

Abstract
Thymine DNA glycosylase (TDG) belongs to the superfamily of uracil DNA glycosylases (UDG) and is the first enzyme in the base-excision repair pathway (BER) that removes thymine from G:T mismatches at CpG sites. This glycosylase activity has also been found to be critical for active demethylation of genes involved in embryonic development. Here we show that wild-type p53 transcriptionally regulates TDG expression. Chromatin immunoprecipitation (ChIP) and luciferase assays indicate that wild-type p53 binds to a domain of TDG promoter containing two p53 consensus response elements (p53RE) and activates its transcription. Next, we have used a panel of cell lines with different p53 status to demonstrate that TDG mRNA and protein expression levels are induced in a p53-dependent manner under different conditions. This panel includes isogenic breast and colorectal cancer cell lines with wild-type or inactive p53, esophageal squamous cell carcinoma cell lines lacking p53 or expressing a temperature-sensitive p53 mutant and normal human bronchial epithelial cells. Induction of TDG mRNA expression is accompanied by accumulation of TDG protein in both nucleus and cytoplasm, with nuclear re-localization occurring upon DNA damage in p53-competent, but not -incompetent, cells. These observations suggest a role for p53 activity in TDG nuclear translocation. Overall, our results show that TDG expression is directly regulated by p53, suggesting that loss of p53 function may affect processes mediated by TDG, thus negatively impacting on genetic and epigenetic stability.
Keywords: TDG, p53, base-excision repair, glycosylase, transcriptional regulation, mutator phenotype, epigenetic stability
Introduction
The most frequent type of mutation in the human genome is G to A transition at CpG sites, representing 23% of mutations in human hereditary diseases and 24% of human single nucleotide polymorphisms (SNP).1,2 In human cancer, CpG transitions account for about 24% of tumor-suppressor gene TP53 mutations, including all mutation hotspots.3 The predominance of these mutations is due, to a large extent, to the highly mutable characteristics of G:C to A:T base pairs within CpG sites. They originate from non-repaired G:T mismatches resulting from the spontaneous deamination of 5′methylcytosine (5mC) at CpG sites. The 5mC is less stable than cytosine and undergoes spontaneous deamination into thymine at a rate five times higher than the unmethylated base.4
TDG is the most proximal enzyme in the BER pathway that removes thymine from G:T mismatches at CpG sites. TDG hydrolyzes the carbon-nitrogen bond between the sugar-phosphate backbone of DNA and the mispaired thymine, thus removing the thymine moiety from G:T mismatches and creating an abasic site which is repaired by subsequent enzymatic steps in this BER pathway.5,6 The enzyme methyl-CpG-binding domain protein 4 (MBD4) is also implicated in repairing G:T mismatches at CpG sites.7 MBD4 belongs to a family of nuclear proteins containing a methyl-CpG-binding domain (MBD) and also presents a COOH-terminal glycosylase domain.8,9 Knocking out the Mb4 locus in mice results in a 3-fold increase in the rate of G to A transition at CpG sites.10,11 By comparison, homozygous Tdg-knockout mice are not viable,12,13 and in vitro repair of G:T mismatches is abolished in nuclear extracts of Tdg −/− mouse embryo fibroblast.12,13 These data suggest that TDG is the most important enzyme for this DNA repair pathway, and that MBD4 cannot make up for the lack of its activities.
Additionally to TDG anti-mutagenic role in DNA repair, it has been shown that TDG is involved in the regulation of epigenetic events. Recently, TDG was found to interact with transcription factors, de novo DNA methyltranferases and histone acetyltransferases, to protect CpG islands from hypermethylation.14-17 Through its glycosylase activity, it plays an active role in the removal of 5mC and, thus, in the activation by demethylation of developmentally and hormonally regulated gene promoters.13 This evidence supports the notion that TDG plays an important role in regulating and maintaining appropriate genetic and epigenetic states. However, the the mechanisms involved in the regulation of TDG expression and activity are still poorly known.
The tumor-suppressor gene TP53 has essential roles in preventing abnormal cell growth and maintaining genomic stability. Its product, p53, accumulates under stress signals and triggers the transcription of p53 target genes, leading to a wide range of suppressive responses, including growth arrest, apoptosis, DNA repair and senescence. The type of biological response is highly dependent upon cell type and stress-inducing signals. Together, these responses have a protective effect toward genetic stability by preventing the survival and proliferation of cells with damaged DNA.18 Specifically, p53 exerts a control over DNA-repair pathways, including BER, either directly (by regulating the transcription of hOGG1 or MGMT) or indirectly (by interacting with and exerting non-transcription-dependent effects on a wide range of repair effectors such as WRN, ERCC1/2, ERF1/APE1 or 53BP1).19-21 However, so far, there is no information on whether p53 may control TDG expression. Therefore, in this study we have investigated whether TDG may be transcriptionally regulated by wild-type p53.
Results
Wild-type p53 regulates the expression of thymine DNA glycosylase (TDG)
To assess the effects of p53 status on TDG expression, we first used an esophageal squamous cell carcinoma (ESCC) cell line, TE1, previously shown to constitutively express a temperature-sensitive TP53 mutant, p.V272M.22,23 At 32°C (permissive temperature), the mutant protein adopts an essentially “wild-type” immunological conformation, binds to p53REs and transactivates many p53-dependent genes. However, at 37°C (restrictive temperature), the protein folds into a “mutant” form unable to specifically bind DNA and to regulate gene expression in a p53-dependent manner.24 Although the conformational and functional shift is only partial (at 32°C a significant fraction of the protein remains “mutant-like”), this cell line has been previously used as a model to identify p53-regulated genes in an ESCC background.25,26Figure 1A shows a 4-fold higher level of TDG mRNA in TE-1 cells at 32°C, than at 37°C. siRNA silencing of p53 in TE-1 at 32°C resulted in a 45% decrease in TDG mRNA level, indicating that the effect induced by temperature shift was p53-dependent rather than being a fortuitous consequence of temperature change. Western blot analysis with an anti-TDG antibody (Fig. 1A, center panel) shows that p53 silencing also reduced TDG protein levels. By contrast, the amount of TDG protein in TE-1 cells at 37°C containing “mutant” p53 was not affected by silencing of p53.

