

Abstract
The manganese transport regulator (MntR) represses the expression of genes involved in manganese uptake in Bacillus subtilis. It selectively responds to Mn2+ and Cd2+ over other divalent metal cations including Fe2+, Co2+ and Zn2+. Previous work has shown that MntR forms binuclear complexes with Mn2+ or Cd2+ at two binding sites, labeled A and C, that are separated by 4.4 Å. Zinc activates MntR poorly and binds only to the A site, forming a mononuclear complex. The difference in metal binding stoichiometry suggested a mechanism for selectivity in MntR. Larger metal cations are strongly activating because they can form the binuclear complex, while smaller metal ions cannot bind with the geometry needed to fully occupy both metal-binding sites. To investigate this hypothesis, structures of MntR in complex with two other non-cognate metal ions, Fe2+ and Co2+, have been solved. Each metal forms a mononuclear complex with MntR with the metal ion bound in the A site, supporting the conclusions drawn from the Zn2+ complex. Additionally, we investigated two site-specific mutants of MntR, E11K and H77A, that contain substitutions to metal binding residues in the A site. While metal binding in each mutant is significantly altered relative to wild-type MntR, both mutants retain activity and selectivity for Mn2+ in vitro and in vivo. That observation, coupled with previous studies, suggests that the A and C sites both contribute to the selectivity of MntR.
Keywords: Metalloregulation, Metal ion binding, Transcriptional Regulator
While many metal ions are essential to life, all are toxic at a high enough concentration. Cells must carefully regulate internal levels of metals to avoid the opposing hazards of deficiency and excess. In bacteria, functional responsibility for this balancing act typically falls to transcriptional regulators known as metalloregulatory proteins. Functioning as sensors, metalloregulators respond to variations in metal ion concentration in order to maintain homeostasis (1–3). Metalloregulators manage concentrations of the essential metal ions Mn2+, Fe2+, Co2+, Ni2+, Cu+ and Zn2+ as well as the toxic metals Cd2+, Hg2+ and Pb2+ (1, 4). Each protein must be exclusively sensitive to its cognate metal ion(s) in the physiological context, so as not to respond to the wrong signal.
Among first row transition metals, manganese presents a particular challenge to specific recognition. The common cellular form of manganese, Mn2+, is a high spin d5 species and lacks a strong preference for a given coordination geometry. Additionally, the Irving-Williams series identifies Mn2+ as the divalent first-row transition metal ion that forms the weakest complexes with ligands in aqueous solution, due to its relatively large ionic radius and its lack of crystal field stabilization energy (5). As a result Mn2+ generally competes poorly with other metal ions for binding sites in proteins. That difficulty can be avoided in some cases by maintaining relatively high local concentrations of manganese (6), but such an approach is not universally appropriate. Manganese can be present at lower concentrations than other transition metals, such as iron, in the cell (7). Therefore, sensing mechanisms for manganese may need to overcome its relatively low affinity for proteins.
The manganese transport regulator of Bacillus subtilis (MntR) appears to have developed an effective mechanism for generating specificity. A member of the DtxR family of transcriptional regulators, MntR negatively regulates the expression of two manganese uptake transporters in response to elevated concentrations of Mn2+ (8, 9). The protein functions as a homodimer of 142-residue subunits and binds a 21-base pair operator when cellular Mn2+ concentrations are high, blocking transcription. MntR is also sensitive to Cd2+ in vivo, which has a physiological function since Cd2+ is taken up by Mn2+ transporters (8, 10), but it is unresponsive to other first row transition metals added to the growth medium (8). In vitro MntR shows superior activation by Cd2+ over Mn2+, followed by Co2+ and Fe2+ and then Ni2+ and Zn2+ (11, 12). Despite that trend, metal-binding assays indicate that MntR binds other metals more tightly than Mn2+ (13), in keeping with the Irving-Williams series. Though selectively activated by Mn2+, MntR does not bind Mn2+ selectively.
The structure of MntR suggests an explanation for this behavior (14–16). Each subunit is composed of two domains: an N-terminal DNA-binding domain (residues 1–74) and a C-terminal dimerization domain (residues 75–142). The C-terminal domains partner to form the structural core of the dimer, while the DNA-binding domains can adopt a variety of positions with respect to that core, due to flexibility in a long α helix (α4; residues 68–88) that links the two domains (16–18) (Figure 1). MntR binds two Mn2+ ions per subunit, separated by 4.4 Å, at the interface of the N- and C-terminal domains (14, 15). The two metal-binding sites, labeled A and C, are linked by bridging carboxylates from Glu99 and Glu102. In addition to the bridging residues, the A site metal is also coordinated a solvent molecule and by the side chains of His77 and Glu11. Including bidentate interactions from Glu11 and Glu102, the A site Mn2+ possesses heptacoordinate geometry. The hexacoordinate C site includes the bridging carboxylates of Glu99 and Glu102, the backbone carbonyl of Glu99, the side chains of Asp8 and His103 and a solvent molecule as ligands, yielding near octahedral geometry (Figure 1). Although the MntR•Mn2+ complex retains some conformational flexibility, the bound manganese ions serve to fix the positions of the DNA-binding domains with respect to the core in a conformation suitable for DNA binding (15–17).
Figure 1.
