

Abstract
Background
The genera Aspergillus and Penicillium include some of the most beneficial as well as the most harmful fungal species such as the penicillin-producer Penicillium chrysogenum and the human pathogen Aspergillus fumigatus, respectively. Their mitochondrial genomic sequences may hold vital clues into the mechanisms of their evolution, population genetics, and biology, yet only a handful of these genomes have been fully sequenced and annotated.
Results
Here we report the complete sequence and annotation of the mitochondrial genomes of six Aspergillus and three Penicillium species: A. fumigatus, A. clavatus, A. oryzae, A. flavus, Neosartorya fischeri (A. fischerianus), A. terreus, P. chrysogenum, P. marneffei, and Talaromyces stipitatus (P. stipitatum). The accompanying comparative analysis of these and related publicly available mitochondrial genomes reveals wide variation in size (25–36 Kb) among these closely related fungi. The sources of genome expansion include group I introns and accessory genes encoding putative homing endonucleases, DNA and RNA polymerases (presumed to be of plasmid origin) and hypothetical proteins. The two smallest sequenced genomes (A. terreus and P. chrysogenum) do not contain introns in protein-coding genes, whereas the largest genome (T. stipitatus), contains a total of eleven introns. All of the sequenced genomes have a group I intron in the large ribosomal subunit RNA gene, suggesting that this intron is fixed in these species. Subsequent analysis of several A. fumigatus strains showed low intraspecies variation. This study also includes a phylogenetic analysis based on 14 concatenated core mitochondrial proteins. The phylogenetic tree has a different topology from published multilocus trees, highlighting the challenges still facing the Aspergillus systematics.
Conclusions
The study expands the genomic resources available to fungal biologists by providing mitochondrial genomes with consistent annotations for future genetic, evolutionary and population studies. Despite the conservation of the core genes, the mitochondrial genomes of Aspergillus and Penicillium species examined here exhibit significant amount of interspecies variation. Most of this variation can be attributed to accessory genes and mobile introns, presumably acquired by horizontal gene transfer of mitochondrial plasmids and intron homing.
Background
The genera Aspergillus and Penicillium contain some of the most beneficial as well as the most harmful fungal species. They include many industrial producers of important antibiotics, enzymes and pharmaceuticals, which have brought about a massive transformational impact on human health [1]. Several other species are extensively used in production of foods or other useful compounds. Pathogenic fungi, on the other hand, can significantly affect livestock and crops, both as agents of infection and by means of contamination with mycotoxins, and they commonly cause allergic and occasionally life-threatening infections in humans [2]. Fungal infections in humans are notoriously difficult to diagnose and treat, especially in immunocompromised patients. To achieve a better understanding of their biology, nuclear genomes of several Aspergillus and Penicillium species have been sequenced, yet only a handful of mitochondrial genomes have been sequenced and annotated.
With a few notable exceptions such as Podospora anserina[3], Chaetomium thermophilum[4], and Rhizoctonia solani (S. Pakala, unpublished), fungal mitochondrial genomes are small, with an average size of 44,681 bp (based on the fungal mitochondrial genomes in the NCBI organelle database, January 2012). For a fraction of the cost required to sequence a nuclear genome, mitochondrial genomic sequences may provide vital clues into the evolution, population genetics, and biology of these fungi. Aside from energy metabolism, mutations in mitochondrial genes have been linked to cellular differentiation, cell death and senescence pathways, as well as drug resistance and hypovirulence [5-9]. The widespread uniparental inheritance and high copy number of these organelles make them promising markers for cost-effective species identification and for studying fungal population structure [10]. Mitochondrial DNA can be a rich source of novel genotyping markers due to the presence of highly mobile introns in many fungal mitochondria [11]. Finally, fungal mitochondria may serve as valuable experimental models for studies of human heart and muscle diseases linked to mitochondrial dysfunction [12]. Mitochondrial biology is gaining notice with the advent of so called “three parent in vitro fertilization” as a means of producing disease free children from a mother with an inherited mitochondrial genetic disease [13]. In Aspergillus fumigatus the advent of the ability to perform sexual crosses will potentially allow for the genetic analysis of mitochondrial mutations and their phenotypes.
Despite their importance, only a few complete Aspergillus and Penicillium mitochondrial genomes have been reported [14-17]. In A. fumigatus, the ratio of mitochondrial to nuclear genomes is 12:1, based on optical mapping [18]. Although mitochondrial sequence reads are generated in every eukaryotic genome sequencing project, most studies only report nuclear genomes. As a result, little is known about the mitochondrial genome organization in many important fungi, such as the human pathogen A. fumigatus and the penicillin producer P. chrysogenum, which impedes their functional studies. Here we report the complete sequence and annotation of mitochondrial genomes of six Aspergillus and three Penicillium species. The accompanying comparative analysis of these and related publicly available genomes provides insight into mitochondrial genome organization, distribution of group I introns and plasmid-encoded genes, and phylogenetic relationships among these fungi.
Results and discussion
Assembly and annotation of mitochondrial genomes
The following mitochondrial genomic DNAs were sequenced, assembled, and/or annotated in this study: Aspergillus fumigatus AF293, Aspergillus fumigatus A1163, Aspergillus fumigatus 210, Aspergillus clavatus NRRL 1, Aspergillus oryzae RIB40, Aspergillus flavus NRRL 3357, Neosartorya fischeri NRRL 181 (teleomorph of Aspergillus fischerianus), Aspergillus terreus NIH 2624, Penicillium chrysogenum 54–1255, Penicillium marneffei ATCC 18224, and Talaromyces stipitatus ATCC 10500 (teleomorph of Penicillium stipitatum) (Table 1 and Additional file 1). Additional strains in the process of being sequenced were also analyzed with respect to SNPs and DIPs. Sequence reads for A. fumigatus AF293, A. fumigatus A1163, A. clavatus, N. fischeri and P. chrysogenum were generated in the course of nuclear genome sequencing projects [18-21]. The A. oryzae mitochondrial genome [16] was re-annotated in this study to include protein coding genes. The A. terreus mitochondrial genome was assembled using Sanger reads obtained from GenBank [GenBank:AAJN00000000]. After being trimmed and rotated, mitochondrial sequences were processed through the standard J. Craig Venter Institute (JCVI) annotation pipeline to ensure annotation consistency.
Table 1.