Figure 1. Wild-type p53 regulates thymine DNA glycosylase (TDG) expression. (A) siRNA targeting p53 (p53 siRNA) or TDG (TDG siRNA) or control siRNA (Scramble, SCR) was transfected in TE-1 cells which express the temperature-sensitive p53 mutant, V272M. Cells maintained at 32°C (permissive temperature) present most of the p53 in an active form, and those maintained at 37°C (restrictive temperature) present most of the p53 in an inactive form. Upper panel: detection of TDG mRNA levels by reverse-transcription quantitative PCR (RT-qPCR). Middle panel: western blotting analysis of TDG, using Ku-80 as loading control. Lower panel: detection of TDG and p53 by confocal microscopy. sip53 and siTDG: siRNA to p53 and TDG, respectively. Green fluorescence, TDG; red fluorescence, p53; blue fluorescence, nucleus (ToPro). (B) The ESCC p53-null cell line TE-13 was transfected with either 0.5, 1.0, 1.5 or 2.0 μg of DNA of an expression vector for p53 protein (p53wt-pcDNA3) or with an empty vector (pCDNA3-empty), used as control (0). Upper panel: detection of TDG mRNA levels by RT-qPCR. Lower panel: western blotting analysis of TDG, using Ku-80 as loading control. (C) TE-1 cells, cultured at either 32°C or 37°C, were treated with 0.25, 0.5, 1.0 or 2.0 mM of MMS for 3 h or not (-). Upper panel: detection of TDG mRNA levels by RT-qPCR. Middle panel: western blotting analysis of TDG, using Ku-80 as loading control. Lower panel: detection of TDG and p53 by confocal microscopy. Green fluorescence, TDG; red fluorescence, p53. (D) The isogenic breast cancer cell lines, MN1 and MDD2, were treated with 500 ng/mL doxorubicin (Dox) for 24 h andTDG mRNA levels were detected by RT-qPCR. (E) The non-transformed bronchial cells, NHBE, were treated with 0.25, 0.5 or 1.0 mM of MMS for 3 h or not (-) and TDG mRNA expression levels were assessed by RT-qPCR. Stars indicate statistical significance: *p < 0.05 (Student’s t-test using with software GraphPad Prism 4, GraphPad Software Inc.).
The TDG protein in TE-1 cells appears in this western blot as a doublet with an apparent molecular weight in the range of 75–80 kDa. Previous reports have shown that TDG may exist as two forms: the native protein with an apparent size around 60 kDa, and as a complex with small ubiquitin-like modifiers (SUMOs).27-29 Sumoylated forms of TDG have apparent sizes around 75‒80 kDa. Probing these western blots with anti-SUMO-1 antibodies revealed that the doublet in Figure 1A corresponds to sumoylated TDG (Fig. S1). Thus, in TE-1 cells, TDG appears to exist predominantly, if not exclusively, in a sumoylated form.
We next used confocal microscopy to detect TDG in TE-1 cells. Figure 1A (lower panel) shows that a fluorescent TDG signal was detectable in both nuclear and cytoplasmic compartments. This signal was essentially removed by silencing either TDG or p53 in TE-1 cells cultured at 32°C, but only by silencing TDG in cells maintained at 37°C. Another ESCC cell line, TE-13, that does not express p53 due to a mutation into the splice acceptor site of intron 4 was used.23,30 Transfection of a wild-type p53 expression vector into TE-13 produced a strong increase in both TDG mRNA expression and in the amount of TDG protein in the cells (Fig. 1B).
To determine whether TDG may be upregulated by p53 as part of its response to DNA-damage, we used methyl methanesulfonate (MMS), a DNA alkylating agent, to activate p53 in TE-1 cells cultured at 32°C or 37°C. At 32°C, the expression of TDG increased in a concentration-dependent manner up to 1 mM MMS, reaching a maximum induction of about 2-fold in TDG mRNA levels (Fig. 1C, upper panel). By contrast, no significant effect on TDG expression was detected in cells cultured at 37°C. A parallel effect of induction was detected at the protein level (Fig. 1C, middle panel). Figure 1C (lower panel) shows that MMS treatment of TE-1 cells at 32°C, but not at 37°C, not only led to an induction of TDG, but also promoted its nuclear re-localization.
To further assess whether the effect of p53 on TDG expression is a general phenomenon, we treated a pair of isogenic cell lines derived from MCF7 cells with doxorubicin. MN1 is stably transfected with an empty vector, whereas MDD2 expresses a small peptide that binds to the C terminus of p53 and prevents its assembly into a transcriptionally active tetramer.31 There was a clear and significant induction of 2.2-fold in TDG mRNA in MN1, but not in MDD2 cells (Fig. 1D) after doxorubicin treatment. Finally, we used non-transformed bronchial cells, NHBE, and showed that exposure of these cells to MMS also increased TDG mRNA expression (Fig. 1E). Overall, these results provide evidence that TDG expression is regulated by wild-type p53 in several cell types.