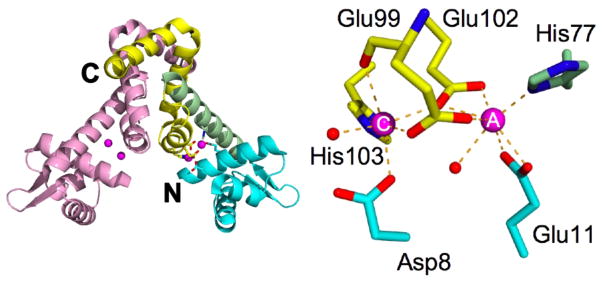
Structure of the MntR dimer and metal binding region. One subunit of the dimer is colored pink and the other is colored according to region: the N-terminal DNA-binding domain is cyan, the dimerization domain is yellow and the linking helix (α4) is colored green. The N-and C-termini of multi-colored subunit are labeled. The bound Mn2+ ions are magenta. In the magnified view of the metal binding site the A and C metal ions are labeled and metal-ligand interactions are indicated by dashed lines.
The coordination geometry of the MntR•Mn2+ complex appears to be essential in generating the selective response to cognate metals. Cadmium, which coordinates to MntR in a similar fashion to Mn2+, also binds with micromolar affinity and strongly stabilizes the active conformation (13, 15, 17). In contrast Zn2+, which binds to MntR more tightly than Mn2+, is a weaker activator (11–13). While Mn2+ and Cd2+ occupy the A site by accepting ligand interactions with seven lone pair donors, Zn2+ binds with tetrahedral geometry due in part to the displacement of the bridging residues, Glu99 and Glu102. We have hypothesized that the selective activation of MntR is achieved through the geometry of the A site (15). Larger metal cations, like Mn2+ and Cd2+, are capable of supporting the expanded coordination geometry, permitting appropriate positioning of the bridging residues for participation in the C site. The smaller, non-cognate zinc ion adopts a different geometry with a lower coordination number in the A site. The bridging residues are displaced, reducing their ability to bind a metal in the C site. Without a C-site metal, MntR is not fully activated. Evidence to support this view comes from the study of the Asp8 to Met (D8M) mutant, which has a disrupted C site. The D8M protein binds only a single metal ion per subunit, in the A site (14), and is poorly activated for DNA-binding. As predicted by this model, D8M possesses reduced specificity for Mn2+ both in vivo and in vitro (12, 19). So, while MntR does not bind Mn2+ more strongly than other metal cations, the geometry adopted by Mn2+ in the A site appears to be essential for full activation.
Here, we test this model for the specific activation of MntR by extending our structural studies to complexes of wild-type MntR with Co2+ and Fe2+, two weakly activating metals. Also, we have investigated two mutant forms of the protein in which the A site has been modified. The H77A mutant has the A site ligand, His77, replaced by alanine, a non-coordinating residue, and the E11K mutant has had the non-bridging carboxylate residue Glu11 replaced by lysine. The H77A mutant was expected to be functionally impaired due to the loss of a ligand from the A site, while the side chain ammonium group of lysine in the E11K mutant has the potential to functionally substitute for an A site metal (concomitant with C site occupancy) (20). We hypothesized that the selectivity of each mutant would be reduced due to the absence of a geometrically sensitive A site, but instead we found that both mutants retain selectivity. The biochemical and structural characterization of these two mutant proteins provides new insight into the mechanism of activation of MntR by Mn2+ and Cd2+. The C site is unexpectedly found to possess specificity independent of the A site, although non-cognate metals at the A site actively disrupt C site function.
EXPERIMENTAL METHODS
Mutagenesis and protein purification
The plasmid pHB7506 harbors the mntR gene under the control of the T7 promoter and lacO operator (8). Mutants of the mntR gene were prepared by the primer extension method of site-directed mutagenesis (21) and verified by DNA sequencing. For expression of mutant proteins, the resulting plasmids were transformed into BL21(DE3)-AI cells, which place expression under the dual control of LacI and AraC. Protein was typically purified from cells harvested from 2 L of culture using sequential anion and cation exchange chromatography columns (14). Purified H77A was generally stored in Buffer B (25 mM HEPES 7.5, 200 mM NaCl, 10% glycerol), but Buffer C (25 mM HEPES 8.0, 500 mM NaCl, 10% glycerol) was chosen if the sample was to be used in calorimetric experiments (see below). Due to solubility problems at low salt concentrations, all samples of E11K were prepared in Buffer C.
Crystallization and Structure Solution
Crystallization of H77A and E11K was attempted in the presence of Mn2+, Cd2+ and Co2+. In addition, wild-type MntR was co-crystallized with Co2+ and Fe2+. All crystals of MntR and its mutants complexed to metal ion effectors were obtained following broad screening of conditions using commercial kits. The pale green crystals of MntR•Fe2+ proved to be air sensitive and were grown and handled in an anaerobic glove box (Coy Labs, Lansing, MI). The optimized conditions for each crystal form are given in Table S1 (see Supporting Information). Data were collected at the Advanced Light Source from flash-frozen crystals, mounted on nylon loops (22) prepared with the addition of up to 30% glycerol to the mother liquor or by immersing crystals in paratone oil. Integration was performed with either Mosflm (23) or d*trek (24). Data reduction was performed using Scala as implemented in the CCP4 suite (25) and molecular replacement, as implemented in CNS (26) or EPMR (27), was used to obtain starting models of each complex. For monoclinic or orthorhombic crystals forms, the starting model in molecular replacement was the MntR•Mn2+ complex (PDB ID: 2F5D) and for trigonal crystal forms, the starting model was the D8M•Mn2+ complex (PDB ID: 1ON2). Refinement proceeded using CNS, followed by refinement using Refmac5 (28) or PHENIX (29) to take advantage of TLS modeling of domain motion. TLS domains were identified using the TLSMD web server (30). Metal-ligand geometries were unrestrained during refinement. The stereochemical quality of the models was evaluated with Molprobity (31).
