRNA pseudouridine synthase required for normal growth of Escherichia coli. RNA. 2004;10:231–239. doi: 10.1261/rna.5187404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano WL. MacPyMOL: A PyMOL-based Molecular Graphics Application for MacOS X. South San Francisco, CA, USA: DeLano Scientific LLC; 2005. [Google Scholar]
- Durant PC, Davis DR. Stabilization of the anticodon stem-loop of tRNAlys,3 by an A+-C base-pair and by pseudouridine. J Mol Biol. 1999;285:115–131. doi: 10.1006/jmbi.1998.2297. [DOI] [PubMed] [Google Scholar]
- Foster PG, Huang L, Santi DV, Stroud RM. The structural basis for RNA recognition and pseudouridine formation by pseudouridine synthase I. Nat Struct Biol. 2000;7:23–27. doi: 10.1038/71219. [DOI] [PubMed] [Google Scholar]
- Gu X, Liu Y, Santi DV. The mechanism of pseudouridine synthase I as deduced from its interaction with 5-fluorouracil-tRNA. Proc Natl Acad Sci USA. 1999;96:14271–14275. doi: 10.1073/pnas.96.25.14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Yu M, Ivanetich KM, Santi DV. Molecular Recognition of tRNA by tRNA Pseudouridine 55 Synthase. Biochemistry. 1998;37:339–343. doi: 10.1021/bi971590p. [DOI] [PubMed] [Google Scholar]
- Guda C, Lu S, Scheeff ED, Bourne PE, Shindyalov IN. CE-MC: a multiple protein structure alignment server. Nucleic Acids Res. 2004;32:W100–W103. doi: 10.1093/nar/gkh464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutgsell NS, Deutscher MP, Ofengand J. The pseudouridine synthase RluD is required for normal ribosome assembly and function in Escherichia coli. RNA. 2005;11:1141–1152. doi: 10.1261/rna.2550105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton CS, Spedaliere CJ, Ginter JM, Johnston MV, Mueller EG. The roles of the essential Asp-48 and highly conserved His-43 elucidated by the pH dependence of the pseudouridine synthase TruB. Arch Biochem Biophys. 2005;433:322–334. doi: 10.1016/j.abb.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Hamma T, Reichow SL, Varani G, Ferre-D'Amare AR. The Cbf5-Nop10 complex is a molecular bracket that organizes box H/ACA RNPs. Nat Struct Biol. 2005;12:1101–1107. doi: 10.1038/nsmb1036. [DOI] [PubMed] [Google Scholar]
- Hoang C, Chen J, Vizthum CA, Kandel JM, Hamilton CS, Mueller EG, Ferre-D'Amare AR. Crystal structure of pseudouridine synthase RluA: Indirect Sequence readout through protein-induced rna structure. Mol Cell. 2006;24:535–545. doi: 10.1016/j.molcel.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Hoang C, Ferre-D'Amare AR. Cocrystal structure of a tRNA U55 pseudouridine synthase: Nucleotide Flipping by an RNA-modifying enzyme. Cell. 2001;107:929–939. doi: 10.1016/s0092-8674(01)00618-3. [DOI] [PubMed] [Google Scholar]
- Hoang C, Ferre-D'Amare AR. Crystal structure of the highly divergent pseudouridine synthase TruD reveals a circular permutation of a conserved fold. RNA. 2004;10:1026–1033. doi: 10.1261/rna.7240504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Pookanjanatavip M, Gu X, Santi DV. A conserved aspartate of tRNA pseudouridine synthase is essential for activity and a probable nucleophilic catalyst. Biochemistry. 1998;37:344. doi: 10.1021/bi971874+. [DOI] [PubMed] [Google Scholar]
