

Abstract
Autophagy is a catabolic process that sequesters intracellular proteins and organelles within membrane vesicles called autophagosomes with their subsequent delivery to lyzosomes for degradation. This process involves multiple fusions of autophagosomal membranes with different vesicular compartments; however, the role of vesicle fusion in autophagosomal biogenesis remains poorly understood. This study addresses the role of a key vesicle fusion regulator, soluble N-ethylmaleimide-sensitive factor attachment protein α (αSNAP), in autophagy. Small interfering RNA-mediated downregulation of αSNAP expression in cultured epithelial cells stimulated the autophagic flux, which was manifested by increased conjugation of microtubule-associated protein light chain 3 (LC3-II) and accumulation of LC3-positive autophagosomes. This enhanced autophagy developed via a non-canonical mechanism that did not require beclin1-p150-dependent nucleation, but involved Atg5 and Atg7-mediated elongation of autophagosomal membranes. Induction of autophagy in αSNAP-depleted cells was accompanied by decreased mTOR signaling but appeared to be independent of αSNAP-binding partners, N-ethylmaleimide-sensitive factor and BNIP1. Loss of αSNAP caused fragmentation of the Golgi and downregulation of the Golgi-specific GTP exchange factors, GBF1, BIG1 and BIG2. Pharmacological disruption of the Golgi and genetic inhibition of GBF1 recreated the effects of αSNAP depletion on the autophagic flux. Our study revealed a novel role for αSNAP as a negative regulator of autophagy that acts by enhancing mTOR signaling and regulating the integrity of the Golgi complex.
Keywords: Beclin1, NSF, mTOR, Bif-1, vesicle trafficking, Golgi disruption
Introduction
Autophagy is an evolutionarily conserved process that involves sequestration of intracellular organelles or parts of free cytoplasm within double-membrane vesicles, autophagosomes, with their subsequent delivery to lysosomes for degradation.1,2 Autophagy plays an important homeostatic role by eliminating misfolded/aggregated proteins and damaged organelles and by providing nutrients under conditions of metabolic stress.1,2 It also has specialized functions including mediation of cell death, antigen processing and unconventional protein secretion.3-5 Autophagy is usually described as a cascade of events triggered by various environmental stimuli and mediated by a number of proteins encoded by autophagy-related genes (Atgs).2,5,6 A classical autophagosome formation pathway in starved mammalian cells is initiated by inhibition of mammalian target of rapamycin (mTOR) and involves several distinct steps, such as nucleation, elongation, maturation and fusion.2,7-9 The nucleation leads to formation of pre-autophagosomal structures, isolation membranes or phagophores, which happens either de novo or from preexisting membranes of the endoplasmic reticulum (ER), Golgi, mitochondria and plasma membrane.9-15 The nucleation step is regulated by a conserved macromolecular complex composed by Vps34 phosphatidylinositol 3-kinase, beclin-1 and p150 proteins.5,7,8 The elongation of phagophores is driven by two ubiquitination-like reactions. In the first reaction, covalent binding of Atg12 and Atg5 results in the formation of a large oligomeric complex transiently associated with the phagophore.2,6 The second reaction involves covalent modification of a microtubule-associated protein light chain 3 (LC3). This protein exists as an equilibrium of two isoforms, LC3-I and LC3-II, with LC3-II resulting from the conjugation of LC3-I to phosphatidylethanolamine with assistance of Atg7.2,6 While LC3-I remains in the cytoplasm, LC3-II is specifically recruited to the elongating autophagosomal membrane, where it persists with the mature autophagosomes until fusion with lysosomes. Determination of LC3-II levels by immunoblotting or visualization of LC3-II-positive vesicles by immunofluorescence are the most reliable methods to measure autophagy in cultured cells and tissue samples.16,17
While the specific multiprotein machinery that nucleates and elongates autophagosomal membranes has been studied in great detail, the involvement of fundamental cellular processes, such as vesicle trafficking, cell adhesion and cytoskeletal remodeling in autophagic pathways remains incompletely understood. Vesicle trafficking is an emerging crucial regulator of autophagy that can control multiple steps of autophagosome biogenesis.13,15,18,19 For example, both secretory and endocytic trafficking pathways can provide membranes for autophagosomal precursors.13,15,18,19 Additionally, maturation of mammalian autophagosomes involves their fusion with different endosomal compartments, whereas delivery of autophagosomal cargo for degradation requires fusion with lysosomes.5,11,19 These fusion reactions involve assembly of classic SNARE (soluble N-ethylmaleimide-sensitive factor associated receptor) complexes. SNARE proteins are located on interacting membranes, and by participating in specific trans-interactions, they bring two membranes into close opposition and drive their fusion.20-22 Recent studies have found that several mammalian SNAREs such as syntaxins-5, 7 and 8, Sec22b, VAMP7 and Vti1b are essential for a steady-state and starvation-induced autophagy.23-26
The hexameric ATPase, N-ethylmaleimide-sensitive factor (NSF) and its adaptor soluble N-ethylmaleimide-sensitive factor-attachment protein α (αSNAP) are key regulators of SNARE-mediated vesicle fusion.20,21,27-29 NSF hydrolyzes ATP to generate the energy required for disassembly and recycling of SNARE complexes, whereas αSNAP acts as a linker and force transducer from NSF to the SNAREs. Interestingly, αSNAP has a number of additional binding partners27 and appears to play important NSF-independent functions in epithelial cells.30,31 Although a key role of NSF in regulating yeast autophagy has been recently reported,32 the involvement of αSNAP in autophagosomal biogenesis remains unexplored. The present study examines the role of αSNAP in regulating autophagy in model human epithelia. We report that loss of αSNAP induces non-conventional beclin-1-independent autophagy by mechanisms involving downregulation of mTOR signaling and desintegration of the Golgi.
Results
Depletion of αSNAP altered the autophagic flux in human epithelial cells
In order to examine the involvement of αSNAP in autophagic pathways, we downregulated expression of this protein by RNA interference. SK-CO15 human colonic epithelial cells33 were transfected with either control siRNA or two αSNAP-specific siRNA duplexes (D1 and D2) followed by measuring the cellular levels of key autophagic markers, LC3, p62 and NBR1, by immunoblotting. Both siRNA duplexes caused a dramatic decrease in the αSNAP protein level at 24–72 h post-transfection (Fig. 1A). This was accompanied by a significant increase in the amount of conjugated LC3 (LC3-II) that became evident at 48 h and was further enhanced at 72 h of αSNAP depletion (Fig. 1A). By contrast to a continuous increase in LC3 conjugation, loss of αSNAP had a biphasic effect on the expression of NBR1 and p62, proteins that bind ubiquitinated cargo and recruit it into autophagosomes.16,34 Indeed, the levels of these proteins initially decreased at 48 h post-transfection, but subsequently were elevated after 72 h of αSNAP knockdown (Fig. 1A). To rule out the impact of altered transcription on the observed changes in the protein markers of autophagy, we next examined the mRNA expression of LC3, p62 and NBR1. A real-time RT-PCR analysis did not find effects of αSNAP depletion on the expression of LC3 and NBR1 mRNAs (Fig. S1). In contrast, the p62 mRNA level was significantly increased after 72 h of αSNAP knockdown (Fig. S1). This result indicates that p62 expression can be regulated by several mechanisms and thereby cannot be considered a reliable marker of the autophagy pathway in αSNAP-depleted epithelial cells.

Figure 1. siRNA-mediated downregulation of αSNAP alters the autophagic flux. (A) SK-CO15 cells were transfected with either control or two different αSNAP-specific siRNA duplexes (D1 and D2). Expression of αSNAP and autophagic markers, LC3, NBR1 and p62 in total-cell lysates was determined by immunoblotting. (B) siRNA depletion of αSNAP was performed in either control SK-CO15 cells (SK-neo) or cells with stable expression of siRNA-resistant bovine αSNAP (SK-αSNAP). Expression of αSNAP and autophagic markers in cell lysates was determined by immunoblotting 48 h post-transfection. (C and D) HeLa-GFP-LC3 cells were transfected with either control or αSNAP-specific siRNAs and formation of autophagosomes was visualized by confocal microscopy analysis of GFP fluorescence in fixed cells 72 h post-transfection. Data in this and other figures are presented as mean ± SEM of three independent experiments. *p < 0.001 compared with control siRNA-transfected cells. Scale bar, 20 µm.