Isothermal Titration Calorimetry
Metal ion binding to MntR mutants was evaluated by isothermal titration calorimetry (ITC) as described previously (15). Protein and metal ion solutions were prepared in Buffer C and degassed prior to use. Metal ion solutions (typically 0.7–2 mM) were titrated into solutions of the protein (typically 80–110 μM) and thermal data were fit to binding models within Origin 7.0 software. In the case of Co2+ titrations of the wild-type and E11K proteins, the thermograms did not return to baseline values. In those instances, a linear baseline was calculated from the final six injections and subtracted from the overall titration. Binding models were then tested against the adjusted data.
Visible Spectroscopy
Solutions of wild-type and mutant MntR were prepared in Buffer C at a concentration of roughly 300 μM in subunits. One hundred microliters of solution were placed in a 50 μL microcuvette (Starna) with 1 cm path length, and spectra were recorded at room temperature in a Shimadzu UV1610 or Cary 100 spectrophotometer. A concentrated stock of CoCl2 in Buffer C was used in titrations so that ten additions would lead to an excess of Co2+ relative to MntR subunits. Spectra were recorded at least twice following each addition to be sure of consistent absorbance.
DNA-binding Assays Monitored by Fluorescence Anisotropy
The activation of MntR and its mutants was measured using a fluorescence anisotropy based DNA-binding assay as described previously (15). A 21-base oligodeoxynucleotide with the sequence GAGTTTCCTTAAGGCAAATTG, which contains the mntH operator sequence, was labeled with Alexa Fluor 488 (Integrated DNA Technologies) at the 5′ end and annealed with a 10% molar excess of its complement in Buffer A by heating to 80°C and then slowly cooling to room temperature, creating a DNA duplex containing a single fluorescent label. The DNA-binding activity was measured by titrating solutions of MntR and its mutants into 1 mL of solution containing labeled DNA (1 nM) in FA buffer (25 mM HEPES 7.5, 300 mM NaCl) in the presence or absence of a fixed concentration of the activating metal(s). Anisotropy was measured on a Beacon 2000 fluorescence polarization instrument (Panvera, Madison, WI). Data were analyzed assuming a 1:1 binding stoichiometry between the MntR dimer and labeled DNA using equations 1 or 2. The latter was employed when the measured Kd was less than 10 nM.
| (Eq. 1) |
| (Eq. 2) |
In these equations r is the measured anisotropy, while rmin and rmax are the anisotropies of unbound and fully bound DNA, respectively. P is the total concentration of protein dimers, D is the total concentration of fluoresceinated DNA and Kd is the dissociation constant of MntR from the duplex DNA.
Beta-galactosidase expression assays
Wild-type, H77A or E11K mntR with the native promoter were incorporated into Bacillus subtilis mntR− strain (HB7503) at the amyE locus as described (8, 9). SPβ lysates from strain HB7510 were used for transduction to create corresponding strains with mntH promoter-lacZ fusions, named HB15091, HB15092 and HB15093, respectively (8). For β-galactosidase assays, B. subtilis cells were grown in metal limiting minimal medium (MLMM) (32) supplemented with 0.1 μM Mn and 1 μM Fe (in the form of ultrapure MnCl2 and FeCl3) to assure good growth overnight. Cells were then harvested and washed twice with MLMM and diluted 1:5 into MLMM with no metal, 1 μM Mn, 10 μM Mn or 10 μM Fe. Cells were grown to log phase and were analyzed for β-galactosidase activity as described (33). All experiments were done in triplicate.
RESULTS
Interactions of Fe2+ and Co2+ with MntR
To further define the basis of selectivity in wild-type MntR, we investigated the complexes formed with Fe2+ and Co2+ by x-ray crystallography and solution techniques. Crystals of MntR grown in the presence of 1 mM Fe2+ or Co2+ were obtained from comparable conditions at pH 7 (Table S1 in Supporting Information). To maintain iron in the ferrous oxidation state, MntR•Fe2+ crystals were grown and manipulated in the presence of 10 mM ascorbate under anaerobic conditions. Exposure to air caused darkening of the crystals and loss of diffraction. The blue-violet color of MntR•Co2+ crystals is consistent with the presence of protein-bound high-spin divalent cobalt ion (34). Diffraction data were collected at synchrotron sources, and initial models were obtained using molecular replacement (Table 1). The overall structures of the MntR•Co2+ and MntR•Fe2+ complexes superimpose closely to the structures of the MntR•Mn2+ and MntR•Zn2+ (15), with root mean square deviations (rmsd) between α carbon positions of each of the four dimeric proteins ranging from 0.2 to 0.6 Å
Table 1.