p53-dependent regulation of thymine DNA glycosylase (TDG) promoter
To determine whether p53 may activate TDG transcription, we used MatInspector software to search for potential p53-binding sites in a region of 5 Kb extending from about 2.5 Kb of upstream of the transcription initiation site of TDG to the first third on intron 1.32 Three regions containing at least a consensus p53RE half-site [(5′- RRRCWWGYYY-3′)] were found (Fig. 2).33 We then derived two fragments of this promoter, one containing RE1 (838 bp, corresponding to the distal 5′ part of the promoter) and one containing RE2 and RE3 (1,498 bp, containing the part of the promoter directly upstream of the transcription initiation site). These two fragments were cloned separately into luciferase reporter vectors. These vectors were transfected together with either p53 expression vector or control (empty) vector in TE-13 cells or alone in TE-1 (Fig. 3A and B). In TE-13, both promoter fragments conferred dependence of luciferase expression upon p53, with a 5‒10-fold increase for the RE1 promoter fragment and a 50‒250-fold increase with the RE2/RE3 promoter fragment. In TE-1 cells cultured at 32°C, treatment with 0.5 mM MMS significantly activated the expression from both promoter fragments by 2.5‒3-fold, whereas in cells cultured at 37°C, only a slight and non-significant activation was observed. Similar results were observed in p53-competent (HCT116 +/+) and -deficient (HCT116 −/−) colon tumor cells treated with 0.5 mM MMS (data not shown). We then used site-directed mutagenesis to delete either RE2, RE3 or both (Fig. 3C). Removal of either sequence abrogated the induction by p53 in TE-13 cells transfected by wild-type p53, whereas removal of both REs decreased the basal level of reporter gene expression (Fig. 3C). Next, we performed chromatin immunoprecipitation (ChIP) assay to evaluate whether p53 may bind to a promoter fragment encompassing RE2 and RE3 (Fig. 4). By transfection of wild-type p53 in TE-13, we found that p53 can bind to this region of TDG promoter in conditions comparable to the p21/WAF1 promoter, a well-characterized p53-responsive gene (Fig. S2). Therefore, these experiments provide evidence that TDG is directly regulated by p53 at the transcriptional level, and that p53 may play an important role in the induction of TDG transcription by DNA damage.
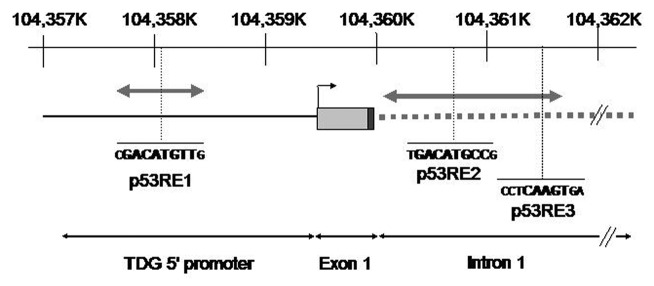
Figure 2. Thymine DNA glycosylase promoter. Schematic representation of the position of putative p53 response elements (p53RE) in the 5′ regulatory region of TDG. The interval 104,357K‒104,362K on Chr 12p is represented (reference: GRCh37.p5). The position of TDG transcription initiation site and of exon 1 are shown. The proximal part of intron 1 is represented by a gray hatched bar. The motif p53RE1 is located about 1.4 kb upstream of transcription initiation site. The p53R2 and p53R3 motifs are located in intron 1. The size and position of fragments used for cloning in luciferase vectors are shown by gray double-arrowed bars. p53R1 fragment, 0.84 kb; p53R2/R3 fragment, 1.5 kb.

Figure 3. p53 transcriptional regulation of TDG. (A) TE-13 cells were co-transfected with TDG-luciferase reporters and either increasing amounts of the expression vector for p53 (p53wt-pcDNA3) or with 2 μg of the control (empty) vector pcDNA3 (Ø). (B) TE-1 cells cultured at either 32°C or 37°C were treated with 0.25, 0.5 or 1.0 mM of methyl methanesulfonate (MMS) for 3 h and transfected with TDG-luciferase reporters and the luciferase assays were performed. Basic, promoterless luciferase reporter (pGL3 basic); p53RE1, luciferase reporter under the control of the 838 bp segment of TDG promoter containing the first p53RE; p53RE2/3, luciferase reporter under the control of the 1.5 kb segment of TDG promoter containing the second and the third p53REs. (C) The TDG-luciferase construct containing p53RE2/p53RE3 had either p53RE2, p53RE3 or both deleted by site-directed mutagenesis (as schematically shown in right panel) and mutated plasmids were co-transfected with 2 μg of expression vector for p53 (p53wt-pcDNA3) in TE-13 cells and the luciferase assays performed. Del p53RE2, plasmids harboring the deletion of p53RE2; Del p53RE3, plasmids harboring the deletion of p53RE3; Del p53RE2/3, plasmids harboring the deletion of p53RE2 and 3. Stars indicate statistical significance: *p < 0.05 (Student’s t-test was performed with software GraphPad Prism 4, GraphPad Software Inc.).
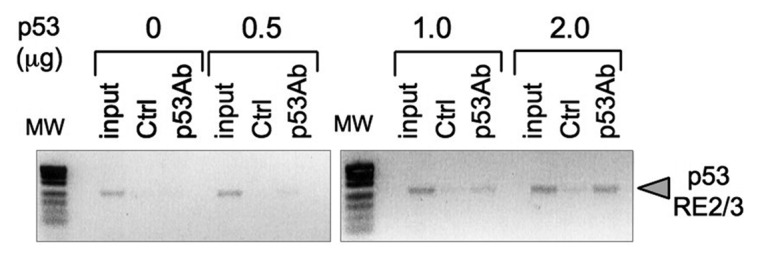
Figure 4. Binding of p53 to TDG promoter region. TE-13 cells were transfected with increasing amounts p53 expression vector (p53wt-pcDNA3) as indicated. Cross-linked chromatin was immunoprecipited with anti-p53 antibody CM1 (p53Ab) or control antibody (Ctrl) and analyzed by polymerase chain reaction (PCR) using primers specific for TDG promoter region encompassing p53RE2/3. MW, molecular weight markers; input, non-precipitated cross-linked chromatin.
Discussion
This study provides evidence that wild-type p53 regulates the expression of TDG, the most proximal enzyme in the BER pathway that processes both G > T and G > U mismatches at CpG sites.34-36 Consequently, TDG transcription is induced by p53 and by DNA damage in a p53-dependent manner, although we cannot rule out the participation of other DNA damaging signaling mediators in this regulation. Our results document an effect of p53 on TDG expression in several types of cells (esophageal, breast and colon cancer cell lines and non-transformed bronchial cells). We have identified three p53RE located in the TDG promoter, and luciferase reporter assays demonstrated their effectiveness in driving p53-dependent expression. Overall, these results support that TDG is a direct target for transcriptional regulation by p53. These observations suggest that TDG is part of a network of BER factors that are under the direct control of p53.