Aspergillus and Penicillium mitochondrial genome statistics
| Genomea | length of mitochondrial genome (bp) | GC % | Protein-coding genes | Protein-coding genes with introns | Length (and number) of introns in protein-coding genes (bp) | Length of intron in large subunit rRNA (bp) | tRNAs | intronic ORFS | Length (and number) of non-intronic accessory genes (bp) | length of nuclear genome (bp) |
|---|---|---|---|---|---|---|---|---|---|---|
|
A. terreus NIH 2624 |
24,658 |
27.1 |
15 |
0 |
0 (0) |
1,705 |
27 |
1 |
0 (0) |
29,331,195 |
|
P. chrysogenum 54-1255 |
27,017 |
24.7 |
16 |
0 |
0 (0) |
1,678 |
28 |
1 |
1,227 (1) |
32,223,735 |
|
A. oryzae RIB40b |
29,202 |
26.2 |
17 |
1 |
1,780 (1) |
1,703 |
26 |
2 |
2,010 (2) |
37,088,582 |
|
A. flavus NRRL 3357 |
29,205 |
26.2 |
17 |
1 |
1,776 (1) |
1,703 |
27 |
2 |
2,004 (1) |
36,892,344 |
|
A. fumigatus A1163 |
30,696 |
25.5 |
19 |
1 |
2,020 (1) |
1,720 |
31 |
3 |
1,410 (2) |
29,205,420 |
|
A. niger N909 |
31,103 |
26.9 |
16 |
2 |
1,502 (2) |
1,800 |
25 |
0 |
1,674 (2) |
not available |
|
A. fumigatus AF210 |
31,762 |
25.4 |
20 |
1 |
2,020 (1) |
1,720 |
31 |
3 |
2,253 (3) |
not available |
|
A. fumigatus AF293 |
31,765 |
25.4 |
20 |
1 |
2,020 (1) |
1,720 |
31 |
3 |
2,253 (3) |
29,384,958 |
|
A. nidulans FGSC A4 |
33,227 |
24.9 |
20 |
2 |
4,108 (5) |
1,689 |
28 |
4 |
1,524 (2) |
29,828,291 |
|
A. tubingensis 0932 |
33,656 |
26.8 |
16 |
2 |
3,690 (4) |
1,794 |
25 |
0 |
1,647 (2) |
not available |
|
N. fischeri NRRL 181 |
34,373 |
25.4 |
22 |
1 |
3,429 (2) |
1,721 |
28 |
4 |
2,559 (4) |
31,770,017 |
|
A. clavatus NRRL 1 |
35,056 |
25.0 |
21 |
2 |
4,304 (3) |
1,719 |
26 |
4 |
4,509 (3) |
27,859,441 |
|
P. marneffei ATCC 18224 |
35,432 |
24.6 |
26 |
3 |
9,775 (9) |
1,672 |
30 |
10 |
768 (2) |
28,643,865 |
|
P. marneffei MP1 |
35,438 |
24.6 |
25 |
3 |
9,776 (9) |
1,672 |
30 |
9 |
1,647 (2) |
not available |
| T. stipitatus ATCC 10500 | 36,351 | 24.9 | 26 | 3 | 12,140 (11) | 1,713 | 27 | 12 | 0 (0) | 35,685,443 |
aGenomes annotated at JCVI are shown in bold.
bProtein coding genes were annotated at JCVI.
Mitochondrial genome size variation and sources of genome expansion
The sequenced Aspergillus and Penicillium mitochondrial genomes showed remarkable variation in size, ranging from 24,658 bp to 36,351 bp (Table 1 and Figure 1). The differences in length can be primarily attributed to the number and length of the introns present in the genomes. For example, protein coding genes in the two smallest genomes, A. terreus and P. chrysogenum, do not contain introns, whereas the larger genomes contain one or more introns within ORFs. In contrast, the largest mitochondrial genome, T. stipitatus, contains a total of 11 introns. The presence and number of accessory genes also contribute to the larger size of some of the mitochondrial genomes. Repeat content within the analyzed species was determined to be insignificant (0 – 1% of the genomes, data not shown). Comparison of nuclear genome sizes shows no correlation with the mitochondrial DNA sizes (Table 1).
Figure 1.

Contributions from core and accessory genes, ncRNAs, intronic and intergenic regions, to mitochondrial genomes. Each vertical bar represents the length of a mitochondrial genome.
Comparative analysis of 11 A. fumigatus strains showed little intraspecies variation in their mitochondrial DNA (Additional file 2). To identify single nucleotide polymorphisms (SNPs) and deletion insertion polymorphisms (DIPs), Illumina reads of 10 A. fumigatus strains, generated in the course of a genome sequencing study (Nierman, unpublished), were aligned against the AF293 mitochondrial DNA. The analysis showed few SNPs and no significant DIPs. In total, we identified 15 candidate SNPs. The number of SNPs in individual strains varied from zero (F15861 and F15767) to nine (AF210). Six SNPs were located in intergenic regions, and nine were located in coding regions including eight non-synonymous SNPs. Five of these eight non-synonymous SNPs were found in cob, cox1, nad1, and nad2 genes.
A 1.1 kb deletion was detected in strain A1163 (30,696 bp) compared to strains AF293 (31,765 bp) and AF210 (31,762 bp). The deletion in A1163 was confirmed by PCR using primers flanking the missing region. In AF293 and AF210, the region contains an 843-bp open reading frame (AFUA_m0390, AFUD_m0390; hypothetical protein), which is missing in A1163. A putative homolog of this hypothetical protein (NFIA_m0370) is present in the closely-related N. fischeri mitochondrial genome, suggesting a recent gene loss in A. fumigatus A1163.
Likewise, only minor differences were found between mitochondrial genomes of A. oryzae and A. flavus as well as between two strains of P. marneffei (ATCC 18224 and MP1). The P. marneffei genomes are 99.9% identical and their sizes differ only by 6 bp (Table 1). The genomes of A. flavus and A. oryzae differ in size by 3 bp and are also 99.9% identical. It should be noted that some of the strains analyzed were genetically modified during strain development. For example, the P. chrysogenum strain was developed as a result of an extensive strain improvement programs, which may have affected its mitochondrial DNA as well [20].
Our results are consistent with previous studies of mitochondrial intraspecies polymorphism. With a few exceptions, most Pezizomycotina mitochondrial genomes show little variation. By contrast, Aspergillus japonicus, P. anserina, Neurospora crassa and some other fungi exhibit significant mitochondrial intraspecies polymorphism and genome size variation, which has been attributed to mobile introns [11]. Thus, population surveys based on RFLP have demonstrated the presence of different mitochondrial haplotypes in wild-type subpopulations of P. anserina[22].