Data and Refinement Statistics for structures of metal complexes with MntR.
| MntR•Co2+ | MntR•Fe2+ | H77A | E11K•Mn2+ | E11K•Cd2+ | apo-E11K | |
|---|---|---|---|---|---|---|
|
| ||||||
| Data Source | ALS 4.2.2 | ALS 4.2.2 | ALS 4.2.2 | ALS 8.3.1 | ALS 8.3.1 | ALS 8.3.1 |
|
| ||||||
| Wavelength (Å) | 1.000 | 1.2131 | 1.2400 | 1.1159 | 1.1159 | 1.1159 |
|
| ||||||
| Space Group | P212121 | P212121 | P3121 | P21 | P21 | P212121 |
| a (Å) | 45.9 | 46.0 | 51.7 | 49.4 | 49.1 | 38.9 |
| b (Å) | 75.0 | 75.2 | 51.7 | 46.1 | 46.3 | 86.7 |
| c (Å) | 96.2 | 97.4 | 175.6 | 74.9 | 74.1 | 91.2 |
| β (°) | 93.0 | 93.0 | ||||
|
| ||||||
| Resolution (Å)a | 41.45–1.90 (2.00–1.90) | 22.0–1.90 (2.00–1.90) | 44.81–2.30 (2.42–2.30) | 74.79–1.65 (1.74–1.65) | 74.04–1.90 (2.00–1.90) | 45.60–2.00 (2.11–2.00) |
|
| ||||||
| Number of reflections | ||||||
|
| ||||||
| Total | 193256 | 163597 | 66440 | 143151 | 95213 | 139077 |
|
| ||||||
| Unique | 26945 | 26399 | 12767 | 40214 | 25216 | 21377 |
|
| ||||||
| Completeness | 100.0 (100.0) | 96.8 (97.4) | 99.4 (100.0) | 98.8 (97.4) | 95.4(78.4) | 99.2 (94.5) |
|
| ||||||
| I/σ(I) | 18.3 (3.2) | 9.2 (2.7) | 11.2 (5.4) | 22.2 (2.2) | 6.4 (2.5) | 17.4 (5.7) |
|
| ||||||
| Rmerge(%)2 | 8.2 (63.3) | 10.9 (59.9) | 8.1 (21.4) | 5.2 (36.0) | 9.3 (35.7) | 5.9 (27.7) |
|
| ||||||
| Number of protein atoms | 2157 | 2083 | 2202 | 2158 | 2121 | 2187 |
|
| ||||||
| Number of metal ions | 2 | 4 | 3 | 2 | 2 | 0 |
|
| ||||||
| Number of solvent atoms | 122 | 160 | 14 | 113 | 58 | 51 |
|
| ||||||
| Number of solute atoms | 30 | 30 | 0 | 0 | 0 | 0 |
|
| ||||||
| Rcryst/Rfree(%)c | 21.4/25.8 | 23.5/28.9 | 25.2/31.7 | 23.4/26.4 | 24.4/28.6 | 23.5/29.9 |
|
| ||||||
| Bond length deviation (Å) | 0.007 | 0.007 | 0.011 | 0.008 | 0.007 | 0.008 |
|
| ||||||
| Bond angle deviation (°) | 0.86 | 0.915 | 1.08 | 1.12 | 0.881 | 1.01 |
|
| ||||||
| B Factors (Å2) | ||||||
| Main chain atoms | 36.9 | 27.2 | 60.0 | 13.4 | 21.6 | 41.5 |
| Side chain atoms | 43.3 | 34.6 | 67.9 | 15.1 | 27.9 | 50.4 |
| Solvent molecules | 39.2 | 35.7 | 56.5 | 17.3 | 26.8 | 37.5 |
| Metal ions | 24.7 | 29.5 | 62.6 | 10.6 | 20.2 | |
|
| ||||||
| Ramachandran Plot | ||||||
| Favored region (%) | 100 | 99.2 | 98.1 | 100 | 99.6 | 99.6 |
| Allowed region (%) | 0.8 | 1.5 | 0.4 | 0.4 | ||
| Outliers (%) | 0.4 | |||||
|
| ||||||
| PDB Identifier | 3R61 | 4HV5 | 4HV6 | 4HX4 | 4HX7 | 4HX8 |
Numbers in parentheses reflect highest resolution shell.
Rmerge = ΣhΣj|Ih,j−<Ih>|/ΣjΣh|Ih,j|, where Ih,j is the jth observation of reflection h.
Rcryst = Σh||Fo|−|Fc||/Σh|Fo|, where Fo and Fc are the observed and calculated structure factors for reflection h. Rfree is calculated similarly for 5–10% of the data not used in refinement.