To ensure that the observed changes in the autophagic flux following αSNAP knockdown did not represent off-target effects of siRNAs, we performed rescue experiments involving overexpression of bovine αSNAP, which lacks complementation for human siRNA sequences. Remarkably, expression of this siRNA-resistant protein completely reversed the increase in LC3 conjugation and decrease in NBR1 levels caused by αSNAP knockdown (Fig. 1B). These results strongly suggest that altered expression of autophagic markers represents a specific consequence of αSNAP depletion in SK-CO15 cells.
We next asked if loss of αSNAP can induce formation of autophagosomes. We used HeLa cells expressing GFP-labeled LC3, which enables visualization of autophagosomes in situ.16 Similarly to SK-CO15 cells, knockdown of αSNAP in HeLa-GFP-LC3 cells enhanced LC3 conjugation at 48–72 h post-transfection (Fig. S2A). Examination of these cells by fluorescence microscopy revealed dramatic changes in the intracellular distribution of labeled LC3. Indeed, the majority of control siRNA-treated cells demonstrated diffused GFP-LC3 labeling, while more than 80% of αSNAP-depleted cells showed a prominent dot-like GFP-LC3 labeling pattern (Fig. 1C, arrows, and Fig. 1D). These GFP-LC3-positive dots accumulated NBR1 and p62 (Fig. S2B, arrows), thereby demonstrating typical features of autophagosomes.
It is well-recognized that elevated LC3-II expression and increased number of autophagosomes can be a common consequence of two opposite events: increased autophagic flux or decreased autophagy due to inhibition of lysosomal degradation of autophagosomal markers.17 To distinguish between these possibilities, we used known pharmacologic inhibitors of lysosomal functions, bafilomycin A and chloroquine. Only if autophagy is activated in αSNAP-depleted cells will lysosome inhibition be expected to further increase LC3-II level and the number of autophagosomes.17 SK-CO15 cells were transfected with control or αSNAP-specific siRNAs and, 48 h post-transfection, were incubated for an additional 6 h with either vehicle, bafilomycin A (0.2 µM) or chloroquine (100 µM). Immunoblotting analysis showed that both lysosomal inhibitors significantly increased the amount of conjugated LC3 not only in control, but also in αSNAP-depleted cells (Fig. 2A and B). Thus, bafilomycin A and chloroquine treatment increased the LC3-II/LC3-I ratio in αSNAP-depleted cells from 3.5 to 10 and 14, respectively (Fig. 2B). Similarly, inhibition of lysosomal functions in αSNAP-depleted HeLa-GFP-LC3 cells markedly increased formation of autophagosomes that became fused into large perinuclear clusters (Fig. 2C, arrows). These data strongly suggest that loss of αSNAP stimulates autophagy in human epithelial cells at least at early times of the knockdown. It is still possible that the autophagic flux could be inhibited at late times of αSNAP depletion, which would explain the elevated level of NBR1 in SK-CO15 cells at 72 h post-transfection (Fig. 1A). However, in subsequent experiments we focused on mechanisms of the early induction of autophagy in αSNAP-deficient epithelia.

Figure 2. Lysosomal inhibitors exaggerate LC3 conjugation and accumulation of autophagosomes in αSNAP-depleted epithelial cells. SK-CO15 (A and B) or HeLa-GFP-LC3 (C) cells transfected with either control or αSNAP duplex 1 siRNAs were treated for 4 h with either vehicle or lysosomal inhibitors bafilomycin A (0.2 µM) or chloroquine (100 µM). Expression of LC3-II was determined in SK-CO15 cells by immunoblotting, whereas formation of autophagosomes was examined in HeLa-GFP-LC3 cells by fluorescence microscopy. **p < 0.05 compared with the vehicle-treated αSNAP-depleted cells. Scale bar, 20 µm.
Induction of autophagy in αSNAP-depleted cells was associated with inhibition of mTOR signaling and required the elongation but not the nucleation steps of autophagosome formation
To gain insights into molecular mechanisms of elevated autophagy in αSNAP-deficient epithelia, we analyzed which step of autophagosome biogenesis was affected. Specifically, the roles of upstream mTOR signaling, beclin-1-dependent nucleation and Atg5/Atg7-mediated elongation steps of the autophagic cascade were investigated. Immunoblotting analysis revealed decreased mTOR expression in SK-CO15 cells following αSNAP knockdown (Fig. 3). According to densitometric quantification, such expression was downregulated approximately 80% by 72 h post-transfection. Consistently, phosphorylation of downstream mTOR targets, p70 S6 kinase and translational inhibitor 4E-BP1, was diminished in αSNAP-depleted cells; total 4E-BP1 protein levels also decreased (Fig. 3). Collectively, these data highlight inhibition of mTOR signaling as a prominent consequence of αSNAP knockdown.
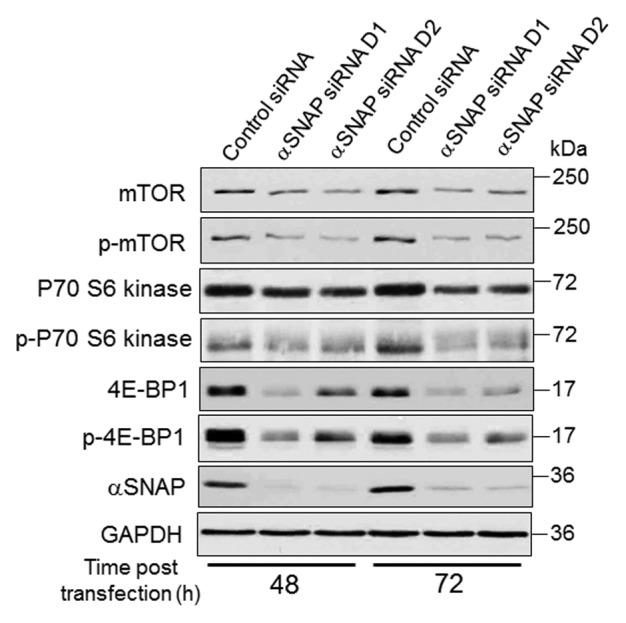
Figure 3. Loss of αSNAP decreases expression of mTOR and its downstream effector, 4E-BP1. SK-CO15 cells were transfected with either control or αSNAP-specific siRNAs. Expression and phosphorylation of mTOR and its downstream effectors p70 S6 kinase and 4E-BP1 in total-cell lysates was determined at different times post-transfection.
A canonical mechanism of nucleation of autophagosomal precursors depends on beclin-1, which interacts with adaptor protein p150 to stimulate activity of PI3 kinase VPS34.5,6,8,35 Therefore, we investigated if the beclin-1-p150-dependent nucleation step can mediate enhanced autophagy in αSNAP-depleted epithelial cells. Surprisingly, co-knockdown of αSNAP with either beclin-1 or p150 in SK-CO15 cells failed to prevent the increase in LC3-II level triggered by αSNAP depletion (Fig. S3A and B). Furthermore, downregulation of either beclin-1 or p150 did not affect accumulation of autophagosomes in αSNAP-deficient HeLa-GFP-LC3 cells (Fig. S3C and D). Collectively, these results suggest that loss of αSNAP triggered an unconventional autophagy that bypassed the beclin-1/p150-dependent nucleation of autophagosomal precursors.