Core mitochondrial genes
All sequenced Aspergillus and Penicillium mitochondrial genomes contain 14 core genes involved in oxidative phosphorylation, ATP synthesis and mitochondrial protein synthesis, all present on the forward strand (Additional file 3). In addition, these genomes carry a complete set of tRNAs, the small and large subunits of ribosomal RNA, and the mitochondrial ribosomal protein S5. Additional file 4 depicts the protein-coding genes and non-coding RNAs in the reference mitochondrial genome of A. fumigatus AF293. The core genes share a high level of sequence conservation (Additional files 5 and 6) and synteny. Figure 2 shows the conservation of gene order of the core genes in all the Aspergillus and most of the Penicillium mitochondrial genomes annotated in this study. The exception is the atp9 gene, which is located between nad2 and cob in P. marneffei ATCC 18224). In all the other genomes, atp9 lies between cox1 and nad3. The number of tRNA genes varies from 25 to 31 with no particular correlation between the number and mitochondrial or nuclear genome size (Table 1).
Figure 2.
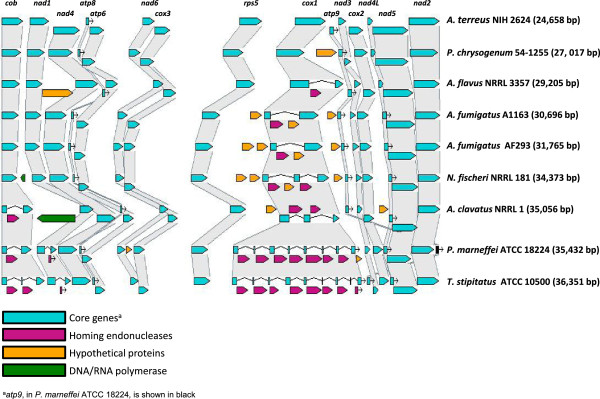
Conservation of gene order in mitochondrial genomes. A SynView representation of the protein-coding genes in the Aspergillus and Penicillium mitochondrial genomes annotated in this study. Clusters of orthologous core genes (blue) are connected by regions shaded in grey. Accessory genes are homing endonucleases (pink), hypothetical proteins (orange) and DNA/RNA polymerases (green). The synteny of the core genes is maintained with the exception of the location of atp9 in P. marneffei 18224 (black).
To explore the possibility that the core genes can provide new insights into Aspergillus evolution, we performed a phylogenetic analysis of 14 concatenated core proteins encoded by 13 Aspergillus and Penicillium mitochondrial genomes (Figure 3). The obtained phylogenetic tree clusters the A. nidulans and A. niger nodes together with a bootstrap support of 83%. Notably, A. terreus and A. oryzae form another group with bootstrap support of 80%. The tree topology also indicates that P. chrysogenum is not closely related to P. marneffei and T. stipitatus. This finding is consistent with morphological observations [20,23,24]. The topology of the Maximum Parsimony based tree is consistent with that of the Maximum-Likelihood tree (data not shown).
Figure 3.

Maximum Likelihood tree showing the phylogenetic relationships among the sequenced Aspergillus and Penicillium species. The tree is based on 14 concatenated core mitochondrial proteins from 13 genomes. Gibberella zeae was used as an outgroup. Branch lengths correspond to substitutions per site calculated using a Maximum Likelihood approach. Identical topology was predicted using the Maximum Parsimony approach.
We have also examined individual mitochondrial genes (cox1, cob, nad5) and rDNA internal-transcribed spacers (ITS), which have been proposed as markers for species identification and classification to complement common nuclear DNA markers [10]. Phylogenetic analysis based on these individual protein coding and non-coding genes did not yield phylogenies with >75% bootstrap support for any of the trees (data not shown) and/or did not have enough power to resolve the relationships between the species. The single-gene trees show incompatible topologies with each other and with the topology obtained from the 14 concatenated proteins (Figure 3). This confirms that individual mitochondrial genes are not suitable for species identification in Aspergillus species. As shown previously [10], the presence of introns makes cox1 and other mitochondrial genes poor candidates for building fungal phylogenies, although cox1 is often used in animal phylogenetic studies.
In contrast, our results suggest that the concatenated core mitochondrial proteins can be of value for species level phylogeny construction. Interestingly, the concatenated tree topology is identical to the topology obtained previously from over 100 concatenated nuclear proteins with identical number of introns among orthologs [20]. The A. terreus and A. oryzae grouping is also supported by previously published studies based on multilocus phylogenetic analysis of both nuclear ribosomal and protein-coding genes [25] and concatenated nuclear protein-coding genes [26,27]. However, the A. terreus and A. oryzae nodes form two separate groups in phylogenies based on only nuclear ribosomal genes [28]. Our analysis highlights the challenges still facing the Aspergillus systematics including the unresolved Aspergillus tree backbone.
Accessory mitochondrial genes
In addition to the core set, most mitochondrial genomes contain accessory genes. The most compact genome (A. terreus), contains only the core set of mitochondrial genes, while other genomes contain at least one accessory gene. Some accessory genes are shared by a subset of closely related mitochondrial genomes, but do not show sequence similarity to other genes in public databases. These genes have been annotated as ‘hypothetical’. Notably, A. fumigatus strains AF293 and A1163 contain 3 hypothetical genes each with > 99% identity.
In contrast, two accessory genes, A. clavatus ACLA_m0040 and N. fischeri NFIA_m0030, share similarity with mitochondrial DNA and RNA polymerase genes from distantly related fungi. They are also the only two protein-coding genes on the reverse strand, and are located between cob and nad1 in their respective genomes. ACLA_m0040 has a potential frame shift and thus appears to be a pseudogene. Homologous DNA polymerase sequences are found within the main mitochondrial genome (as in Glomerella graminicola M1.001) or on linear mitochondrial plasmids (as in pAL2-1 of P. anserina, pClK1 of the phytopathogenic fungus Claviceps purpurea and pFP1 from Fusarium proliferatum). NFIA_m0030 is present as a C-terminal fragment (lacking a start codon) and is most similar to the RNA polymerase genes found on linear plasmids in Blumeria graminis f. sp. hordei (pBgh) and C. purpurea (pClK1).
The similarity to mitochondrial plasmid-encoded genes suggests that both A. clavatus and N. fischeri polymerase genes were acquired by horizontal gene transfer followed by integration of ancestral mitochondrial plasmids into mitochondrial DNA. Indeed, linear fungal mitochondrial plasmids typically encode DNA and RNA polymerases, while circular plasmids have a single gene for a DNA polymerase and a reverse transcriptase [29]. In several studies, plasmids integrated into mitochondrial DNA have been implicated in the mechanism underlying mycelial senescence in fungi [30-32].