TDG was first described as a 55 kDa protein purified from HeLa cell extracts.37 Subsequent studies reported TDG as being a 60 kDa polypeptide, with the 55 kDa form resulting from alternative splicing and/or proteolytic degradation.38,39 Here, we observed that TDG migrated with an apparent molecular weight of 75–80 kDa in various cell lines, although purified human TDG showed a molecular weight of 55–60 KDa (Fig. S1). This difference in size was accounted for by interaction of TDG with SUMO proteins, which are polypeptides of about 11 kDa.40 It has been proposed that binding of SUMO regulates the DNA binding and repair capacity of TDG.27-29 Specifically, SUMO conjugation appears to induce a conformational shift in the N-terminal domain of TDG, reducing the affinity of the enzyme for DNA and abrogating its capacity to repair G > T mismatches. Conversely, conjugation increases the turnover of the enzyme and facilitates the processing of a wide spectrum of substrates including G > U mismatches.28,29 Our results showing that in various cell lines TDG is mostly present as a SUMO conjugate suggest that in these cells, TDG may operate with broad substrate specificity.
TDG is essentially a nuclear enzyme.41 However, conjugation with SUMO may affect its localization and stability.6 Mohan and colleagues investigated TDG subcellular localization and regulation upon SUMO conjugation and observed a shift from nucleus to cytoplasm after deleting the N-terminal domain of TDG (residues 1‒156).42 In the present study, we have observed that in TE-1 cells at 32°C (containing a temperature-sensitive mutant p53 in the “wild-type,” active conformation), MMS induced not only a p53-dependent increase in TDG levels, but also its translocation from cytoplasm to nucleus. However, whether this translocation is accompanied by a change in SUMO conjugation status is not known. Taken together, these results and those on the effects of SUMO on substrate specificity suggest that SUMO exerts a tight control on the nuclear accumulation of TDG and on its capacity to process G > T mismatches.
It is well established that TP53 plays an essential role in maintaining genomic stability, and that the p53 protein, which accumulates under stress signal, works through several mechanisms to ensure the proper functioning of the cell DNA repair machinery.18,20,43,44 Numerous studies have shown that p53 is involved in regulating several different steps in the BER pathway.45-49 BER is critical for the repair of a wide range of DNA lesions. The first step of BER pathway consists in processing chemically modified bases by glycosylases such as MPG (responsible for processing the alkylated bases N7-methylguanine and 3-methyladenine), UNG (which removes uracil arising either by spontaneous deamination of cytosine or by the misincorporation of uracil during replication and repair), hOGG1 (the primary glycoslyase responsible for excision 7,8-dihydro-8-oxoguanine) or TDG (which processes both G > T or G > U mismatches). Glycosylases generate apurinic (AP) sites recognized by the APE endonuclease, which incises the damaged strand immediately 5′ of the AP site. DNA polymerase-β (pol β) then fills the repair patch and participates in removing the overhang created by the displaced strand.50 In long-patch BER, the flap endonuclease FEN-1 then removes the 5′ DNA flap displaced during DNA synthesis. The BER process is completed by a ligation step. In 2005, Chatterjee and colleagues reported that hOGG1 was regulated by p53 at the transcriptional level.51 Furthermore, p53 was shown to enhance BER activity by interacting with APE1 and with pol β. In particular, Seo and collaborators showed that p53-deficient cells had decreased BER capacity, and that this was correlated with low pol β expression.47 Our results showing that p53 regulates the expression of TDG thus parallel those on the regulation of hOGG1 and further support the notion that p53 is essential for the repair of multiple types of lesions processed by BER.
The importance of TP53 in the maintenance of genome stability, its high mutation frequency in human cancer and its role in controlling DNA repair and recombination, cell cycle checkpoints and apoptosis lead us to suggest that TP53 may possess an important role in preventing the “mutator phenotype.”47,52-54 The concept of the “mutator phenotype” states that an initial mutation caused by unrepaired DNA damage can result in clonal expansion and mutation in mutator genes, leading to alterations in genes involved in maintaining the genetic stability, causing a cascade of mutations throughout the genome.55,56 Genes that function primarily to maintain genomic stability would be the most critical genes affected by the initial mutation and responsible for the genetic instability and tumor development by increasing the spontaneous mutation rate.56,57 The “mutator phenotype” is a common characteristic of cancer cells and can explain the high number of mutations found in tumor cells when compared with normal cells.57 Germline mutations found in DNA repair genes in hereditary cancers support the “mutator phenotype” concept. Once these mutations are present in all the cells of the organism, another single event would account for the loss of genomic stability and tumor development.58 Consistent with these views, the inactivation of the mismatch repair genes hMSH2, hMLH1 and hPMS2 leading to a mutator phenotype has already been reported.59,60
Aside from its anti-mutagenic role in BER, TDG was shown to operate as regulator of epigenetic (methylation) status. TDG modulates gene expression through interaction with several different transcription factors, including TTF1 (thyroid transcription factor 1), ER (estrogen receptor), RAR (retinoic acid receptor), RXR (retinoic X receptor) and histone acetyl-transferases p300 and CBP.14,17,38,61 Furthermore, recently, TDG activity has been associated with DNA demethylation in gene promoters, leading to the activation of their transcription during embryonic development.12,13,62-64 Inactivation of TDG in mice caused embryonic lethality due to aberrant epigenetic silencing of developmentally and hormonally regulated promoters/enhancers caused by hypermethylation of CpG islands within these promoters.12,13 TDG protects CpG islands from hypermethylation not only by acting as a co-activator promoting demethylation, but also playing a direct active catalytic role in this process. TDG interacts with the deaminase AID (activation-induced deaminase) and the damage-response protein GADD45a in a complex that regulates the processing of 5-methylated or 5-hydroxymethylated cytosine into thymine and hydroxymethyluracil, respectively, followed by TDG-mediated excision repair.13 The modified 5-methylated and 5-hydroxymethylated cytosine can be further metabolized by Tet dioxygenases to originate 5-carboxylcytosine, a more favorable substrate for TDG.65 Taken together, these studies show that TDG is a central enzyme in the epigenetic events, and cooperates with several factors through different mechanisms to maintain epigenetic stability.