The elongation of precursor membranes driven by Atg5 and Atg7-mediated conjugation reactions represents a key step in the formation of autophagic vesicles.5,6,8,35 Since this elongation step regulates the autophagic flux in many, although not all,36 experimental conditions, we examined if the Atg5 and Atg7 proteins play roles in the induction of autophagy in αSNAP-deficient cells. As expected, co-transfection of either Atg5 siRNA or Atg7 siRNA with control siRNAs decreased basal expression of LC3-II in SK-CO15 cells (Fig. 4A and B). Furthermore, dual Atg5/αSNAP or Atg7/αSNAP knockdowns significantly attenuated the increase in LC3-II levels as compared with αSNAP depletion alone (Fig. 4A and B). In HeLa-GFP-LC3 cells, co-knockdown of Atg5 and αSNAP dramatically decreased the number of cells with autophagosomes when compared with the experimental group co-transfected with control and αSNAP siRNAs (Fig. 4C and D). These data strongly suggest that Atg5 and Atg7 mediate accelerated autophagic flux resulting from loss of αSNAP.
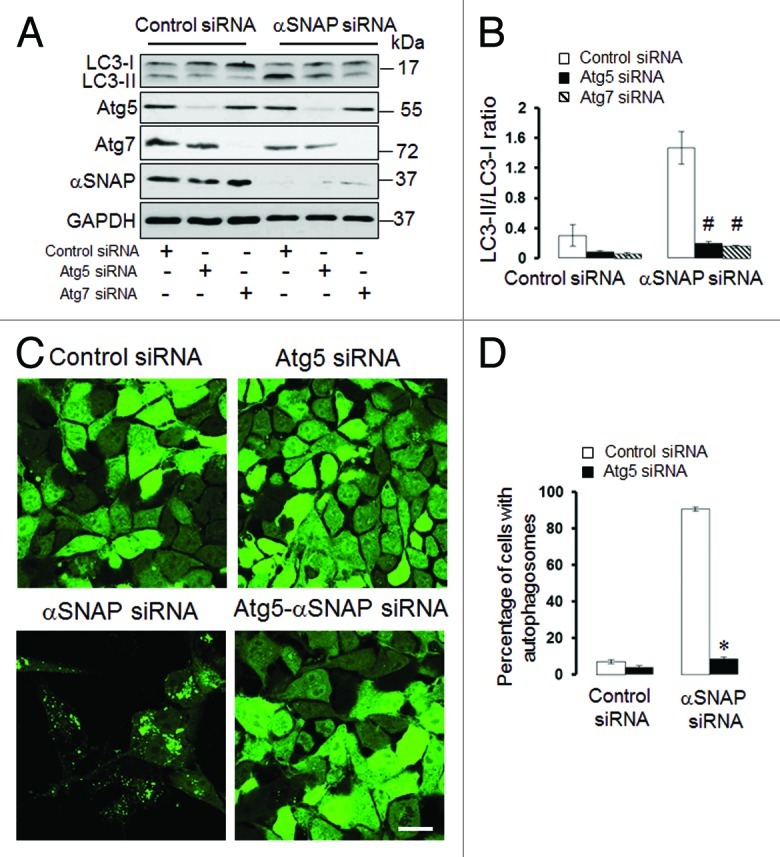
Figure 4. Atg5 and Atg7 play roles in induction of autophagy caused by downregulation of αSNAP. (A and B) SK-CO15 cells were subjected to sequential transfections with one of the following siRNA pairs: control-control, control-Atg5, control-Atg7, control-αSNAP, Atg5-αSNAP or Atg7-αSNAP. Expression of LC3, αSNAP, Atg5 and Atg7 was determined by immunoblotting 48 h after the second transfection. #p < 0.01 compared with control-αSNAP siRNA-transfected cells. (C and D) HeLa-GFP-LC3 cells were sequentially transfected with control-control, control-Atg5, control-αSNAP and Atg5-αSNAP siRNA combinations and formation of autophagosomes was analyzed by fluorescence spectroscopy at 72 h after the second transfection. *p < 0.001 compared with control-αSNAP siRNA-transfected cells. Scale bar, 20 µm.
Autophagy induction in αSNAP-depleted epithelia is independent from NSF and is associated with fragmentation of the Golgi
Since the best-known cellular activity of αSNAP is to assist NSF ATPase in disassembling post-fusion SNARE complexes,27,28 we asked if these binding partners also cooperate in regulating autophagy. Using a specific siRNA SmartPool, we dramatically decreased NSF expression in SK-CO15 without affecting the αSNAP protein level (Fig. S4A). In contrast to αSNAP knockdown, loss of NSF did not alter LC3 conjugation and did not decrease expression of NBR1 (Fig. S4A and B). Likewise, depletion of NSF did not induce formation of autophagosomes in HeLa-GFP-LC3 cells (Fig. S4C and D). These experiments indicate that NSF does not act as a negative regulator of the autophagic flux in epithelial cells.
We previously demonstrated that loss of αSNAP impaired vesicle trafficking between the endoplasmic reticulum (ER) and the Golgi, causing fragmentation of the Golgi.30,31 Since several recent studies suggested that the Golgi can supply membranes to the autophagic pathway,12,14,37-39 we investigated if disintegration of this organelle can contribute to the increased autophagic flux in αSNAP-deficient cells. Using HeLa-GFP-LC3 cells, we observed that formation of autophagosomes after αSNAP knockdown occurred in parallel to fragmentation of both cys and trans-Golgi compartments as visualized by giantin and TGN46 labeling, respectively (Fig. 5, arrows). Interestingly, these Golgi markers were detected in newly formed autophagic vesicles (Fig. 5, inserts), which indicates that fragmented Golgi can provide material to create autophagosomal membranes in αSNAP-depleted cells. If this suggestion is correct, one would expect that Golgi disruption would be sufficient to accelerate autophagy. To test this, we used two pharmacologic agents, Brefeldin A (BFA) and Golgicide A (GA), that are known to inhibit ER to Golgi vesicle trafficking and induce Golgi fragmentation.40,41 Treatment with either BFA (2 µm) or GA (50 µm) for 24 h caused a marked increase in LC3 conjugation in SK-CO15 cells (Fig. 6A and B) and triggered formation of autophagosomes in HeLa-GFP-LC3 cells (Fig. 6C and D, arrows). BFA is known to disrupt the Golgi by inhibiting three guanine nucleotide exchange factors (GEFs) for Arf small GTPases, GBF1, BIG1 and BIG2,42-44 whereas GA is a selective GBF1 inhibitor.41 On the other hand, we recently found that knockdown of epithelial αSNAP downregulated expression of all BFA-sensitive GEFs.30 Therefore, we asked if loss of Golgi-resident GEFs can be responsible for the increased autophagy following αSNAP depletion. First, we confirmed decreased expression of GBF1, BIG1 and BIG2 in αSNAP-deficient SK-CO15 cells (Fig. 7A) and also observed a disappearance of the GBF1 labeling (Fig. 7B, arrowheads) characteristic of intact Golgi under these experimental conditions (Fig. 7B, arrows). Next, we examined the effects of a selective depletion of GBF1 or simultaneous downregulation of BIG1 and BIG2. Immunoblotting analysis revealed a dramatic (> 90%) and specific decrease in expression of the targeted GEF in SK-CO15 cells at 48–72 h post-siRNA transfection (Fig. 8A). Interestingly, GBF1 knockdown increased the LC3-II levels, whereas co-knockdown of BIG1 and BIG2 did not affect LC3 conjugation (Fig. 8A and B). Similarly, GBF1 but not BIG1+BIG2 knockdown triggered a marked formation of autophagosomes in HeLa-GFP-LC3 cells (Fig. 8C and D). Together, these results implicate the Golgi fragmentation in αSNAP-dependent induction of autophagy in epithelial cells and indicate that downregulation of Golgi-resident GBF1 can contribute to this process.