Another set of accessory genes annotated in Aspergillus and Penicillium species encodes putative homing endonucleases. Although these genes (HEGs for “homing endonuclease genes”) can be found within introns and intergenic regions, in this study all HEGs were associated with “homing” group I introns (see next section). Based on sequence conservation, all HEGs were assigned to either LAGLIDADG or GIY-YIG families. The association between HEGs and introns is found in many other species. Homing introns are considered highly mobile, invasive genetic elements common in fungi and plants, but they can be also found in some animals and prokaryotes. They propagate via the double-strand-break-repair pathway into the specific target sequence (“homing site”), which is recognized and cleaved by the endonuclease [33]. Some endonucleases can also function as maturases by facilitating self-splicing of introns [34,35]. HEGs themselves are considered mobile and are typically bounded by two halves of the homing site (15–45 bp). HEGs are considered selfish elements, but may also contribute to mitochondrial DNA integrity [36]. The variability of HEGs in Aspergillus and Penicillium species can be exploited to develop phylogenetic markers that might be useful for differentiation of various species. Variants of HEGs are now being engineered for targeted cleavage of genomic sequences with potential applications in biotechnology, medicine and agriculture [37].
Diversity, evolution and origin of mitochondrial group I intron insertions
Most fungal mitochondrial genomes sequenced to date contain one or more group I or group II introns. In the Pezizomycotina subphylum (to which all these species belong), the largest number of mitochondrial introns, a total of 33, have been documented for P. anserina[3], while Mycosphaerella graminicola is currently the only species identified that lacks mitochondrial introns entirely [38].
The Aspergillus and Penicillium species sequenced here also have variable intron distribution. Similar to previous observations the number and insertion sites of these introns vary, even between closely related species, suggesting cyclical intron gain and loss through horizontal transfer. Our analysis shows that the variation in intron number is the primary source of difference in genome size (Figure 1). Thus, the T. stipitatus genome contains eleven introns, while A. terreus and P. chrysogenum genomes both contain only a single intron.
The mitochondrial DNA sequences analyzed here contain group I introns distributed between three protein coding genes and the large subunit ribosomal RNA (LSU) (Table 1). As observed in a variety of mitochondrial genomes [10], the cox1 gene contains the most variable number of introns. For the Aspergillus species, A. nidulans has three cox1 introns, A. clavatus and N. fischeri each have two, and A. fumigatus, A. flavus and A. oryzae contain a single intron. A. terreus is the only Aspergillus species in this study that does not contain an intron in the cox1 gene. The cox1 intron insertions sites also vary between the Aspergillus species. The cox1 intron insertion site in the A. fumigatus species is seen in the closely related N. fischeri genome and in the more distantly related P. marneffei and T. stipitatus genomes. The cox1 insertion site in A. flavus and A. oryzae is also present in A. clavatus.
For the Penicillium species, P. marneffei and T. stipitatus contain seven and eight cox1 introns, respectively, while P. chrysogenum does not contain any cox1 introns. Between P. marneffei and T. stipitatus, six of the intron insertion sites are identical. All of the cox1 intron insertions sites have been previously observed in other distantly related fungi such as Saccharomyces cerevisiae, P. anserina and N. crassa[3,39,40].
The other two protein coding genes that contain group I introns are the cob and nad1 genes. A. nidulans and A. clavatus contain a common cob intron, while P. marneffei contains a single intron and T. stipitatus contains two cob introns. These two Penicillium species are also the only two in this study with an intron in the nad1 gene. The cox1, cob, and nad1 introns in all the species encode either a LAGLIDADG or GIY-YIG homing endonuclease (as discussed above).
By contrast, the one intron common to all the mitochondrial genomes described here is found at a single location in the LSU rRNA gene. These introns are closely related in secondary structure (Figure 4) with the only major difference between the genera being an additional stem-loop structure (P6c) present in all the Aspergillus introns (except A. nidulans), but absent in the Penicillium species. All Pezizomycotina mitochondria sequenced to date, with the exception of the Diothideomycetes (Phaeosphaeria nodorum and Mycosphaerella graminicola), contain a mitochondrial LSU rRNA intron inserted at this position. Intron insertions are also commonly found in the mitochondrial LSU gene in plants and other fungi.
Figure 4.

Secondary structure of the mitochondrial LSU rRNA intron. This intron is present in all the mitochondrial genomes described here, with the sequence shown being from A. fumigatus. Grey highlighted residues are identical in all species. All of the introns contain a mitochondrial S5 protein ORF in the P8 stem-loop. The primary difference between the Aspergillus and Penicillium species is the presence of an additional stem-loop structure, P6a.1, that is present in the Aspergillus species (except A. nidulans), but absent in the Penicillium species. P. chrysogenum is the only species to contain an extended P9.1 region. 5’SS: 5’ splice site; 3’SS: 3’ splice site.
Our results are consistent with two previously observed characteristics of Pezizomycotina mitochondrial LSU introns. First, these introns do not contain endonuclease ORFs, but do contain the mitochondrial ribosomal protein S5 ORF within the P8 stem-loop of the intron. In S. cerevisiae and other fungi, mitochondrial S5 is nuclear encoded and transported into the mitochondrial matrix [41] but is not encoded in the nuclear genome of any Pezizomycotina fungi sequenced to date. No studies have thoroughly examined the function of the mitochondrial S5 ORF, but decreased expression of mitochondrial S5 from the intron ORF in N. crassa leads to mitochondrial small ribosomal subunit assembly defects and decreased mitochondrial protein expression [42,43]. Second, though group I introns are normally considered self-splicing, the Pezizomycotina mitochondrial LSU introns tested to date cannot self-splice [44-47]. These introns require a mitochondrial tyrosyl-tRNA synthetase (TyrRS) as a structure-stabilizing splicing cofactor found only in Pezizomycotina [47].
The intron distribution in the genera described here suggests two distinct mechanisms of intron evolution within the lineage. The presence of homing endonucleases in all of the cox1, cob1, and nad1 introns suggests they likely follow the previously proposed “omega” cycle of intron gain and loss [48]. In this model, horizontal transmission into an intronless site is promoted by a functional homing endonuclease, followed by endonuclease degradation, and eventually, intron loss. The cycle can then be restarted by a new horizontal transmission event. The sporadic distribution of introns in the protein coding genes analyzed here indicates that horizontal gene transfer may be quite common in the Aspergillus and Penicillium mitochondrial genomes. It also highlights the challenges associated with using cox1 and other mitochondrial genes to build species phylogenies.