In conclusion, regulation of TDG by p53 may provide cells with a dual mechanism that control both genetic and epigenetic stability. It follows that loss of p53 function (e.g., by mutation) may contribute to both a mutator phenotype and an epigenetic instability. The mutator phenotype would result from decreased capacity to repair mismatches resulting from the spontaneous deamination of methylated cytosine, thus leading to accumulation of G:C to A:T transitions. The epigenetic phenotype would be the consequence of increased DNA methylation of promoters during tumor progression. Further studies are needed to determine the impact of p53-mediated regulation of TDG on genetic stability and epigenetic patterns in vivo.
Material and Methods
Cell lines and treatments
ESCC cell lines TE-1 and TE-13 were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (PAA), 1% penicillin/streptomycin/glutamine (Gibco) at 37°C under 10% CO2. MN1 and MDD2 were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (PAA), 1% penicillin/streptomycin/glutamine (Gibco BRL) and 0.4 mg/mL geneticin G418 (geneticin, 0.5 mg/mL, Roche), at 37°C under 5% CO2. HCT116/ p53 [colon carcinoma (ATCC CCL247)] and HCT116/p53−/− (kindly given by B. Vogelstein) were grown in McCoy's 5A modified medium supplemented with 10% fetal bovine serum (PAA) 1% penicillin/streptomycin/glutamine (Gibco), at 37°C under 5% CO2. Normal human bronchial epithelial (NHBE) cells, purchased from Lonza and used at passage 2–3, were maintained in bronchial epithelial cell basal medium (Lonza) supplemented with 0.4% bovine pituitary extract (BPE), 0.1% hydrocortisone, 0.1% human epidermal growth factor (hEGF), 0.1% epinephrine, 0.1% transferrin, 0.1% insulin, 0.1% retinoic acid, 0.1% triiodothyronine, 0.1% gentamicin/amphotericin B (GA-1000) (Lonza), at 37°C under 5% CO2. MMS (methylmethane sulfonate) and doxorubicin (Sigma-Aldrich) were added in culture medium of exponentially growing cells.
Transfections
TE-13 cells were plated in 6-well plates (3 × 105 cells/well/2 mL) and transfected with 0.5, 1.0, 1.5 or 2.0 μg of DNA of a p53 expression vector p53wt-pcDNA366 or a pcDNA3-empty vector (used as a negative control) (Invitrogen) using Fugene (Roche) at 3 μL/μg of vector DNA and harvested 48 h later. Twenty μM of siRNA targeting TP53 (ID:106141) or negative control siRNA (scr catalog:4611) or 12 μM of siRNA targeting TDG (ID:s13950) (Ambion, Applied Biosystems) were transfected twice into TE-1 cells plated in 6-well plates (3 or 4.5 × 105 cells/well/2 mL), at an interval of 24 h, using 6 μL of HiPerfect (QiaGen) following cell harvest 48 h after the second transfection.
RNA isolation and real-time quantitative RT-PCR
Total RNA from the cells used was isolated using the Nucleospin-RNAII kit (Macherey-Nagel) and reverse transcribed with SuperScriptII reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. Real-time RT-PCR was performed in a Stratagene Mx3000 machine using SYBR Green Master Mix (QiaGen) and primers (Table S1). Each sample was analyzed in triplicate. Relative mRNA levels were calculated using the comparative threshold cycle (CT) with the analyzed gene expression levels normalized by those of the GAPDH and using the control of the experiment as the reference.
Protein extraction and western blotting
Proteins were extracted from the cells using RIPA-like buffer (250 mM NaCl, 50 mM TRIS-HCl pH7.4, 0.1% SDS, 2 mM DTT and 0.5% NP-40) containing protease inhibitors (Complete-Mini, Roche). Equal amounts of total proteins were resolved onto a 7.5% SDS-PAGE (PAGE), transferred a PVDF-membrane (Roche) and probed with the appropriate antibodies (Table S2). Detection was performed with enhanced chemiluminescence (ECL Kit, Amersham).
Immunofluorescence
TE-1 (1.5 or 3 × 105 cells/well/mL) cells were cultured on coverslips in a 12-well plates, rapidly washed with PBS and then fixed in a mixture of methanol and acetone (70:30) during 5 min at -20°C. Cells were profoundly washed in PBS, permeabilized by 0.4% of Triton X-100 and blocked in 0.5% blocking reagent in TN buffer (TNB, Perkin Elmer). The immunostaining for TDG was performed using TDG primary antibody (produced against TDG peptide KEEKYDPGYEAA by Davids Biotechnologie; 1/1,000) and goat anti-rabbit HRP-conjugated secondary antibody (Perkin Elmer, 1/200) with the green fluorescence signal enhanced by using the TSATM Plus kit from Perkin-Elmer, following the manufacturer’s instructions. For p53 immunostaining, the primary DO-7 (Dako, 1/500) and the secondary goat anti-mouse conjugated to Alexa Fluor 568 (Invitrogen, 1/150) antibodies were used. Cells nucleus were stainned using either ToPro (Invitrogen, 1/300) or mounting medium with DAPI (Vectashield).
Cloning of TDG promoter, site-directed mutagenesis and dual-luciferase assay
The sequence 5′ of TDG coding regions was analyzed in search of potential p53RE using MatInspector software. The three p53REs contained within TDG promoter were cloned separately into the promoter-less luciferase plasmid pGL3-Basic (Invitrogen), producing two luciferase reporter systems. The sequences of the primers used are described in Table S3. The amplified fragments were first cloned into pDrive cloning vector (Qiagen) and then cloned upstream of the luciferase gene in pGL3-Basic. The restriction enzymes used to release the fragments from pDrive cloning vector and to open pGL3-basic were KpnI and SacI (Fermentas, Thermo Fisher). For site-directed mutagenesis, the TDG-luciferase construct containing p53RE2/p53RE3 had either p53RE2, p53RE3 or both deleted by PCR-like technique using mutagenic primers (Table S4). Either a 32 bp sequence (TGGCATGTCTGGAATCTGATTCTGACATGCCG) encompassing p53RE2 (located 675 bp downstream of initiation) and/or 44 bp sequence (CACCATGTTGGCCAGGCTGGTCTTGAACTCTTGCCCTCAAGTGA) encompassing the p53RE3 (1,406 bp downstream of initiation) were deleted to produce vectors harboring p53RE2 and p53RE3 deletions, respectively, whereas both sequences were deleted to generate a vector lacking both p53RE2 and p53RE3. The activity of the luciferase repoter systems produced was assayed by the dual luciferase reporter assay system kit (Promega). TE-1 and TE-13 cells were plated in a 24-well plate to ~80% of confluence prior to transfection. One μg of DNA of TDG-luciferase reporter systems was transfected into cells using Fugene (Roche). pGL3-basic was used as negative control and the level of firefly luciferase activity was normalized to that of the pRL-TK control vector enconding Renilla luciferase activity. The luciferase activity was measured 48 h after transfection using a TD20/20 luminometer (Optocompl, MGM Instruments).