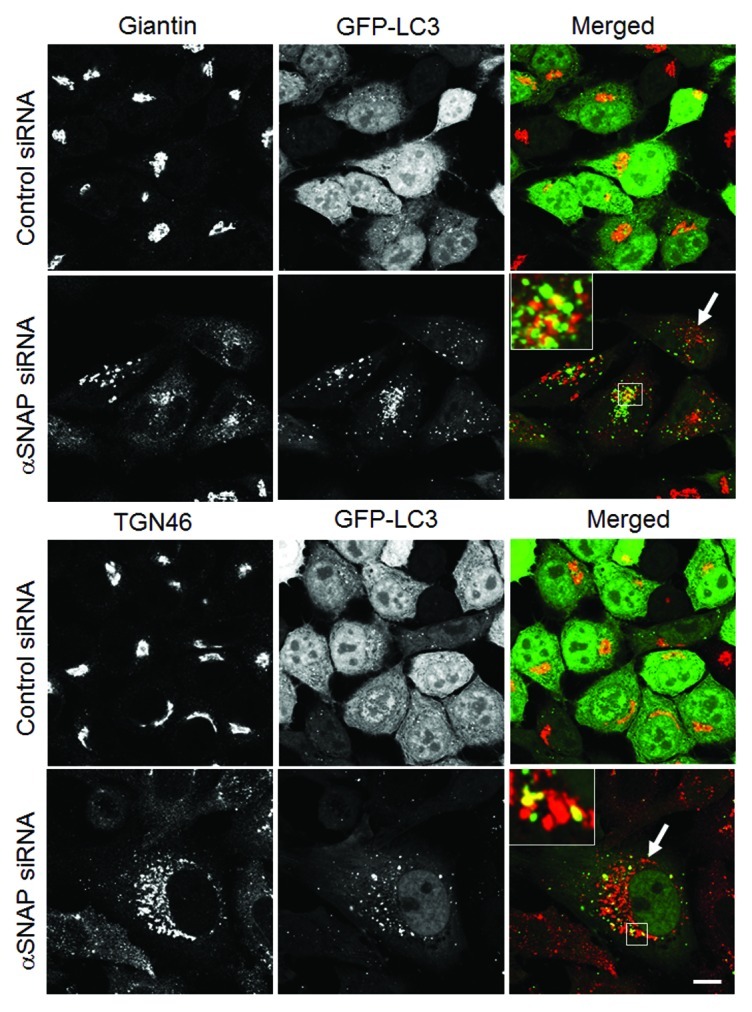
Figure 5. Loss of αSNAP triggers fragmentation of the Golgi in parallel to autophagy induction. Control and αSNAP-depleted HeLa-GFP-LC3 cells were immunofluorescence labeled for Golgi markers Giantin and TGN46 (red) at 72 h post-transfection. Control cells are characterized by the compact perinuclear Golgi complex, whereas αSNAP depletion results in a dramatic fragmentation of the Golgi (arrows) and appearance of Golgi markers in LC3-positive autophagosomes (inserts). Scale bar, 10 µm.

Figure 6. Pharmacological disruption of the Golgi stimulates autophagy. SK-CO15 (A and B) and HeLa-GFP-LC3 (C and D) cells were treated for 24 h with either vehicle, Brefeldin A (2 µM) or Golgicide A (50 µM), and expression of LC3-II and accumulation of autophagosomes were determined by immunoblotting and fluorescence microscopy, respectively. *p < 0.001 compared with the vehicle-treated group (n = 3). Scale bar, 20 µm.
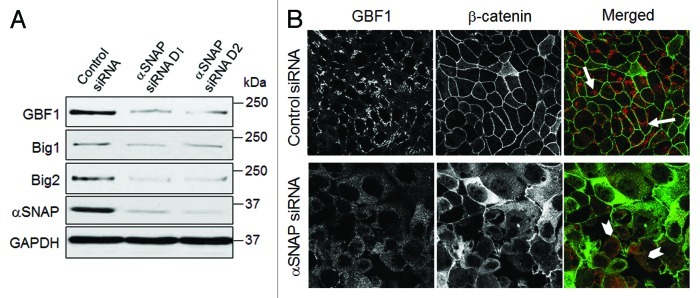
Figure 7. Loss of αSNAP decreases expression of Brefeldin-sensitive guanine nucleotide exchange factors. (A) Expression of Brefeldin-sensitive exchange factors GBF1, BIG1 and BIG2 was examined in control and αSNAP-depleted SK-CO15 cells 48 h after siRNA transfection. (B) Effect of αSNAP knockdown on localization of GBF1 (red) in SK-CO15 cells was analyzed by immunofluorescence labeling and confocal microscopy. Scale bar, 20 µm.

Figure 8. Depletion of GBF1, but not other Brefeldin-sensitive exchange factors, accelerates epithelial cell autophagy. (A and B) SK-CO15 cells were transfected with either control GBF1 or a combination of BIG1 and BIG2 siRNAs, and expression of targeted proteins and LC3 was examined at two different times post-transfection. (C and D) HeLa-GFP-LC3 cells were transfected with either control GBF1 or BIG1, plus BIG2 siRNAs, and formation of autophagosomes was analyzed by fluorescence microscopy. *p < 0.001 compared with control siRNA-transfected cells. Scale bar, 20 µm.
Bif-1 but not BNIP1 is involved in αSNAP-dependent autophagy
Since our results highlight the dispersed Golgi as a possible source of autophagosomal precursors in αSNAP-depleted epithelia, we sought to identify molecular mechanisms that mediate transformation of Golgi ciscernae into autophagic vesicles. One such mechanism may involve Bif-1, a membrane-curving protein that is known to be located at the TGN and to mediate autophagosome formation from the fragmented TGN in starved cells.14 To probe the role of Bif-1, we decreased its expression in epithelial cells and analyzed the effects of Bif-1 depletion on αSNAP-dependent autophagy. Immunoblotting analysis revealed that dual Bif-1/αSNAP knockdown in SK-CO15 cells significantly attenuated the increased LC3 conjugation as compared with downregulation of αSNAP alone (Fig. 9A and B). Likewise, depletion of Bif-1 inhibited formation of autophagosomes in αSNAP-depleted HeLa-GFP-LC3 cells (Fig. 9C and D).

Figure 9. Bif-1 is involved in the enhanced autophagy caused by downregulation of αSNAP. SK-CO15 (A and B) and HeLa-GFP-LC3 cells were subjected to sequential transfections with one of the following siRNA pairs: control-control, control-Bif-1, control-αSNAP and Bif-1-αSNAP. Levels of LC3 and Bif-1, as well as accumulation of autophagosomes, were determined by immunoblotting and fluorescence microscopy, respectively. #p < 0.01 compared with the control-αSNAP siRNA-transfected group. Scale bar, 20 µm.
An additional mechanism of autophagy induction in αSNAP-deficient cells may involve a known αSNAP binding partner, BNIP1.45 BNIP1 is a member of the pro-apoptotic BH3-only protein family that localizes to the ER and Golgi and mediates vesicle trafficking between these compartments.46 Since a novel role for BNIP1 as a positive regulator of autophagy has been recently suggested,47 we hypothesized that loss of αSNAP would stimulate the pro-autophagic activity of this protein. To test this hypothesis, we examined the effects of BNIP1 knockdown on autophagy in control and αSNAP-deficient cells. In contrast to our prediction, we found that loss of BNIP1 significantly increased LC3 conjugation in control SK-CO15 cells (Fig. 10A and B). Furthermore, a dual knockdown of BNIP1 and αSNAP resulted in a higher LC3-II/LC3-I ratio as compared with αSNAP depletion alone (Fig. 10A and B). In control HeLa-GFP-LC3 cells, loss of BNIP1 caused accumulation of autophagosomes (Fig. 10C, arrows, and Fig. 10D), thereby suggesting that BNIP1 acts as a negative regulator of autophagy in epithelial cells. We also found that knockdown of αSNAP did not alter BNIP1 expression (Fig. 10A) and BNIP1 was absent in newly formed autophagosomes in αSNAP-depleted cells (Fig. 10E). Collectively, these results argue that BNIP1 does not mediate enhanced autophagy in αSNAP-deficient epithelia.