The mitochondrial LSU intron likely follows a slightly different evolutionary trajectory. The widespread distribution of mitochondrial LSU introns in Pezizomycotina that contain the S5 ORF suggests that this intron insertion event occurred after the divergence from the yeasts, and became fixed within the lineages. Fixation was likely due to the selective advantage from the S5 gene, but also from accumulated mutations in the intron, which resulted in its dependence on the nuclear encoded mitochondrial TyrRS splicing factor. This idea is supported by the observation that the Dithideomycetes, the only Pezizomycotina group that lacks mitochondrial LSU introns, contain degraded adaptations of the mitochondrial TyrRS splicing factor necessary for intron splicing [46]. A plausible scenario is that this degeneration occurred after mitochondrial LSU intron loss. It remains unclear how the Dithideomycetes have compensated for the loss of the S5 protein [38,49].
Conclusions
We report here the complete sequence and annotation of mitochondrial genomes of six Aspergillus and three Penicillium species, which represent the two most significant genera among filamentous fungi. The study expands the genomic resources available to fungal biologists by providing mitochondrial genomes with consistent annotations. The accompanying comparative analysis of these and related publicly available genomes provides insight into genome organization and phylogenetic relationships among these organisms. By clustering together A. terreus and A. oryzae, the phylogenetic tree based on 14 concatenated core mitochondrial proteins has a different topology from some previously published single protein trees, but is similar to trees built using multiple nuclear proteins. This suggests that core genes in mitochondrial and nuclear genomes co-evolved in the Aspergillus lineage.
Despite the conservation of the core genes, mitochondrial genomes of Aspergillus and Penicillium species exhibit significant amount of interspecies variation consistent with experimental evidence for intraspecies horizontal transfer and recombination in mitochondrial DNA [50]. Most of this variation can be attributed to accessory genes and mobile introns, presumably acquired via swapping and integration of mitochondrial plasmid and intron homing followed by gene or intron loss. Annotated core and accessory genes can serve as complementary markers in future population genetics and evolution studies.
Methods
Sequencing and assembly
The following genomes were sequenced at JCVI using the Sanger technology: A. fumigatus AF293, A. fumigatus A1163, A. clavatus NRRL 1, A. flavus NRRL 3357, N. fischeri NRRL 181, P. chrysogenum 54–1255, P. marneffei ATCC 18224, and T. stipitatus ATCC 10500 (Additional file 1). The A. fumigatus AF210, genome was sequenced using a combination of 454 GS FLX Titanium instrument (Roche) and Illumina Genome Analyzer II (Illumina). The reads were assembled at JCVI using the Celera Assembler [51]. The P. chrysogenum mitochondrial genome was previously reported [20], but the sequence was not trimmed. We therefore trimmed, rotated, and re-annotated the original sequence to ensure annotation consistency. The A. terreus NIH 2624 genome was assembled at JCVI using Sanger reads or traces obtained from NCBI [GenBank:AAJN00000000], which were deposited by the Broad Institute. The A. nidulans FGSC A4 genome was assembled at JCVI using reads provided by the Broad Institute (with additional sequencing performed at JCVI). The remaining complete mitochondrial genome sequences used in this study were either obtained from NCBI (http://www.ncbi.nlm.nih.gov) or sequenced and assembled at JCVI (see Genome closure section below). The following mitochondrial genome sequences were obtained from NCBI: A. niger N909 [Genbank:DQ207726], A. tubingensis 0932 [Genbank:DQ217399], A. oryzae RIB 40 [Genbank:AP007176], P. marneffei MP1 [GenBank:AY347307] and A. nidulans FGSC A4 [GenBank:JQ435097].
Genome closure
Mitochondrial contigs were completed using available de novo contigs and/or mapping to mitochondrial reference genomes. Seed contigs from de novo assemblies of all genomic data were analyzed for high coverage and for the presence of common mitochondrial genes. Simultaneously, sequence reads were mapped to known references to generate a read set for de novo assembly. Resulting mitochondrial contigs were iteratively extended through recruiting sequence data by aligning to contig edges until no gaps remained. All contigs were manually examined for quality. All contain at least 2 fold high quality coverage of every base and had evidence of circularity based on mate pairing and/or overlapping contig edges. The resulting scaffolds were trimmed and rotated to facilitate comparative analysis. The deletion in A. fumigatus A1163 was confirmed by PCR using primers flanking the missing region (Primers: AF1163C16842: 5′-ATTGTTCATTATTCTACAGTTAAGCC-3′ and AF1163C17705: 5′-AATTAGTATCCTCATCTTCCTTAGG-3′). Annotated scaffolds have been deposited in NCBI [GenBank:JQ346807, GenBank:JQ346808, GenBank:JQ346809, GenBank:JQ354994, GenBank:JQ354995, GenBank:JQ354996, GenBank:JQ354997, GenBank:JQ354998, GenBank:JQ354999, GenBank:JQ355000 and GenBank:JQ355001].
SNP and DIP identification in A. fumigatus mitochondrial DNA
SNPs and small DIPs were predicted using CLC Genomics Workbench from CLC Bio (http://www.clc-bio.com). Illumina reads from target strains were mapped to the reference A. fumigatus AF293 mitochondrial genome. The mapping parameters used were 0.9 for Length fraction and 0.9 for Similarity. Non specific matches were ignored. Default cost parameters were used. To call SNPs and small DIPs, we used stringent parameters that were obtained from extensive manual evaluation of alignments. The following cut-offs were used in CLC to call SNPs: (i) read coverage is equal to or above 10; and (ii) variants supported by at least 99% of the reads. The quality of read alignments and the regions surrounding the called SNP locations were manually inspected. Low complexity regions were ignored. The SNPs that passed these filtering criteria were retained. MUMmer [52] and BLASTn [53] were used to check for the presence of large scale rearrangements and insertions/deletions.
Repetitive regions identification
RepeatMasker (http://www.repeatmasker.org/) was used to check for the presence of high copy interspersed repeats and low complexity DNA sequences. PrintRepeats [54] (http://www.genome.ou.edu/miropeats.html) was used to identify low copy repeats.