Metal ions were placed at the A sites in both models to fit peaks above 10 σ in σA-weighted Fo-Fc maps, but no comparable peaks were visible in the C sites. However, maps derived from the MntR•Fe2+ crystal contained unexplained electron density in a solvent-exposed region adjacent to the C site. Calculation of an anomalous Fourier difference map showed peaks at that position as well as stronger peaks in the A-site (Figure 2), indicating the presence of additional bound iron atoms. We labeled the new site “E”, reflecting its external position 1.8 Å from the C site. The identities of the bound metals were later confirmed as iron by collecting data at energies below (6900 eV) and above (7200 eV) the Fe K edge at 7112 eV from a second iron-containing crystal (Table S2 in Supporting Information). The anomalous Fourier difference map from the low energy data set lacked any peaks above 3.6 σ in the metal binding region, while the high energy data set gave a map with peaks above 18 σ for the A-site metals and 5.4 and 7.8 σ for the E site metal ions. Iron atoms placed at the E site were refined to 76 and 37% occupancy in the two subunits. Two protein ligands, Asp8 and His103, are recruited from the C site to coordinate the E-site Fe2+ ions and two additional solvent molecules were modeled as ligands to the E-site iron in one subunit. The partial occupancy of the E site in crystals grown with 1 mM Fe2+ suggests that the site lacks physiological relevance, since the concentration of free iron available in the cell is much lower (35).
Figure 2.

Metal binding sites of iron and cobalt complexes of MntR. (A) Stereo image of the metal binding region in the MntR•Fe2+ complex. Carbon atoms are colored light tan and Fe2+ ions are brown. Metal-ligand interactions are shown in green, and the H-bond between Glu99 and a solvent ligand is shown in orange. A σA weighted 2Fo-Fc electron density map is shown contoured at 1.5 σ in gray and the anomalous difference map, calculated from data reported in Table 1, is contoured at 4 σ is shown in magenta. (B) Stereo image of the metal binding region of MntR•Co2+. The image is prepared as in A, except that carbons are violet and the Co2+ ion is pink. The tentatively assigned metal-ligand interaction involving Glu99 is in orange. (C) Stereo view of the overlay of metal binding regions in the Mn2+, Fe2+, Co2+ and Zn2+ complexes (structurally aligned by residues 80–120). All atoms of the Fe2+ and Co2+ complexes are colored as in A and B. Carbons of the zinc complex is shown in gray, and carbons of the Mn2+ complex are shown in light green. The A, C and E metal binding positions are labeled.
The coordination geometries of Fe2+, Co2+ and Zn2+ in the A site of MntR vary slightly from one another. Even as mononuclear complexes they retain the interactions made by Mn2+ with a solvent molecule ligand and the side chains of Glu11 and His77, but they each subtly differ from the Mn2+ complex in the positioning of the bridging residues Glu99 and Glu102 (Figure 2). In the presence of Fe2+ and Co2+ there is a minor shift (0.2–0.3 Å) in the position of the side chain carboxylate of Glu102 away from the vacant C site, but it maintains the bidentate coordination observed with Mn2+ (Figure 2). In the Zn2+ complex, Glu102 has shifted further from its position with Mn2+ (0.5 Å) and acts as a monodentate ligand. The side chain carboxylate of Glu99 is shifted 0.7 to 1.0 Å in the Fe2+ and Zn2+ complexes from the position it occupies with bound Mn2+ and no longer coordinates the metal ion, though it does hydrogen bond with the metal-bound solvent molecule (Figure 2, Table S3). In the Co2+ complex, Glu99 is refined to a position closer to the position observed in the Mn2+ complex, and could either ligate the bound metal (at a separation measured at 2.2 and 2.4 Å measured in the two subunits) or hydrogen bond with a metal-bound solvent molecule (at a distance of 2.2 and 2.5 Å in the two subunits). The B-factors for the side chain carboxylate of Glu99 in the Co2+ complex, 50–60 Å2, are high relative to those of other coordinating groups, between 20–35 Å2, suggesting dynamic behavior that may permit both interactions.
To confirm the metal-binding stoichiometry observed in the MntR•Co2+ structure, we performed spectroscopic titrations with cobalt. Additions of Co2+ to solutions of 200–300 μM MntR subunits gave rise to absorbance in the visible spectrum centered between 520 and 560 nm (Figure 3). A plot of the absorbance at 520 nm vs. the molar ratio of Co2+ to MntR shows an endpoint at 1.5 equivalents of added cobalt per subunit (Figure 3), or three metal ions per dimer. Each MntR dimer contains two A sites, but the third metal likely binds at a location that is unique within the dimer, a so-called “dimer site”. Previous evidence exists for this site. The structure of the MntR•Cd2+ complex (PDB ID 2EV0) includes a cadmium ion bound at the interface of the two subunits, coordinating dyad-related His104 residues (15) (Additional text and Figure S1 in Supporting Information). Calorimetric titrations of MntR with Co2+ are also consistent with the 1.5:1 ratio of metal ions to protein dimer, but the experiment was complicated by a failure of the titration to return to baseline. Curve-fitting was only successful following a baseline correction as described in the Experimental Procedures (Figure S2).
Figure 3.

Titrations of (A) wild-type MntR and (B) H104A with CoCl2, monitored by visible spectroscopy. Protein subunit concentrations were 300 μM, and spectra were collected after additions of 0.25 equivalents of Co2+ per subunit. Insets plot molar absorptivity of the protein complex with Co2+ vs. the molar ratio of Co2+ to protein subunits. Lines are fit to data preceding and following inflection points that were identified by visual inspection.