Figure 10. Enhanced autophagy in αSNAP-depleted epithelial cells does not depend on BNIP1. (A and B) SK-CO15 cells were subjected to sequential transfections with one of the following siRNA pairs: control-control, control-BNIP1, control-αSNAP and BNIP1-αSNAP. Expression of targeted proteins and autophagic markers was determined by immunoblotting at 48 h after the second transfection. #p < 0.01 compared with control siRNA-transfected cells. (C and D) HeLa-GFP-LC3 cells were transfected with either control or BNIP1-specific siRNAs and accumulation of autophagosomes was monitored by fluorescence spectroscopy. Scale bar, 20 µm; *p < 0.001 compared with control siRNA-transfected cells. (E) HeLa-GFP-LC3 cells subjected to αSNAP depletion were immunolabeled for BNIP1 (red) at 72 h post-transfection. Scale bar, 10 µm.
Autophagy and apoptosis represent two independent functional consequences of αSNAP knockdown
We have previously shown that knockdown of αSNAP has multiple effects on cultured epithelial cells, including induction of apoptosis.31 Autophagy and apoptosis appeared to be orchestrated in SK-CO15 cells, as they occur simultaneously at 48 h of αSNAP knockdown (Fig. 1A). Since both pro-death and pro-survival roles of autophagy have been observed under different experimental conditions,3,48,49 we were interested in the possible functional interplay between autophagy and apoptosis in αSNAP-depleted epithelia. We inhibited autophagy by siRNA-mediated depletion of either Atg5 or Atg7 and analyzed the effects of such inhibition on apoptosis caused by loss of αSNAP. Surprisingly, a dual knockdown of Atg proteins and αSNAP did not attenuate induction of apoptotic markers, such as cleaved PARP, active caspase 3 and Annexin V/Propidium iodide (PI) labeling, as compared with SK-CO15 cells subjected to αSNAP knockdown alone (Fig. S5). Furthermore, blocking apoptosis with a pan-caspase inhibitor, Z-VAD-fmk, did not affect LC3 conjugation in αSNAP-depleted cells (data not shown). Together, these results strongly suggest that induction of autophagy and apoptosis represent two independent effects of αSNAP depletion in human epithelia.
Discussion
Intracellular vesicle trafficking is considered a crucial regulator of autophagy controlling multiple steps of autophagosome biogenesis. A number of vesicle trafficking proteins have been shown to act as positive regulators of autophagy, since their depletion inhibited the autophagic flux.25,26,32,38,50 In the present study, we describe an unusual example of negative regulation of autophagy by a common membrane fusion protein, αSNAP. This anti-autophagic activity was uncovered by siRNA-mediated depletion of αSNAP, which resulted in enhanced LC3 conjugation, decreased NBR1 level and accumulation of autophagic vesicles (Fig. 1), altogether reflecting the increase in the autophagic flux (Fig. 2).
Our data indicate that induction of autophagy in αSNAP-depleted epithelia involved a non-canonical molecular pathway. This pathway required the classic Atg5-Atg7 conjugation machinery (Fig. 4), but was independent of beclin-1 and its adaptor, p150 (Fig. S3). The beclin-1-p150-Vsp34 complex is considered as a core component of the autophagy machinery that drives de novo formation (nucleation) of autophagosomal membranes.5,6,8,35 Nevertheless, a number of studies have described beclin-independent mechanisms of autophagy induction.51 For example, this non-canonical pathway was shown to be activated by reactive oxygen species,52 resveratrol,53 mitochondrial toxins54 and bacterial products.55 So far no common theme has emerged to explain mechanisms of different types of non-canonical autophagy, and it remains to be investigated if the beclin-1-independent pathway activated by loss of αSNAP bears similarities with previously described unconventional autophagic events.
The beclin-1-independent nature of autophagy in αSNAP-depleted cells implies that under these conditions, autophagosomal biogenesis bypasses the nucleation step, and that Atg proteins were recruited to the preexisting membranes to drive the subsequent maturation of autophagosomes. Our data suggest that these membranes are likely to have originated from the dispersed Golgi. Indeed, we found that loss of αSNAP triggered Golgi fragmentation in parallel to autophagy induction and that newly formed autophagosomes contained Golgi markers (Fig. 5). Furthermore, either pharmacological or genetic inhibition of Golgi-resident exchange factors for Arf GTPases resulted in orchestrated Golgi fragmentation and increased epithelial cell autophagy (Figs. 6–8). These findings are in good agreement with previous studies that highlighted the Golgi as a crucial regulator of autophagy. For example, knockout of several proteins that mediate intra-Golgi or ER-Golgi vesicle trafficking inhibited starvation- or rapamycin-induced autophagy in yeasts.37,38,56,57 Interestingly, Golgi appeared to be involved in expansion of the yeast phagophore, but was dispensable for the nucleation of this autophagosomal precursor.39 In mammalian cells, the Golgi-resident proteins Rab32 and Rab33B were implicated in basal- and starvation-induced autophagy.11,50,58 Furthermore, mammalian Atg9 protein was localized in the TGN under nutrient-rich conditions, but was relocated from the dispersed Golgi to autophagosomes during nutrient deprivation.59 Finally, inhibition of Bif-1-mediated fission of Golgi membranes was shown to attenuate starvation-induced autophagy.14 In the present study, loss of Bif-1 also attenuated autophagosome formation in αSNAP-depleted epithelial cells (Fig. 9), thereby revealing the role of Bif-1-dependent fission of the Golgi membranes in this enhanced autophagic pathway.
Since our results suggest a functional link between Golgi fragmentation and induction of autophagy in αSNAP-depleted epithelia, it is important to understand how loss of this membrane fusion protein can disrupt the Golgi architecture. αSNAP is known to be associated with two distinct SNARE complexes involving either syntaxin-560,61 or syntaxin-1846,62 that, respectively, mediate the anterograde or retrograde vesicle trafficking between the ER and the Golgi.63 It is expected, therefore, that depletion of αSNAP would inhibit SNARE-mediated fusion events at the Golgi and the ER, resulting in structural and functional impairments of these organelles. However, it is unclear if inhibition of Golgi/ER SNAREs alone is sufficient to stimulate autophagy given the recent data that syntaxin-5 knockdown interrupted the autophagic flux in HeLa cells.26 However, αSNAP depletion is likely to have deeper effects on Golgi biogenesis by also causing downregulation of the expression of Golgi-associated GEFs such as GBF1, BIG1 and BIG2 (Fig. 7). These GEFs are known to activate Arf GTPases, which are essential for the formation of COPI-coated vesicles.64,65 As a result, loss of αSNAP impairs two critical trafficking steps at the Golgi, viz, Arf-dependent assembly of coated vesicles and SNARE-mediated vesicle fusion. Furthermore, decreased expression of Golgi GEFs can stimulate autophagy via trafficking independent mechanisms that involve inactivation of mTOR signaling. This suggestion is based on a recent study that implicated Arf1 in the regulation of mTOR activity.66 Interestingly, our results point to defective mTOR signaling in αSNAP-depleted epithelial cells based on the decreased expression of mTOR and its downstream target, 4E-BP1 (Fig. 3). Since both mTOR and 4E-BP1 are known suppressors of autophagy,5,8,67 their simultaneous downregulation would generate powerful pro-autophagic signals. These upstream signals combined with the increased membrane supply from the fragmented Golgi are likely to play major roles in stimulating the autophagic pathway in αSNAP-deficient cells. At present, we do not understand how loss of αSNAP results in the decreased expression of mTOR or 4E-BP1 as well as some other proteins (Bcl-2 and p120-catenin) described in our previous publications.30,31 Future studies are required to address this important question.