Mitochondrial genome annotation
All mitochondrial genomes analyzed in this study were annotated at JCVI, except for A. niger, A. tubingensis, A. nidulans and P. marneffei MP1 (Additional file 1). Mitochondrial A. oryzae tRNA genes were obtained from NCBI. Open reading frames (ORFs) were identified using Artemis [55] with genetic code 4. Functional assignments were made based on sequence similarity to characterized fungal mitochondrial proteins using BLASTp searches against NCBI databases. ORFs containing more than 100 amino acids, and no sequence homology to known genes, were designated as hypothetical genes. tRNA genes were identified using tRNAscan-SE and ribosomal RNA genes were identified using BLASTn [53]. Core mitochondrial-encoded genes were identified by all-against-all comparison using BLASTp.
Group I intron annotation
Approximate group I intron insertion boundaries were initially established as interruptions in the protein-coding genes and LSU rRNA gene identified through BLASTp and BLASTn searches. Precise 5′ intron boundaries were determined by identifying the conserved U-G or C-G within the intron’s P1 stem. The 3′ intron boundaries were determined by identifying the G at the end of the intron and the ability for the downstream sequence to form the P10 guide sequence stem [47,56]. The identified boundaries were confirmed by tBLAST searches of the putative spliced products. The mitochondrial LSU group I intron secondary structures were constructed from the previously described A. nidulans secondary structure [47].
Synteny analysis of core genes
OrthoMCL [57] was used to identify the orthologous relationships between the 15 core protein-coding genes. The first step was an all-against-all BLASTp search with an expect value of 1e-05. This was followed by the MCL clustering algorithm using default parameters and the main inflation value (−I) set to 1.5. The orthologous clusters were displayed in Figure 2 using SynView [58].
Phylogenetic analysis
To generate phylogenetic trees, 14 core proteins encoded by 13 genomes were first concatenated and then aligned using Muscle [59]. Regions with poor alignments were removed with Gblocks using default settings [60]. Maximum-Likelihood (ML) trees were generated using the Randomized Axelerated Maximum Likelihood (RAxML) program [61]. Multiple ML trees were generated and the best-scoring tree was identified. 100 boot-strapped trees were generated and used to assign the boot strap support values to the best-scoring ML tree. The JTT amino acid substitution model was used with the Gamma model of rate heterogeneity. Gibberella zeae (anamorph Fusarium graminearum) was used as an outgroup. A Maximum Parsimony based tree was generated using the Protpars program of the PHYLIP package [62].
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VJ and NFA performed the comparative analysis and interpretation of data and drafted the manuscript. VJ and OOA annotated the mitochondrial genomes. JH coordinated genome assembly and closure. PJP conducted intron annotation and analysis. SP, SBP and NZ performed phylogenetic, repeat content, comparative genomics, and other analyses. GP, AA, DWD and WCN made substantial contributions to the study conception and design and to the preparation of the manuscript. All authors have read and approved the final manuscript.
Supplementary Material
Sequence data sources.
SNPs predicted in A.fumigatus strains with respect to the AF293 reference mitochondrial DNA.
Core and accessory protein-coding genes in Aspergillus and Penicillium mitochondrial genomes.
The A.fumigatus AF293 mitochondrial genome showing the protein-coding genes, and the non-coding RNAs.
Protein sequences of the core mitochondrial genes.
Multiple sequence alignment of the core mitochondrial proteins.
Contributor Information
Vinita Joardar, Email: vinita@jcvi.org.
Natalie F Abrams, Email: natalie.abrams@nih.gov.
Jessica Hostetler, Email: jessica.hostetler@nih.gov.
Paul J Paukstelis, Email: paukstel@umd.edu.
Suchitra Pakala, Email: supakala@jcvi.org.
Suman B Pakala, Email: spakala@jcvi.org.
Nikhat Zafar, Email: nzafar@jcvi.org.
Olukemi O Abolude, Email: kabolude@som.umaryland.edu.
Gary Payne, Email: gary_payne@ncsu.edu.
Alex Andrianopoulos, Email: alex.a@unimelb.edu.au.
David W Denning, Email: ddenning@manchester.ac.uk.
William C Nierman, Email: wnierman@jcvi.org.
Acknowledgements
We would like to thank Jennifer Wortman and Qiandong Zeng at the Broad Institute for annotation of the A. nidulans mitochondrial genome, and Stephanie Mounaud and Jaya Onuska at JCVI for their superb technical assistance. This project has been funded in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contract numbers N01-AI-30071 and HHSN272200900007C and from the US Department of Agriculture grant USDA2007-04744.
References
- Hoffmeister D, Keller NP. Natural products of filamentous fungi: enzymes, genes, and their regulation. Nat Prod Rep. 2007;24(2):393–416. doi: 10.1039/b603084j. [DOI] [PubMed] [Google Scholar]
- Enoch DA, Ludlam HA, Brown NM. Invasive fungal infections: a review of epidemiology and management options. J Med Microbiol. 2006;55(Pt 7):809–818. doi: 10.1099/jmm.0.46548-0. [DOI] [PubMed] [Google Scholar]
- Cummings DJ, McNally KL, Domenico JM, Matsuura ET. The complete DNA sequence of the mitochondrial genome of Podospora anserina. Curr Genet. 1990;17(5):375–402. doi: 10.1007/BF00334517. [DOI] [PubMed] [Google Scholar]
- Amlacher S, Sarges P, Flemming D, van Noort V, Kunze R, Devos DP, Arumugam M, Bork P, Hurt E. Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell. 2011;146(2):277–289. doi: 10.1016/j.cell.2011.06.039. [DOI] [PubMed] [Google Scholar]
- Osiewacz HD, Brust D, Hamann A, Kunstmann B, Luce K, Muller-Ohldach M, Scheckhuber CQ, Servos J, Strobel I. Mitochondrial pathways governing stress resistance, life, and death in the fungal aging model Podospora anserina. Ann N Y Acad Sci. 2010;1197:54. doi: 10.1111/j.1749-6632.2010.05190.x. [DOI] [PubMed] [Google Scholar]