ChIP assay
TE-13 cells (2 × 106 cells/plate/10 mL) were plated in 10 mm plates and, 24 h later, 1% formaldehyde was added to the culture medium for 10 min at room temperature to cross-link proteins to DNA and then neutralized by the addition of 125 mM glycine pH 2.5. Cells were lysed for 15 min at 4°C in SDS-lysis buffer [1% SDS, 10 mM ethylene diamin tetraacetic acid (EDTA), 50 mM TRIS-HCl] containing protease inhibitors. Lysates were sonicated [twice: 12 X (5s on: 5s off) at 21% amplitude] (Vibra cell 75041), and immunoprecipitation was performed using 4 μg of CM1 (anti-p53) antibody (Novocastra) and magnetic beads (ChIP-Adem-Kit), according to the manufacturer’s protocol. Pre-immune serum was used as a negative control. Cross-linking was reversed by adding 200 mM of NaCl for 4 h at 65°C; DNA was recovered by phenol chloroform extraction. Immunoprecipitated DNA was amplified by PCR (Table S5) and visualized on a 2% agarose gel.
Statistical analysis
Student’s t-test and Fisher’s exact test were performed using the software GraphPad Prism 4 (GraphPad Software Inc.) and a p value of < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported with grants from CNPq (INCT and PQID) and Faperj (CNE and PPSUS) to L.F. Ribeiro Pinto. N.M. was supported by an ICRETT fellowship of UICC (www.uicc.org/).
Glossary
Abbreviations:
- TDG
thymine DNA glycosylase
- UDG
uracil DNA glycosylases
- BER
base-excision repair
- ChIP
chromatin immunoprecipitation
- p53RE
p53 consensus response element
- SNP
single nucleotide polymorphism
- 5mC
5′methylcytosine
- MBD4
enzyme methyl-CpG-binding domain protein 4
- MBD
methyl-CpG-binding domain
- ESCC
esophageal squamous cell carcinoma
- SUMO
small ubiquitin-like modifier
- MMS
methyl methanesulfonate
- Dox
doxorubicin
- NHBE
normal human bronchial epithelial
- TTF1
thyroid transcription factor 1
- ER
estrogen receptor
- RAR
retinoic acid receptor
- RXR
retinoic X receptor
- AID
activation-induced deaminase
- BPE
bovine pituitary extract
- hEGF
human epidermal growth factor
- CT
comparative threshold cycle
- NaCl
sodium chloride
- Tris
tris hydroxymethyl aminomethane
- DTT
Dithiothreitol
- NP-40
Nonidet P-40
- SDS
sodium dodecyl sulfate
- PAGE
polyacrylamide gel electrophoresis
- PVDF
polyvinylidene difluoride
- PBS
phosphate buffered saline
- TNB
Tris-NaCl blocking buffer
- TSA
tyramide signal amplification
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22843
References
- 1.Krawczak M, Ball EV, Cooper DN. Neighboring-nucleotide effects on the rates of germ-line single-base-pair substitution in human genes. Am J Hum Genet. 1998;63:474–88. doi: 10.1086/301965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomso DJ, Bell DA. Sequence context at human single nucleotide polymorphisms: overrepresentation of CpG dinucleotide at polymorphic sites and suppression of variation in CpG islands. J Mol Biol. 2003;327:303–8. doi: 10.1016/S0022-2836(03)00120-7. [DOI] [PubMed] [Google Scholar]
- 3.Waters TR, Swann PF. Thymine-DNA glycosylase and G to A transition mutations at CpG sites. Mutat Res. 2000;462:137–47. doi: 10.1016/S1383-5742(00)00031-4. [DOI] [PubMed] [Google Scholar]
- 4.Kumar S, Subramanian S. Mutation rates in mammalian genomes. Proc Natl Acad Sci USA. 2002;99:803–8. doi: 10.1073/pnas.022629899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiebauer K, Jiricny J. In vitro correction of G.T mispairs to G.C pairs in nuclear extracts from human cells. Nature. 1989;339:234–6. doi: 10.1038/339234a0. [DOI] [PubMed] [Google Scholar]
- 6.Cortázar D, Kunz C, Saito Y, Steinacher R, Schär P. The enigmatic thymine DNA glycosylase. DNA Repair (Amst) 2007;6:489–504. doi: 10.1016/j.dnarep.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 7.Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401:301–4. doi: 10.1038/45843. [DOI] [PubMed] [Google Scholar]
- 8.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–47. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Genuardi M, Karbowski M, et al. Investigation of the substrate spectrum of the human mismatch-specific DNA N-glycosylase MED1 (MBD4): fundamental role of the catalytic domain. J Cell Physiol. 2000;185:473–80. doi: 10.1002/1097-4652(200012)185:3<473::AID-JCP19>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 10.Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B, et al. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science. 2002;297:403–5. doi: 10.1126/science.1073354. [DOI] [PubMed] [Google Scholar]
- 11.Kondo E, Gu Z, Horii A, Fukushige S. The thymine DNA glycosylase MBD4 represses transcription and is associated with methylated p16(INK4a) and hMLH1 genes. Mol Cell Biol. 2005;25:4388–96. doi: 10.1128/MCB.25.11.4388-4396.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cortázar D, Kunz C, Selfridge J, Lettieri T, Saito Y, MacDougall E, et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011;470:419–23. doi: 10.1038/nature09672. [DOI] [PubMed] [Google Scholar]
- 13.Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen D, Lucey MJ, Phoenix F, Lopez-Garcia J, Hart SM, Losson R, et al. T:G mismatch-specific thymine-DNA glycosylase potentiates transcription of estrogen-regulated genes through direct interaction with estrogen receptor alpha. J Biol Chem. 2003;278:38586–92. doi: 10.1074/jbc.M304286200. [DOI] [PubMed] [Google Scholar]