The H104A mutant was prepared to allow investigation of cobalt binding without interference from the dimer site. The interaction of Co2+ with H104A, monitored by isothermal titration calorimetry, reflects the loss of the dimer site, with a binding stoichiometry of 0.94 ± 0.01 Co2+ ions per subunit (Figure S2). Likewise, when visible spectroscopy is used to monitor titrations of H104A with Co2+, an endpoint is observed at 0.9 equivalents of Co2+ per H104A subunit (Figure 3 and Figure S1). The molar absorptivity of 150 M−1cm−1 for the H104A•Co2+ complex is consistent with pentacoordinate binding geometry in the A site (36), suggesting that Glu99 is forming a hydrogen bond to the metal bound solvent molecule, but is not coordinating the cobalt ion (Figure 2B).
The above results support our hypothesis that the ability of MntR to differentiate cognate metal ions (Mn2+ and Cd2+) from non-cognates (Fe2+, Co2+ and Zn2+) relies on the inability of the latter to form binuclear complexes. The results are also consistent with the notion that the A site discriminates between strongly and poorly activating metals on the basis of ionic radius. When ions smaller than Mn2+ bind to the A site, the bridging residues Glu99 and Glu102 are displaced from positions that support binding of a C-site metal, essential for full activation of the protein. To further examine that phenomenon, we prepared two mutations to A-site residues to more closely examine the role of the A site in specificity.
Activity of the E11K and H77A Mutants
To test the importance of the A site in discriminating against weakly activating metal ions, we prepared the E11K and H77A mutants of MntR. Both mutations were expected to reduce the selectivity of MntR, either by generating a constitutively occupied A site (E11K) or by significantly impairing metal binding in the A site by removing a ligand (H77A). To assess selectivity, two activity assays were performed: an in vitro assay of DNA binding monitored by fluorescence anisotropy and an in vivo assay that monitors regulation of a β-galactosidase reporter gene under the control of MntR in Bacillus subtilis cells.
Fluorescence anisotropy has been used previously to evaluate the capacity of various metal ions to activate MntR for DNA binding (11, 15, 17). Studies in the Cohen lab compared activation of wild-type MntR by a variety of divalent metal ions at 1 mM concentration and shown that the order of activation is Cd2+ > Mn2+ > Fe2+, Co2+ > Ni2+, Zn2+ > apo-MntR (11, 12). Our assay was performed with the strongly activating metal ions, Mn2+ and Cd2+, as well as the weakly activating Co2+ ion. All three yield binding isotherms that can be modeled assuming 1:1 binding stoichiometry between MntR dimers and the duplex DNA (Figure S3 of the Supporting Information, Table 2).
Table 2.
Dissociation constants obtained from titrations of fluoresceinated DNA with MntR variants, monitored by fluorescence anisotropy measurements. For each metal ion listed, 1 mM of the chloride salt was present during the titration. The average and standard deviation are reported for three trials under each condition.
| Kd (nM) for MntR•DNA complex in the presence of 1 mM metal ion | Ratios of Kd | ||||
|---|---|---|---|---|---|
| Mn2+ | Cd2+ | Co2+ | Co2+ + Mn2+ | Mn: Cd: Co | |
| Wild-typea | 5.6 ± 0.8 | 6.5 ± 0.5 | 250 ± 20 | 90 ± 20 | 1 : 1 : 40 |
| Wild-typeb | 30.4 ±1.3 | 24.3 ±4.6 | 81.5 ±13.8 | - | 1 : 0.8 : 3 |
| H77Aa | 15.4 ± 1.4 | 11.7 ± 0.3 | 510 ± 80 | 330 ± 60 | 1: 0.8 : 30 |
| E11Ka | 6.5 ± 1.3 | 2.4 ± 0.1 | 370 ± 60 | 240 ± 30 | 1 : 0.4 : 60 |
This study. Titration buffer included 25 mM HEPES 7.5, 300 mM NaCl using a 21-base pair DNA duplex.
Data from the Cohen laboratory (12). Titration buffer included 20 mM HEPES (pH 7.2), 500 mM NaCl and 5% glycerol, with a 26-base pair DNA duplex (mntA26).
Under our assay conditions wild-type MntR, E11K and H77A are each activated to varying extents by Mn2+, Cd2+ and Co2+ (Table 2) but do not measurably bind DNA in the absence of added metal (data not shown). Wild-type MntR binds a 21-base pair DNA fragment with equal affinity in the presence of 1 mM Mn2+ or Cd2+ (dissociation constants of 5.6 ± 0.8 nM and 6.5 ± 0.5 nM, respectively) while it is 40-fold less active in the presence of 1 mM Co2+ (Kd of 250 ± 20 nM). E11K is as active as the wild-type protein in the presence of Mn2+, possesses higher affinity for DNA in the presence of Cd2+ (Kd of 2.4 ± 0.1 nM), and exhibits 60-fold selectivity against activation by cobalt (Table 2). The activation of H77A exhibits a similar pattern. It is two- to three-fold less active than wild-type MntR in the presence of 1 mM Mn2+ and Cd2+, but shows even lower activity in the presence of 1 mM Co2+, retaining 30-fold selectivity in activation by manganese over cobalt. In addition, the anisotropy of the H77A•Co2+•DNA complex is reduced in comparison to wild-type MntR and E11K complexes with DNA. Dissociation constants are obtained from anisotropy data without being constraints by the absolute values of anisotropy measured in a given experiment. However, the low value observed for H77A•Co2+ complexed with DNA suggests a distinct conformation for that complex (Figure S3).