- Sanglard D, Ischer F, Bille J. Role of ATP-binding-cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob Agents Chemother. 2001;45(4):1174–1183. doi: 10.1128/AAC.45.4.1174-1183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari S, Sanguinetti M, Torelli R, Posteraro B, Sanglard D. Contribution of CgPDR1-regulated genes in enhanced virulence of azole-resistant Candida glabrata. PLoS One. 2011;6(3):e17589. doi: 10.1371/journal.pone.0017589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins VP, Dinamarco TM, Soriani FM, Tudella VG, Oliveira SC, Goldman GH, Curti C, Uyemura SA. Involvement of an alternative oxidase in oxidative stress and mycelium-to-yeast differentiation in Paracoccidioides brasiliensis. Eukaryot Cell. 2011;10(2):237–248. doi: 10.1128/EC.00194-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheckhuber CQ, Hamann A, Brust D, Osiewacz HD. Cellular homeostasis in fungi: impact on the aging process. Sub-cellular biochemistry. pp. 233–250. [DOI] [PubMed]
- Santamaria M, Vicario S, Pappada G, Scioscia G, Scazzocchio C, Saccone C. Towards barcode markers in fungi: an intron map of Ascomycota mitochondria. BMC Bioinforma. 2009;10(Suppl 6):S15. doi: 10.1186/1471-2105-10-S6-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamari Z, Juhasz A, Kevei F. Role of mobile introns in mitochondrial genome diversity of fungi (a mini review) Acta Microbiol Immunol Hung. 2002;49(2–3):331–335. doi: 10.1556/AMicr.49.2002.2-3.22. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen. 2010;51(5):440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- Tavare A. Scientists are to investigate “three parent IVF” for preventing mitochondrial diseases. BMJ (Clinical research ed) 2012;344:e540. doi: 10.1136/bmj.e540. [DOI] [PubMed] [Google Scholar]
- Juhasz A, Pfeiffer I, Keszthelyi A, Kucsera J, Vagvolgyi C, Hamari Z. Comparative analysis of the complete mitochondrial genomes of Aspergillus niger mtDNA type 1a and Aspergillus tubingensis mtDNA type 2b. FEMS Microbiol Lett. 2008;281(1):51–57. doi: 10.1111/j.1574-6968.2008.01077.x. [DOI] [PubMed] [Google Scholar]
- Juhasz A, Engi H, Pfeiffer I, Kucsera J, Vagvolgyi C, Hamari Z. Interpretation of mtDNA RFLP variability among Aspergillus tubingensis isolates. Antonie Van Leeuwenhoek. 2007;91(3):209–216. doi: 10.1007/s10482-006-9110-x. [DOI] [PubMed] [Google Scholar]
- Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, Kusumoto K, Arima T, Akita O, Kashiwagi Y. et al. Genome sequencing and analysis of Aspergillus oryzae. Nature. 2005;438(7071):1157–1161. doi: 10.1038/nature04300. [DOI] [PubMed] [Google Scholar]
- Woo PC, Zhen H, Cai JJ, Yu J, Lau SK, Wang J, Teng JL, Wong SS, Tse RH, Chen R. et al. The mitochondrial genome of the thermal dimorphic fungus Penicillium marneffei is more closely related to those of molds than yeasts. FEBS Lett. 2003;555(3):469–477. doi: 10.1016/s0014-5793(03)01307-3. [DOI] [PubMed] [Google Scholar]
- Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, Berriman M, Abe K, Archer DB, Bermejo C. et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature. 2005;438(7071):1151–1156. doi: 10.1038/nature04332. [DOI] [PubMed] [Google Scholar]
- Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, Batzoglou S, Lee SI, Basturkmen M, Spevak CC, Clutterbuck J. et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature. 2005;438(7071):1105–1115. doi: 10.1038/nature04341. [DOI] [PubMed] [Google Scholar]
- van den Berg MA, Albang R, Albermann K, Badger JH, Daran JM, Driessen AJ, Garcia-Estrada C, Fedorova ND, Harris DM, Heijne WH. et al. Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat Biotechnol. 2008;26(10):1161–1168. doi: 10.1038/nbt.1498. [DOI] [PubMed] [Google Scholar]
- Fedorova ND, Khaldi N, Joardar VS, Maiti R, Amedeo P, Anderson MJ, Crabtree J, Silva JC, Badger JH, Albarraq A. et al. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 2008;4(4):e1000046. doi: 10.1371/journal.pgen.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Diepeningen AD, Goedbloed DJ, Slakhorst SM, Koopmanschap AB, Maas MF, Hoekstra RF, Debets AJ. Mitochondrial recombination increases with age in Podospora anserina. Mech Ageing Dev. 2010;131(5):315. doi: 10.1016/j.mad.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Samson RA, Pitt JI. Advances in Penicillium and Aspergillus systematics. New York: Plenum Press; 1985. [Google Scholar]
- Berbee M, Yoshimura A, Sugiyama J, Taylor JW. Is Penicillium monophyletic? An evaluation of phylogeny in the family Trichocomaceae from 18S, 5.8S And ITS ribosomal DNA sequence data. Mycologia. 1995;87(2):210–222. [Google Scholar]
- Geiser DM, Samson RA, Varga J, Rokas A, Witiak SM. In: Aspergillus in the genomics era. Varga J, Sampson RA, editor. Wageningen: Wageningen Academic Publishers; 2008. A review of molecular phylogenetics in Aspergillus, and prospects for a robust genus-wide phylogeny; pp. 17–32. [Google Scholar]
- Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, de Vries RP, Albang R, Albermann K. et al. Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nature biotechnology. 2007;25(2):221–231. doi: 10.1038/nbt1282. [DOI] [PubMed] [Google Scholar]
- Rokas A, Galagan JE. In: The aspergilli: genomics, medical aspects, biotechnology, and research methods. Osmani SA, Goldman GH, editor. Boca Raton: CRC Press; 2007. Aspergillus nidulans genome and a comparative analysis of genome evolution in Aspergillus; pp. 43–55. [Google Scholar]
- Peterson SW. Phylogenetic analysis of Aspergillus species using DNA sequences from four loci. Mycologia. 2008;100(2):205–226. doi: 10.3852/mycologia.100.2.205. [DOI] [PubMed] [Google Scholar]
- Griffiths AJ. Natural plasmids of filamentous fungi. Microbiol Rev. 1995;59(4):673–685. doi: 10.1128/mr.59.4.673-685.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshwari R, Navaraj A. Senescence in fungi: the view from Neurospora. FEMS Microbiol Lett. 2008;280(2):135–143. doi: 10.1111/j.1574-6968.2007.01027.x. [DOI] [PubMed] [Google Scholar]
- Maas MF, Sellem CH, Hoekstra RF, Debets AJ, Sainsard-Chanet A. Integration of a pAL2-1 homologous mitochondrial plasmid associated with life span extension in Podospora anserina. Fungal Genet Biol. 2007;44(7):659–671. doi: 10.1016/j.fgb.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Court DA, Griffiths AJ, Kraus SR, Russell PJ, Bertrand H. A new senescence-inducing mitochondrial linear plasmid in field-isolated Neurospora crassa strains from India. Curr Genet. 1991;19(2):129–137. doi: 10.1007/BF00326294. [DOI] [PubMed] [Google Scholar]