- 15.Li YQ, Zhou PZ, Zheng XD, Walsh CP, Xu GL, LI YQ Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 2007;35:390–400. doi: 10.1093/nar/gkl1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallais R, Demay F, Barath P, Finot L, Jurkowska R, Le Guével R, et al. Deoxyribonucleic acid methyl transferases 3a and 3b associate with the nuclear orphan receptor COUP-TFI during gene activation. Mol Endocrinol. 2007;21:2085–98. doi: 10.1210/me.2006-0490. [DOI] [PubMed] [Google Scholar]
- 17.Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell. 2002;9:265–77. doi: 10.1016/S1097-2765(02)00453-7. [DOI] [PubMed] [Google Scholar]
- 18.Lu C, El-Deiry WS. Targeting p53 for enhanced radio- and chemo-sensitivity. Apoptosis. 2009;14:597–606. doi: 10.1007/s10495-009-0330-1. [DOI] [PubMed] [Google Scholar]
- 19.Adimoolam S, Ford JM. p53 and regulation of DNA damage recognition during nucleotide excision repair. DNA Repair (Amst) 2003;2:947–54. doi: 10.1016/S1568-7864(03)00087-9. [DOI] [PubMed] [Google Scholar]
- 20.Hussain SP, Harris CC. p53 biological network: at the crossroads of the cellular-stress response pathway and molecular carcinogenesis. J Nippon Med Sch. 2006;73:54–64. doi: 10.1272/jnms.73.54. [DOI] [PubMed] [Google Scholar]
- 21.Bocangel D, Sengupta S, Mitra S, Bhakat KK. p53-Mediated down-regulation of the human DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) via interaction with Sp1 transcription factor. Anticancer Res. 2009;29:3741–50. [PMC free article] [PubMed] [Google Scholar]
- 22.Nishihira T, Hashimoto Y, Katayama M, Mori S, Kuroki T. Molecular and cellular features of esophageal cancer cells. J Cancer Res Clin Oncol. 1993;119:441–9. doi: 10.1007/BF01215923. [DOI] [PubMed] [Google Scholar]
- 23.Barnas C, Martel-Planche G, Furukawa Y, Hollstein M, Montesano R, Hainaut P. Inactivation of the p53 protein in cell lines derived from human esophageal cancers. Int J Cancer. 1997;71:79–87. doi: 10.1002/(SICI)1097-0215(19970328)71:1<79::AID-IJC14>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 24.Guimarães DP, Oliveira IM, de Moraes E, Paiva GR, Souza DM, Barnas C, et al. Interferon-inducible guanylate binding protein (GBP)-2: a novel p53-regulated tumor marker in esophageal squamous cell carcinomas. Int J Cancer. 2009;124:272–9. doi: 10.1002/ijc.23944. [DOI] [PubMed] [Google Scholar]
- 25.Cortes U, Moyret-Lalle C, Falette N, Duriez C, Ghissassi FE, Barnas C, et al. BTG gene expression in the p53-dependent and -independent cellular response to DNA damage. Mol Carcinog. 2000;27:57–64. doi: 10.1002/(SICI)1098-2744(200002)27:2<57::AID-MC1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 26.Guimarães DP, Hainaut P. TP53: a key gene in human cancer. Biochimie. 2002;84:83–93. doi: 10.1016/S0300-9084(01)01356-6. [DOI] [PubMed] [Google Scholar]
- 27.Minty A, Dumont X, Kaghad M, Caput D. Covalent modification of p73alpha by SUMO-1. Two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif. J Biol Chem. 2000;275:36316–23. doi: 10.1074/jbc.M004293200. [DOI] [PubMed] [Google Scholar]
- 28.Hardeland U, Steinacher R, Jiricny J, Schär P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002;21:1456–64. doi: 10.1093/emboj/21.6.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinacher R, Schär P. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr Biol. 2005;15:616–23. doi: 10.1016/j.cub.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 30.North S, Pluquet O, Maurici D, El-Ghissassi F, Hainaut P. Restoration of wild-type conformation and activity of a temperature-sensitive mutant of p53 (p53(V272M)) by the cytoprotective aminothiol WR1065 in the esophageal cancer cell line TE-1. Mol Carcinog. 2002;33:181–8. doi: 10.1002/mc.10038. [DOI] [PubMed] [Google Scholar]
- 31.Bacus SS, Yarden Y, Oren M, Chin DM, Lyass L, Zelnick CR, et al. Neu differentiation factor (Heregulin) activates a p53-dependent pathway in cancer cells. Oncogene. 1996;12:2535–47. [PubMed] [Google Scholar]
- 32.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, et al. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–42. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 33.Menendez D, Inga A, Snipe J, Krysiak O, Schönfelder G, Resnick MA. A single-nucleotide polymorphism in a half-binding site creates p53 and estrogen receptor control of vascular endothelial growth factor receptor 1. Mol Cell Biol. 2007;27:2590–600. doi: 10.1128/MCB.01742-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waters TR, Swann PF. Kinetics of the action of thymine DNA glycosylase. J Biol Chem. 1998;273:20007–14. doi: 10.1074/jbc.273.32.20007. [DOI] [PubMed] [Google Scholar]
- 35.Abu M, Waters TR. The main role of human thymine-DNA glycosylase is removal of thymine produced by deamination of 5-methylcytosine and not removal of ethenocytosine. J Biol Chem. 2003;278:8739–44. doi: 10.1074/jbc.M211084200. [DOI] [PubMed] [Google Scholar]
- 36.Hardeland U, Bentele M, Jiricny J, Schär P. The versatile thymine DNA-glycosylase: a comparative characterization of the human, Drosophila and fission yeast orthologs. Nucleic Acids Res. 2003;31:2261–71. doi: 10.1093/nar/gkg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neddermann P, Jiricny J. The purification of a mismatch-specific thymine-DNA glycosylase from HeLa cells. J Biol Chem. 1993;268:21218–24. [PubMed] [Google Scholar]