Surprisingly, the observed effects of αSNAP on autophagy appear to be independent of its major binding partner, NSF. Indeed, these two proteins did not regulate each other’s expression, and loss of NSF had no effect on LC3 conjugation and autophagosome formation in human epithelial cells (Fig. S4). A current paradigm that considers NSF as an important regulator of autophagosomal biogenesis is based on observations in yeast cells where loss of NSF (Sec18p) either impaired formation of autophagosomes32 or interrupted their fusion to the vacuole.68 Evidence supporting the role of NSF in mammalian autophagy relies exclusively on the experiments with pharmacological inhibition of this ATPase by N-ethylmaleimide.25 However N-ethylmaleimide is known to promiscuously modify the free thiol groups of many cellular proteins and, as such, is a far less selective tool than the siRNA-mediated NSF knockdown used in the present study. Our results strongly suggest that NSF is not an obligate regulator of mammalian autophagy. They reinforce recently published data that other cellular functions of αSNAP, such as regulation of cell-cell adhesions and cell survival, do not depend on its interactions with NSF.30,31
What is the biological significance of the enhanced autophagy in αSNAP-depleted cells? According to our results, it does not counteract the simultaneously induced apoptosis (Fig. S5) and therefore cannot be considered as an adaptive pro-survival response. We speculate that such autophagy linked to fragmentation of the Golgi can mimic events occurring during epithelial colonization by intracellular pathogens. It is well recognized that many bacteria and viruses (Coxiella, Listeria, Legionella, Chlamidya, poliovirus, HIV, etc.) can subvert host cell autophagy to either create a protective intracellular niche or provide nutrient supply for their replication.69,70 On the other hand, some of autophagy-hijacking pathogens can also perturb ER-Golgi trafficking,71,72 and they very likely use membranes generated from these organelles for creating their own autophagic compartments. It would be interesting to investigate if intracellular pathogens can modulate αSNAP activity in order to disperse the Golgi and induce autophagy during mucosal infection. It should be noted that both SK-CO15 and HeLa cell lines used in our study are tumor-derived cells, and they may have different mechanisms of autophagy regulation compared with non-transformed epithelial cells. However, we found that loss of αSNAP induced Golgi fragmentation and altered the autophagic flux in non-tumorigenic 293 human embryonic kidney cells (data not shown). This finding indicates that the observed regulatory mechanism is not a peculiar response of cancer cells. Furthermore, it would be interesting to know if αSNAP can regulate autophagy in vivo. The existence of viable αSNAP hypomorphic hyh mice73 provides the opportunity for future studies to address this important question.
In summary, our study reveals a novel role for αSNAP as a negative regulator of epithelial cell autophagy. This regulatory role involves multiple mechanisms, most notably the inhibition of mTOR-related signaling and αSNAP-dependent control of the Golgi integrity, which limits membrane supply for autophagosomal biogenesis. This novel function of αSNAP could be essential for maintenance of normal epithelial homeostasis and control of mucosal inflammation and infection.
Materials and Methods
Antibodies and chemicals
The following primary monoclonal (mAb) and polyclonal antibodies (pAb) were used to detect the signaling and autophagy-related proteins: anti-αSNAP, p150, NBR1 and GFP mAbs (Abcam); anti-cleaved PARP, active caspase-7, Atg5, Atg7, LC3, beclin-1, phospho-mTOR, p70 S6 kinase, phospho-p70 S6 kinase. 4E-BP1, phospho-4E-BP1 pAbs and anti-GAPDH mAb (Cell Signaling Technology); anti-NSF, BNIP1, p62, GBF1, mTOR and β-catenin mAbs (BD Biosciences); anti-giantin pAb (Covance); anti-Bif-1 mAb (Imgenex), anti BIG1 and BIG2 pAb (Bethyl Laboratories, Inc.) and anti-TGN46 pAB (AbD Serotec). Horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse secondary antibodies were purchased from Jackson Immunoresearch Laboratories. Alexa-488 or Alexa-555 dye conjugated donkey anti-rabbit and goat anti-mouse secondary antibodies were obtained from Invitrogen. Brefeldin A, Golgicide A, bafilomycin A and chloroquine were obtained from Sigma-Aldrich.
Cell culture
SK-CO15 (a gift from Dr. Enrique Rodriguez-Boulan, Weill Medical College of Cornell University) and HeLa-LC3-GFP cells (a gift from Dr. Ramnik Xavier, Massachusetts General Hospital) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% of fetal bovine serum. SK-CO15 cells with stable overexpression of bovine αSNAP and their appropriate controls were generated as described elsewhere.30 For biochemical and immunolabeling experiments, the cells were cultured on 6-well plastic plates and collagen-coated coverslips, respectively.
Immunoblotting
Cells were homogenized in a radioimmunoprecipitation lysis buffer [20 mM Tris, 50 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1% sodium deoxycholate, 1% Triton X-100 (TX-100) and 0.1% SDS, pH 7.4], containing a protease inhibitor cocktail (1:100, Sigma) and phosphatase inhibitor cocktails 1 and 2 (both at 1:200, Sigma). Lysates were cleared by centrifugation, diluted with 2x SDS sample buffer and boiled. SDS-PAGE and immunoblotting were conducted by standard protocols with an equal amount of total protein (10 or 20 μg) per lane. Protein expression was quantified by densitometry of three immunoblot images, each representing an independent experiment, with a Kodak Image Station 2000R and Kodak Molecular Imaging software V 4.0 (Eastman Kodak). Data are presented as normalized values assuming the expression levels in control siRNA-treated groups were at 100%. Statistical analysis was performed with raw densitometric data using the Microsoft Excel program.
Immunofluorescence labeling
To examine formation of autophagosomes, HeLa-GFP-LC3 cells were fixed in 4% paraformaldehyde and mounted on slides without permeabilization. For colocalization analysis, cells were in 4% paraformaldehyde followed by Triton X-100 permeabilization and were immunolabeled for the Golgi and autophagy markers as described previously.74-76 Labeled cells were examined using an Olympus FluoView 1000 confocal microscope (Olympus America). The Alexa Fluor 488 and 555 signals were imaged sequentially in frame-interlace mode to eliminate crosstalk between channels. The images were processed using the Olympus FV10-ASW 2.0 Viewer software and Adobe Photoshop. All images were obtained at room temperature using UPLAN APO oil x100 objective with an N.A. of 1.4 in H2O, and immersion oil Type F (Olympus) with a refractive index of 1.518. Images shown are representative of at least three experiments, with multiple images taken per slide. Induction of autophagy was quantified by manually counting HeLa-GFP-LC3 cells with more than 10 autophagic puncta per cell and expressing their number as a percentage of all cells in the microscopic field. A minimum 116 cells from three independent experiments were counted in each experimental group.
RNA interference
Small interfering (si) RNA-mediated knockdown of αSNAP, NSF, beclin-1, p150, Atg5, Atg7, BIG1, BIG2, GBF1, BNIP1 and Bif-1 was performed as previously described.31,74,76 Individual siRNA duplexes GAAGGUGGCUGGUUACGCU (duplex 1) and CAGAGUUGGUGGACAUCGA (duplex 2; Dharmacon) were used to downregulate αSNAP expression, whereas knockdown of other targets was performed by using gene-specific siRNA SmartPools (Dharmacon). A noncoding siRNA duplex-2 (Dharmacon) served as a control. SK-CO15 cells were transfected using the DharmaFect 1 while HeLa LC3-GFP cells were transfected with DharmaFect 3 reagent (Dharmacon) in Opti-MEM I medium (Invitrogen) according to the manufacturer’s protocol with a final siRNA concentration of 50 nM. For dual knockdown, cells were first transfected with either BNIP1 or Bif-1 or Atg siRNAs, then cultured for 12 h in complete DMEM followed by the second transfection with αSNAP siRNA, duplex 1. Cells were analyzed 48–72 h after the second transfection.