- Belfort M, Roberts RJ. Homing endonucleases: keeping the house in order. Nucleic Acids Res. 1997;25(17):3379–3388. doi: 10.1093/nar/25.17.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delahodde A, Goguel V, Becam AM, Creusot F, Perea J, Banroques J, Jacq C. Site-specific DNA endonuclease and RNA maturase activities of two homologous intron-encoded proteins from yeast mitochondria. Cell. 1989;56(3):431–441. doi: 10.1016/0092-8674(89)90246-8. [DOI] [PubMed] [Google Scholar]
- Wenzlau JM, Saldanha RJ, Butow RA, Perlman PS. A latent intron-encoded maturase is also an endonuclease needed for intron mobility. Cell. 1989;56(3):421–430. doi: 10.1016/0092-8674(89)90245-6. [DOI] [PubMed] [Google Scholar]
- Basse CW. Mitochondrial inheritance in fungi. Curr Opin Microbiol. 2010;13(6):712–719. doi: 10.1016/j.mib.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Stoddard BL. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure. 2011;19(1):7–15. doi: 10.1016/j.str.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torriani SF, Goodwin SB, Kema GH, Pangilinan JL, McDonald BA. Intraspecific comparison and annotation of two complete mitochondrial genome sequences from the plant pathogenic fungus Mycosphaerella graminicola. Fungal Genet Biol. 2008;45(5):628–637. doi: 10.1016/j.fgb.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Foury F, Roganti T, Lecrenier N, Purnelle B. The complete sequence of the mitochondrial genome of Saccharomyces cerevisiae. FEBS Lett. 1998;440(3):325–331. doi: 10.1016/s0014-5793(98)01467-7. [DOI] [PubMed] [Google Scholar]
- Field DJ, Sommerfield A, Saville BJ, Collins RA. A group II intron in the Neurospora mitochondrial coI gene: nucleotide sequence and implications for splicing and molecular evolution. Nucleic Acids Res. 1989;17(22):9087–9099. doi: 10.1093/nar/17.22.9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M. et al. Life with 6000 genes. Science New York, NY. 996;274(5287):546. doi: 10.1126/science.274.5287.546. 563–547. [DOI] [PubMed] [Google Scholar]
- LaPolla RJ, Lambowitz AM. Mitochondrial ribosome assembly in Neurospora crassa. Purification of the mitochondrially synthesized ribosomal protein, S-5. J Biol Chem. 1981;256(13):7064–7067. [PubMed] [Google Scholar]
- Lapolla RJ, Lambowitz AM. Mitochondrial ribosome assembly in Neurospora. Structural analysis of mature and partially assembled ribosomal subunits by equilibrium centrifugation in CsCl gradients. J Cell Biol. 1982;95(1):267–277. doi: 10.1083/jcb.95.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo QB, Akins RA, Garriga G, Lambowitz AM. Structural analysis of the Neurospora mitochondrial large rRNA intron and construction of a mini-intron that shows protein-dependent splicing. J Biol Chem. 1991;266(3):1809–1819. [PubMed] [Google Scholar]
- Kamper U, Kuck U, Cherniack AD, Lambowitz AM. The mitochondrial tyrosyl-tRNA synthetase of Podospora anserina is a bifunctional enzyme active in protein synthesis and RNA splicing. Mol Cell Biol. 1992;12(2):499–511. doi: 10.1128/mcb.12.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur M, Geese WJ, Waring RB. Self-splicing activity of the mitochondrial group-I introns from Aspergillus nidulans and related introns from other species. Curr Genet. 1997;32(6):399–407. doi: 10.1007/s002940050294. [DOI] [PubMed] [Google Scholar]
- Paukstelis PJ, Lambowitz AM. Identification and evolution of fungal mitochondrial tyrosyl-tRNA synthetases with group I intron splicing activity. Proc Natl Acad Sci U S A. 2008;105(16):6010–6015. doi: 10.1073/pnas.0801722105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard MR, Burt A. Recurrent invasion and extinction of a selfish gene. Proc Natl Acad Sci U S A. 1999;96(24):13880–13885. doi: 10.1073/pnas.96.24.13880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hane JK, Lowe RG, Solomon PS, Tan KC, Schoch CL, Spatafora JW, Crous PW, Kodira C, Birren BW, Galagan JE. et al. Dothideomycete plant interactions illuminated by genome sequencing and EST analysis of the wheat pathogen Stagonospora nodorum. Plant Cell. 2007;19(11):3347–3368. doi: 10.1105/tpc.107.052829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamari Z, Toth B, Beer Z, Gacser A, Kucsera J, Pfeiffer I, Juhasz A, Kevei F. Interpretation of intraspecific variability in mtDNAs of Aspergillus niger strains and rearrangement of their mtDNAs following mitochondrial transmissions. FEMS Microbiol Lett. 2003;221(1):63–71. doi: 10.1016/S0378-1097(03)00165-4. [DOI] [PubMed] [Google Scholar]
- Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A, Johnson J, Li K, Mobarry C, Sutton G. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics (Oxford, England) 2008;24(24):2818–2824. doi: 10.1093/bioinformatics/btn548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30(11):2478–2483. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Parsons JD. Miropeats: graphical DNA sequence comparisons. Comput Appl Biosci. 1995;11(6):615–619. doi: 10.1093/bioinformatics/11.6.615. [DOI] [PubMed] [Google Scholar]
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics (Oxford, England) 2000;16(10):944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Michel F, Westhof E. Modelling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. J Mol Biol. 1990;216(3):585–610. doi: 10.1016/0022-2836(90)90386-Z. [DOI] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Su Y, Mackey AJ, Kraemer ET, Kissinger JC. SynView: a GBrowse-compatible approach to visualizing comparative genome data. Bioinformatics (Oxford, England) 2006;22(18):2308–2309. doi: 10.1093/bioinformatics/btl389. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics (Oxford, England) 2005;21(4):456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP—phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence data sources.
SNPs predicted in A.fumigatus strains with respect to the AF293 reference mitochondrial DNA.
Core and accessory protein-coding genes in Aspergillus and Penicillium mitochondrial genomes.
The A.fumigatus AF293 mitochondrial genome showing the protein-coding genes, and the non-coding RNAs.
Protein sequences of the core mitochondrial genes.
Multiple sequence alignment of the core mitochondrial proteins.