- Huang N, Banavali NK, Mackerell AD., Jr Protein-facilitated base flipping in DNA by cytosine-5-methyltransferase. Proc Natl Acad Sci USA. 2003;100:68–73. doi: 10.1073/pnas.0135427100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr D Biol Crystallogr. 2001;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- Kaya Y, Ofengand J. A novel unanticipated type of pseudouridine synthase with homologs in bacteria, archaea, and eukarya. RNA. 2003;9:711–721. doi: 10.1261/rna.5230603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TT, Agarwalla S, Stroud RM. A Unique RNA fold in the RumA-RNA-Cofactor Ternary complex contributes to substrate selectivity and enzymatic function. Cell. 2005;120:599–611. doi: 10.1016/j.cell.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Li L, Ye K. Nature. Early Press; 2006. Crystal structure of an H/ACA box ribonucleoprotein particle. On line. [DOI] [PubMed] [Google Scholar]
- MacRae IJ, Zhou K, Li F, Repic A, Brooks AN, Cande WZ, Adams PD, Doudna JA. Structural Basis for Double-Stranded RNA processing by Dicer. Science. 2006;311:195–198. doi: 10.1126/science.1121638. [DOI] [PubMed] [Google Scholar]
- Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure Analysis at single nucleotide resolution by selective 2′-Hydroxyl acylation and primer extension. J Am Chem Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- Olejniczak M, Dale T, Fahlman RP, Uhlenbeck OC. Idiosyncratic tuning of tRNAs to achieve uniform ribosome binding. Nat Struct Biol. 2005;12:788–793. doi: 10.1038/nsmb978. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. New York: Academic Press; 1997. [DOI] [PubMed] [Google Scholar]
- Pan H, Agarwalla S, Moustakas DT, Finer-Moore J, Stroud RM. Structure of tRNA pseudouridine synthase TruB and its RNA complex: RNA recognition through a combination of rigid docking and induced fit. Proc Natl Acad Sci USA. 2003;100:12648. doi: 10.1073/pnas.2135585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid R, Liang B, Baker DL, Youssef OA, He Y, Phipps K, Terns RM, Terns MP, Li H. Crystal Structure of a Cbf5-Nop10-Gar1 complex and implications in RNA-Guided Pseudouridylation and Dyskeratosis Congenita. Mol Cell. 2006;21:249–260. doi: 10.1016/j.molcel.2005.11.017. [DOI] [PubMed] [Google Scholar]
- Sherlin LD, bullock TL, Nissan A, Perona JJ, Lariviere FJ, Uhlenbeck OC, Scaringe SA. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA. 2001;7:1671. [PMC free article] [PubMed] [Google Scholar]
- Shi H, Moore PB. The crystal structure of yeast phenylalanine tRNA at 1.93 A resolution: a classic structure revisited. RNA. 2000;6:1091–1105. doi: 10.1017/s1355838200000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaraman J, Sauve V, larocque R, Stura EA, Schrag JD, Cygler M, Matte A. Structure of the 16S rRNA pseudouridine synthase RsuA bound to uracil and UMP. Nat Struct Biol. 2002;9:353–358. doi: 10.1038/nsb788. [DOI] [PubMed] [Google Scholar]
- Sprinzl M, Horn C, Brown M, Ioudovitch A, Steinberg S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998;26:148–153. doi: 10.1093/nar/26.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- Walker SC, Avis JM, Conn GL. General plasmids for producing RNA in vitro transcripts with homogeneous ends. Nucleic Acids Res. 2003;31:e82. doi: 10.1093/nar/gng082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Nameki N, Matsuo-Takasaki M, Nishimur S, Okada N. tRNA recognition of tRNA-guanine transglycosylase from a hyperthermophilic archaeon, Pyrococcus horikoshii. J Bio Chem. 2001;276:2387–2394. doi: 10.1074/jbc.M005043200. [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- Xue S, Calvin K, Li H. RNA recognition and cleavage by a splicing endonuclease. Science. 2006;312:906–909. doi: 10.1126/science.1126629. [DOI] [PubMed] [Google Scholar]
- Yang C, Mcpheeters DS, Yu YT. Psi35 in the Branch Site Recognition Region of U2 Small nuclear RNA is important for pre-mRNA splicing in Saccharomyces cerevisiae. J Biol Chem. 2005;280:6655–6662. doi: 10.1074/jbc.M413288200. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.