In vivo experiments with MntR variants demonstrate retention of selectivity in E11K and H77A for activation by manganese over iron. Expression assays were performed with extracellular addition of manganese and iron (as MnCl2 and FeCl3) to B. subtilis cells containing a reporter gene coupled to the mntH promoter. Cells harboring wild-type MntR show virtually no change in β-galactosidase expression in response to added iron, while 7-fold repression is shown in response to 1 or 10 μM manganese applied to the growth environment (Figure 4). E11K possesses an activity profile similar to wild-type MntR, with slightly enhanced selectivity for Mn2+ in the cellular environment. H77A on the other hand shows lower levels of activity in response to added manganese relative to wild-type MntR, but it maintains selectivity for manganese over iron.
Figure 4.

β-Galactosidase assays performed with B. subtilis cells grown in minimal medium and supplemented with manganese or iron as indicated (n=3).
We were curious if the cobalt complexes of the three MntR variants could be “rescued” by addition of manganese. In wild-type MntR, for example, Mn2+ might bind in the C site while Co2+ occupies the A site to create a mixed metal complex with high affinity for DNA. While slight enhancements in DNA-binding activity were observed for all three MntR variants (Table 2, Figure S3), Mn2+ failed to complement the cobalt complexes to produce a fully active species. Isothermal titration calorimetry was performed to explore Mn2+ binding to MntR•Co2+ by titrating Mn2+ into a solution containing a solution of wild-type MntR mixed with a two-fold molar excess of Co2+. The thermogram (Figure S4 of the Supporting Information) shows a weak endothermic signal that does not saturate, indicating low affinity of the wild-type MntR•Co2+ complex for Mn2+.
Crystal Structures of E11K and H77A
To investigate the unexpected selectivity of E11K and H77A for activation by Mn2+ and Cd2+ over Co2+ and Fe2+, we performed crystallization screens of both MntR variants with Mn2+, Cd2+, and Co2+. In the case of E11K, crystallization screens were also performed in the absence of added metal ion. Diffraction quality crystals were obtained with E11K in its apo-form and with Mn2+ and Cd2+, and the effort also yielded crystals containing both apo- and manganese-bound H77A. Neither protein yielded diffraction quality crystals in the presence of cobalt.
The structure of apo-E11K indicates that our efforts to engineer a constitutively occupied A site via the side chain of Lys11 did not entirely succeed. The metal-free form of E11K bears stronger structural similarity to the metal-free structures of wild-type MntR (16) than to structures of MntR with a single metal ion in the A site. The distance measured between DNA-binding domains is 42 Å (measured at the α carbon of Lys41), comparable to the metal-free form of wild-type MntR (39 Å) and considerably larger than the separations seen in the binuclear metal complexes of wild-type MntR (between 31–33 Å; Figure 5A) (14, 15). This conformation is accessible because the side chain ammonium group of Lys11 does not occupy the position of the A-site metal. The hydrogen-bond accepting groups of His77, Glu99 and Glu102 are separated by 5–8 Å from the lysine ammonium group in subunit 1 and by 3–8 Å in subunit 2 (Figure 5B). The side chain of Lys11 does not constitutively occupy the A site.
Figure 5.
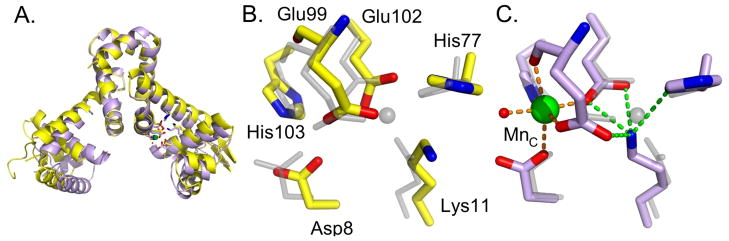
Structures of E11K in metal-free and Mn2+-bound forms. (A) Superposition of apo-E11K in yellow and E11K•Mn2+ in light purple. (B) Structure of the metal-binding region of apo-E11K (carbons in yellow) overlaid on the structure of wild-type MntR•Mn2+ in transparent gray. Note the displacement of residues 8 and 11 in apo-E11K from the wild-type MntR•Mn2+ positions. (C) Structure of the metal-binding region of the E11K• Mn2+ complex (light purple carbons, Mn2+ ion in green) overlaid on the MntR•Mn2+ complex (rendered in transparent gray). Hydrogen bonds to Lys11 are shown in green and metal-ligand bonds to the C-site Mn2+ are shown in orange.