Quantitative real-time RT-PCR
Total RNA was isolated using the RNeasy Mini kit (Qiagen) followed by DNase treatment to remove traces of genomic DNA. Total RNA (1 µg) was reverse transcribed into cDNA using an iScript cDNA synthesis kit (Bio-Rad Laboratories). Quantitative real-time PCR was performed with 2 µl cDNA per reaction using IQ SYBR Green Supermix (Bio-Rad) and Opticon™ DNA Engine Opticon thermocycler (Bio-Rad Laboratories). The levels of gene expression in each sample were determined with the comparative cycle threshold method. The following primers were used: human GAPDH (NM_002046.3) forward-CACCCACTCCTCCACCTTTG reverse-CCACCACCCTGTTGCTGTAG; NBR1 (NM_031862.2) forward GCAGACAGAAGAGCTATGAC, reverse CAAGGGCTTCTATCCCATAC, LC-3B (NM_022818) forward TATCGCCAGAGTCGGATTCG reverse TGCTTCTCACCCTTGTATCG, p62/SQSTM1 (NM_003900) forward TCCAGTGACGAGGAATTGAC reverse AGCCATCGCAGATCACATTG.
Flow cytometry analysis
Control and αSNAP-depleted cells were trypsinized, pooled together with spontaneously detached cells and stained for Annexin-V and propidium iodide using a BD Biosciences Apoptosis Detection Kit (BD Biosciences) according to the manufacturer’s instructions. Labeled cells were analyzed using the Epics XL-MCL flow cytometer (Beckman Coulter) and Expo 32 ADC XL 4 Color software.
Statistics
Numerical values from individual experiments were pooled and expressed as mean ± standard error of the mean (SEM) of three independent experiments throughout. Obtained numbers were compared by either two-tailed Student’s t-test, or one-way ANOVA with Bonferoni’s multiple comparison test, with statistical significance assumed at p < 0.05.
Supplementary Material
Acknowledgments
The authors thank Drs. Enrique Rodriguez-Boulan, Reinhard Jahn and Ramnik Xavier for providing valuable reagents for this study, Dr. Claudia Jasalavich for editing the manuscript. This work was supported by National Institute of Health grants RO1 DK083968 and R01 DK084953 to A.I.I.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22885
References
- 1.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–21. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 3.Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95. doi: 10.1038/cdd.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011;30:4701–11. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 6.Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–56. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- 7.Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–67. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- 8.Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie. 2008;90:313–23. doi: 10.1016/j.biochi.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 9.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–57. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chua CE, Gan BQ, Tang BL. Involvement of members of the Rab family and related small GTPases in autophagosome formation and maturation. Cell Mol Life Sci. 2011;68:3349–58. doi: 10.1007/s00018-011-0748-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohashi Y, Munro S. Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol Biol Cell. 2010;21:3998–4008. doi: 10.1091/mbc.E10-05-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orsi A, Polson HE, Tooze SA. Membrane trafficking events that partake in autophagy. Curr Opin Cell Biol. 2010;22:150–6. doi: 10.1016/j.ceb.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi Y, Meyerkord CL, Hori T, Runkle K, Fox TE, Kester M, et al. Bif-1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy. 2011;7:61–73. doi: 10.4161/auto.7.1.14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12:831–5. doi: 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- 16.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 18.Mari M, Tooze SA, Reggiori F. The puzzling origin of the autophagosomal membrane. F1000 Biol Rep. 2011;3:25. doi: 10.3410/B3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tooze SA, Jefferies HB, Kalie E, Longatti A, McAlpine FE, McKnight NC, et al. Trafficking and signaling in mammalian autophagy. IUBMB Life. 2010;62:503–8. doi: 10.1002/iub.334. [DOI] [PubMed] [Google Scholar]
- 20.Hong W. SNAREs and traffic. Biochim Biophys Acta. 2005;1744:120–44. doi: 10.1016/j.bbamcr.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 21.Malsam J, Kreye S, Söllner TH. Membrane fusion: SNAREs and regulation. Cell Mol Life Sci. 2008;65:2814–32. doi: 10.1007/s00018-008-8352-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ungar D, Hughson FM. SNARE protein structure and function. Annu Rev Cell Dev Biol. 2003;19:493–517. doi: 10.1146/annurev.cellbio.19.110701.155609. [DOI] [PubMed] [Google Scholar]
- 23.Fader CM, Sánchez DG, Mestre MB, Colombo MI. TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta. 2009;1793:1901–16. doi: 10.1016/j.bbamcr.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 24.Furuta N, Fujita N, Noda T, Yoshimori T, Amano A. Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol Biol Cell. 2010;21:1001–10. doi: 10.1091/mbc.E09-08-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC. Autophagosome precursor maturation requires homotypic fusion. Cell. 2011;146:303–17. doi: 10.1016/j.cell.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Renna M, Schaffner C, Winslow AR, Menzies FM, Peden AA, Floto RA, et al. Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J Cell Sci. 2011;124:469–82. doi: 10.1242/jcs.076489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA. A ubiquitous membrane fusion protein α SNAP: a potential therapeutic target for cancer, diabetes and neurological disorders? Expert Opin Ther Targets. 2006;10:723–33. doi: 10.1517/14728222.10.5.723. [DOI] [PubMed] [Google Scholar]
- 28.Burgoyne RD, Morgan A. Analysis of regulated exocytosis in adrenal chromaffin cells: insights into NSF/SNAP/SNARE function. Bioessays. 1998;20:328–35. doi: 10.1002/(SICI)1521-1878(199804)20:4<328::AID-BIES9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 29.Whiteheart SW, Schraw T, Matveeva EA. N-ethylmaleimide sensitive factor (NSF) structure and function. Int Rev Cytol. 2001;207:71–112. doi: 10.1016/S0074-7696(01)07003-6. [DOI] [PubMed] [Google Scholar]
- 30.Naydenov NG, Brown B, Harris G, Dohn MR, Morales VM, Baranwal S, et al. A membrane fusion protein αSNAP is a novel regulator of epithelial apical junctions. PLoS One. 2012;7:e34320. doi: 10.1371/journal.pone.0034320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naydenov NG, Harris G, Brown B, Schaefer KL, Das SK, Fisher PB, et al. Loss of soluble N-ethylmaleimide-sensitive factor attachment protein α (αSNAP) induces epithelial cell apoptosis via down-regulation of Bcl-2 expression and disruption of the Golgi. J Biol Chem. 2012;287:5928–41. doi: 10.1074/jbc.M111.278358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, et al. SNARE proteins are required for macroautophagy. Cell. 2011;146:290–302. doi: 10.1016/j.cell.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Bivic A, Real FX, Rodriguez-Boulan E. Vectorial targeting of apical and basolateral plasma membrane proteins in a human adenocarcinoma epithelial cell line. Proc Natl Acad Sci USA. 1989;86:9313–7. doi: 10.1073/pnas.86.23.9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–97. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- 35.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–8. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 37.Geng J, Nair U, Yasumura-Yorimitsu K, Klionsky DJ. Post-Golgi Sec proteins are required for autophagy in Saccharomyces cerevisiae. Mol Biol Cell. 2010;21:2257–69. doi: 10.1091/mbc.E09-11-0969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo Y, Chang C, Huang R, Liu B, Bao L, Liu W. AP1 is essential for generation of autophagosomes from the trans-Golgi network. J Cell Sci. 2012;125:1706–15. doi: 10.1242/jcs.093203. [DOI] [PubMed] [Google Scholar]