- 38.Um S, Harbers M, Benecke A, Pierrat B, Losson R, Chambon P. Retinoic acid receptors interact physically and functionally with the T:G mismatch-specific thymine-DNA glycosylase. J Biol Chem. 1998;273:20728–36. doi: 10.1074/jbc.273.33.20728. [DOI] [PubMed] [Google Scholar]
- 39.Neddermann P, Gallinari P, Lettieri T, Schmid D, Truong O, Hsuan JJ, et al. Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J Biol Chem. 1996;271:12767–74. doi: 10.1074/jbc.271.22.12767. [DOI] [PubMed] [Google Scholar]
- 40.Su HL, Li SS. Molecular features of human ubiquitin-like SUMO genes and their encoded proteins. Gene. 2002;296:65–73. doi: 10.1016/S0378-1119(02)00843-0. [DOI] [PubMed] [Google Scholar]
- 41.Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–76. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 42.Mohan RD, Rao A, Gagliardi J, Tini M. SUMO-1-dependent allosteric regulation of thymine DNA glycosylase alters subnuclear localization and CBP/p300 recruitment. Mol Cell Biol. 2007;27:229–43. doi: 10.1128/MCB.00323-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Read AP, Strachan T. Cancer Genetics. In: Read AP, Strachan T, eds. Human molecular genetics. 2nd ed. New York: Wiley- Liss Publ, 1999: 427- 44. [Google Scholar]
- 44.Chen F, Wang W, El-Deiry WS. Current strategies to target p53 in cancer. Biochem Pharmacol. 2010;80:724–30. doi: 10.1016/j.bcp.2010.04.031. [DOI] [PubMed] [Google Scholar]
- 45.Offer H, Wolkowicz R, Matas D, Blumenstein S, Livneh Z, Rotter V. Direct involvement of p53 in the base excision repair pathway of the DNA repair machinery. FEBS Lett. 1999;450:197–204. doi: 10.1016/S0014-5793(99)00505-0. [DOI] [PubMed] [Google Scholar]
- 46.Zhou J, Ahn J, Wilson SHE, Prives C. A role for p53 in base excision repair. EMBO J. 2001;20:914–23. doi: 10.1093/emboj/20.4.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seo YR, Fishel ML, Amundson S, Kelley MR, Smith ML. Implication of p53 in base excision DNA repair: in vivo evidence. Oncogene. 2002;21:731–7. doi: 10.1038/sj.onc.1205129. [DOI] [PubMed] [Google Scholar]
- 48.Achanta G, Huang P. Role of p53 in sensing oxidative DNA damage in response to reactive oxygen species-generating agents. Cancer Res. 2004;64:6233–9. doi: 10.1158/0008-5472.CAN-04-0494. [DOI] [PubMed] [Google Scholar]
- 49.de Souza-Pinto NC, Harris CC, Bohr VA. p53 functions in the incorporation step in DNA base excision repair in mouse liver mitochondria. Oncogene. 2004;23:6559–68. doi: 10.1038/sj.onc.1207874. [DOI] [PubMed] [Google Scholar]
- 50.Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nat Struct Biol. 2000;7:176–8. doi: 10.1038/82818. [DOI] [PubMed] [Google Scholar]
- 51.Chatterjee A, Mambo E, Osada M, Upadhyay S, Sidransky D. The effect of p53-RNAi and p53 knockout on human 8-oxoguanine DNA glycosylase (hOgg1) activity. FASEB J. 2006;20:112–4. doi: 10.1096/fj.04-3423fje. [DOI] [PubMed] [Google Scholar]
- 52.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 53.Greenblatt MS, Feitelson MA, Zhu M, Bennett WP, Welsh JA, Jones R, et al. Integrity of p53 in hepatitis B x antigen-positive and -negative hepatocellular carcinomas. Cancer Res. 1997;57:426–32. [PubMed] [Google Scholar]
- 54.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 55.Loeb KR, Loeb LA. Genetic instability and the mutator phenotype. Studies in ulcerative colitis. Am J Pathol. 1999;154:1621–6. doi: 10.1016/S0002-9440(10)65415-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Loeb LA. Mutator phenotype in cancer: origin and consequences. Semin Cancer Biol. 2010;20:279–80. doi: 10.1016/j.semcancer.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci USA. 2003;100:776–81. doi: 10.1073/pnas.0334858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–8. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 59.Parsons R, Li GM, Longley MJ, Fang WH, Papadopoulos N, Jen J, et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75:1227–36. doi: 10.1016/0092-8674(93)90331-J. [DOI] [PubMed] [Google Scholar]
- 60.Bhattacharyya NP, Skandalis A, Ganesh A, Groden J, Meuth M. Mutator phenotypes in human colorectal carcinoma cell lines. Proc Natl Acad Sci USA. 1994;91:6319–23. doi: 10.1073/pnas.91.14.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Missero C, Pirro MT, Simeone S, Pischetola M, Di Lauro R. The DNA glycosylase T:G mismatch-specific thymine DNA glycosylase represses thyroid transcription factor-1-activated transcription. J Biol Chem. 2001;276:33569–75. doi: 10.1074/jbc.M104963200. [DOI] [PubMed] [Google Scholar]
- 62.Zhu B, Benjamin D, Zheng Y, Angliker H, Thiry S, Siegmann M, et al. Overexpression of 5-methylcytosine DNA glycosylase in human embryonic kidney cells EcR293 demethylates the promoter of a hormone-regulated reporter gene. Proc Natl Acad Sci USA. 2001;98:5031–6. doi: 10.1073/pnas.091097298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Métivier R, Gallais R, Tiffoche C, Le Péron C, Jurkowska RZ, Carmouche RP, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 64.Kangaspeska S, Stride B, Métivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, et al. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–5. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 65.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Courtois S, Verhaegh G, North S, Luciani MG, Lassus P, Hibner U, et al. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene. 2002;21:6722–8. doi: 10.1038/sj.onc.1205874. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.