- 39.van der Vaart A, Griffith J, Reggiori F. Exit from the Golgi is required for the expansion of the autophagosomal phagophore in yeast Saccharomyces cerevisiae. Mol Biol Cell. 2010;21:2270–84. doi: 10.1091/mbc.E09-04-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackson CL. Brefeldin A revealing the fundamental principles governing membrane dynamics and protein transport. Subcell Biochem. 2000;34:233–72. doi: 10.1007/0-306-46824-7_6. [DOI] [PubMed] [Google Scholar]
- 41.Sáenz JB, Sun WJ, Chang JW, Li J, Bursulaya B, Gray NS, et al. Golgicide A reveals essential roles for GBF1 in Golgi assembly and function. Nat Chem Biol. 2009;5:157–65. doi: 10.1038/nchembio.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawamoto K, Yoshida Y, Tamaki H, Torii S, Shinotsuka C, Yamashina S, et al. GBF1, a guanine nucleotide exchange factor for ADP-ribosylation factors, is localized to the cis-Golgi and involved in membrane association of the COPI coat. Traffic. 2002;3:483–95. doi: 10.1034/j.1600-0854.2002.30705.x. [DOI] [PubMed] [Google Scholar]
- 43.Morinaga N, Tsai SC, Moss J, Vaughan M. Isolation of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP ribosylation factor (ARF) 1 and ARF3 that contains a Sec7-like domain. Proc Natl Acad Sci USA. 1996;93:12856–60. doi: 10.1073/pnas.93.23.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niu TK, Pfeifer AC, Lippincott-Schwartz J, Jackson CL. Dynamics of GBF1, a Brefeldin A-sensitive Arf1 exchange factor at the Golgi. Mol Biol Cell. 2005;16:1213–22. doi: 10.1091/mbc.E04-07-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakajima K, Hirose H, Taniguchi M, Kurashina H, Arasaki K, Nagahama M, et al. Involvement of BNIP1 in apoptosis and endoplasmic reticulum membrane fusion. EMBO J. 2004;23:3216–26. doi: 10.1038/sj.emboj.7600333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verrier SE, Willmann M, Wenzel D, Winter U, von Mollard GF, Söling HD. Members of a mammalian SNARE complex interact in the endoplasmic reticulum in vivo and are found in COPI vesicles. Eur J Cell Biol. 2008;87:863–78. doi: 10.1016/j.ejcb.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 47.Tang F, Wang B, Li N, Wu Y, Jia J, Suo T, et al. RNF185, a novel mitochondrial ubiquitin E3 ligase, regulates autophagy through interaction with BNIP1. PLoS One. 2011;6:e24367. doi: 10.1371/journal.pone.0024367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–10. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 50.Hirota Y, Tanaka Y. A small GTPase, human Rab32, is required for the formation of autophagic vacuoles under basal conditions. Cell Mol Life Sci. 2009;66:2913–32. doi: 10.1007/s00018-009-0080-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2012;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- 52.Seo G, Kim SK, Byun YJ, Oh E, Jeong SW, Chae GT, et al. Hydrogen peroxide induces Beclin 1-independent autophagic cell death by suppressing the mTOR pathway via promoting the ubiquitination and degradation of Rheb in GSH-depleted RAW 264.7 cells. Free Radic Res. 2011;45:389–99. doi: 10.3109/10715762.2010.535530. [DOI] [PubMed] [Google Scholar]
- 53.Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ. 2008;15:1318–29. doi: 10.1038/cdd.2008.51. [DOI] [PubMed] [Google Scholar]
- 54.Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mestre MB, Fader CM, Sola C, Colombo MI. α-hemolysin is required for the activation of the autophagic pathway in Staphylococcus aureus-infected cells. Autophagy. 2010;6:110–25. doi: 10.4161/auto.6.1.10698. [DOI] [PubMed] [Google Scholar]
- 56.Yen WL, Shintani T, Nair U, Cao Y, Richardson BC, Li Z, et al. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J Cell Biol. 2010;188:101–14. doi: 10.1083/jcb.200904075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zoppino FC, Militello RD, Slavin I, Alvarez C, Colombo MI. Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic. 2010;11:1246–61. doi: 10.1111/j.1600-0854.2010.01086.x. [DOI] [PubMed] [Google Scholar]
- 58.Itoh T, Fujita N, Kanno E, Yamamoto A, Yoshimori T, Fukuda M. Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol Biol Cell. 2008;19:2916–25. doi: 10.1091/mbc.E07-12-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Young AR, Chan EY, Hu XW, Köchl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119:3888–900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 60.Martin HG, Henley JM, Meyer G. Novel putative targets of N-ethylmaleimide sensitive fusion protein (NSF) and α/β soluble NSF attachment proteins (SNAPs) include the Pak-binding nucleotide exchange factor betaPIX. J Cell Biochem. 2006;99:1203–15. doi: 10.1002/jcb.20998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rabouille C, Kondo H, Newman R, Hui N, Freemont P, Warren G. Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell. 1998;92:603–10. doi: 10.1016/S0092-8674(00)81128-9. [DOI] [PubMed] [Google Scholar]
- 62.Aoki T, Kojima M, Tani K, Tagaya M. Sec22b-dependent assembly of endoplasmic reticulum Q-SNARE proteins. Biochem J. 2008;410:93–100. doi: 10.1042/BJ20071304. [DOI] [PubMed] [Google Scholar]
- 63.Jahn R, Scheller RH. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–43. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 64.Donaldson JG, Honda A. Localization and function of Arf family GTPases. Biochem Soc Trans. 2005;33:639–42. doi: 10.1042/BST0330639. [DOI] [PubMed] [Google Scholar]
- 65.Gillingham AK, Munro S. The small G proteins of the Arf family and their regulators. Annu Rev Cell Dev Biol. 2007;23:579–611. doi: 10.1146/annurev.cellbio.23.090506.123209. [DOI] [PubMed] [Google Scholar]
- 66.Li L, Kim E, Yuan H, Inoki K, Goraksha-Hicks P, Schiesher RL, et al. Regulation of mTORC1 by the Rab and Arf GTPases. J Biol Chem. 2010;285:19705–9. doi: 10.1074/jbc.C110.102483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Balakumaran BS, Porrello A, Hsu DS, Glover W, Foye A, Leung JY, et al. MYC activity mitigates response to rapamycin in prostate cancer through eukaryotic initiation factor 4E-binding protein 1-mediated inhibition of autophagy. Cancer Res. 2009;69:7803–10. doi: 10.1158/0008-5472.CAN-09-0910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ishihara N, Hamasaki M, Yokota S, Suzuki K, Kamada Y, Kihara A, et al. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell. 2001;12:3690–702. doi: 10.1091/mbc.12.11.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Campoy E, Colombo MI. Autophagy in intracellular bacterial infection. Biochim Biophys Acta. 2009;1793:1465–77. doi: 10.1016/j.bbamcr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 70.Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–49. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pierini R, Cottam E, Roberts R, Wileman T. Modulation of membrane traffic between endoplasmic reticulum, ERGIC and Golgi to generate compartments for the replication of bacteria and viruses. Semin Cell Dev Biol. 2009;20:828–33. doi: 10.1016/j.semcdb.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Salcedo SP, Holden DW. Bacterial interactions with the eukaryotic secretory pathway. Curr Opin Microbiol. 2005;8:92–8. doi: 10.1016/j.mib.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 73.Chae TH, Kim S, Marz KE, Hanson PI, Walsh CA. The hyh mutation uncovers roles for α Snap in apical protein localization and control of neural cell fate. Nat Genet. 2004;36:264–70. doi: 10.1038/ng1302. [DOI] [PubMed] [Google Scholar]
- 74.Ivanov AI, Bachar M, Babbin BA, Adelstein RS, Nusrat A, Parkos CA. A unique role for nonmuscle myosin heavy chain IIA in regulation of epithelial apical junctions. PLoS One. 2007;2:e658. doi: 10.1371/journal.pone.0000658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ivanov AI, Young C, Den Beste K, Capaldo CT, Humbert PO, Brennwald P, et al. Tumor suppressor scribble regulates assembly of tight junctions in the intestinal epithelium. Am J Pathol. 2010;176:134–45. doi: 10.2353/ajpath.2010.090220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Naydenov NG, Ivanov AI. Adducins regulate remodeling of apical junctions in human epithelial cells. Mol Biol Cell. 2010;21:3506–17. doi: 10.1091/mbc.E10-03-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.