However, the side chain of Lys11 does perform its intended role in the Mn2+ and Cd2+ complexes of E11K. In both structures, a single metal ion is bound per subunit, at the C site, while the ammonium group of Lys11 occupies a position less than 1 Å from the A-site metal position (Figure 5C). The structures of E11K•Mn2+ and E11K•Cd2+ align well with those of MntR•Mn2+ or MntR•Cd2+ (Figure 5C), with root mean square deviations of less than 0.5 Å between α carbons of any pair among the four models. In the metal binding region of E11K the ligating residues (aside from residue 11) adopt positions observed in the wild-type complexes with Mn2+ and Cd2+. The side chain ammonium group of Lys11 interacts with His77, Glu99 and Glu102 in the same manner as a bound A-site metal, though with longer distances appropriate to hydrogen bonding (Table S4). In the metal complexes of E11K, the C site geometry is virtually indistinguishable from those of the comparable wild-type complexes. Both manganese and cadmium are bound in near octahedral geometry, accepting ligating interactions from the two bridging carboxylates, Glu99 and Glu102, the backbone carbonyl of Glu99, Asp8, His103 and a solvent molecule (Table S4).
The H77A mutant of MntR was crystallized in the presence of 1 mM Mn2+ to yield a crystal composed in equal parts of dimers of manganese-free and manganese-bound subunits (Table 1; Supporting information). Each dimer is formed by a crystallographically unique subunit that may be rotated about the 2-fold axis in the P3121 space group to generate the second subunit (Figure S5). The chief structural difference between dimers is the 44.7 Å separation of DNA-binding domains in apo-H77A relative to a 31.4 Å separation in H77A•Mn2+, measured at the α carbon of Lys41. The tertiary structure of H77A•Mn2+ superimposes well upon the structure of wild-type MntR•Mn2+, with a 0.5 Å rmsd between α carbon positions.
The structure of H77A•Mn2+ reveals significant impact of the H77A substitution on metal binding (Figure 6). As with wild-type MntR, H77A binds two manganese ions per subunit, but the separation is decreased from 4.4 Å in the wild-type protein to 3.8 Å in the mutant (Table S4). In H77A, the A-site metal has shifted 1.3 Å away from residue 77 and towards Asp8, presumably due to the loss of lone pair donation from residue 77. In contrast, the C site in the H77A•Mn2+ complex is relatively unperturbed. Each of the five C-site protein ligands that coordinate the C-site metal in wild-type MntR•Mn2+ does so in H77A•Mn2+, though Asp8 adopts an unusual orientation and may be a bidentate ligand (Figure 6, Table S4). The A site metal of H77A•Mn2+, which given the 1.3 Å shift is better described as the “A′ site”, has a novel coordination scheme. In addition to the loss of coordination from residue 77, the side chain carboxylate of Glu99 has also been lost as a ligand, and the shift in position has brought the A site metal closer to the carboxylate of Asp8, normally only involved in C-site coordination. The distance from Asp8 to the A-site Mn2+ is refined to 2.9 Å, so an interaction must be viewed as tentative, but without Asp8 there are only three other electron donors visible in the electron density map – the carboxylates of Glu11 and Glu102 (each monodentate) and a solvent molecule. It is likely that other solvent molecules serve as ligands but are not visible in electron density maps. The H77A mutant succeeds in retaining near-native binding interactions with the C-site Mn2+ despite disruption to A-site geometry.
Figure 6.
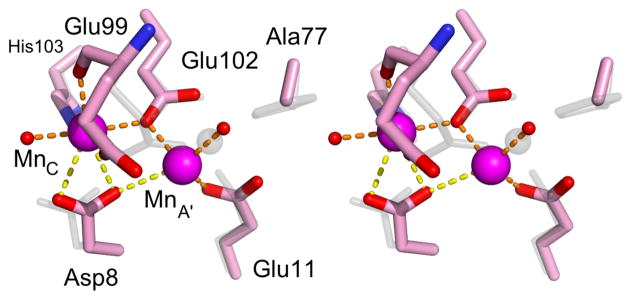
Stereo image of the metal binding region of H77A•Mn2+ (carbons colored pink) and wild-type MntR•Mn2+ (colored gray). Manganese ions in the A′ and C sites are magenta spheres. Metal-ligand interactions are shown in orange with tentatively assigned interactions shown in yellow.
Metal Binding Stoichiometry and Affinity
To confirm the metal-binding stoichiometry observed in the crystal structures of metal complexes of E11K and H77A and to further characterize the behavior of metal complexes that did not yield crystals, we investigated their metal binding stoichiometry using solution-based techniques.
The results from isothermal titration calorimetry are generally consistent with the crystal structures of E11K•Mn2+, E11K•Cd2+ and H77A•Mn2+. Thermograms obtained by titrating Mn2+ into samples of E11K can be fit using a one-site model with a stoichiometry of 1.00 ± 0.16 Mn2+ ions per subunit and a dissociation constant of 22 ± 2 μM, consistent with C-site binding of the manganese ion (Figure S6 of the Supporting Information, Table 3). The titration of E11K with cadmium produces a thermogram that is best fit using a two independent site model in which the two sites exist in a 1:1 and 0.5:1 ratio with E11K subunits (Table 3). That result indicates Cd2+ binding with a dissociation constant of 6 ± 3 μM to the C site of each subunit, as observed crystallographically, and to the dimer site comprised of dyad-related His104 residues (Kd of 150 ± 30 μM). The dimer site was not occupied under the crystallization conditions used for E11K•Cd2+ at pH 7.5, lower than the pH used in the ITC experiments (pH 8) or in the crystallization of MntR•Cd2+ (pH 8.5), where Cd2+ binding to His104 has been observed. Titrations of Mn2+ and Cd2+ into solutions of H77A yield data that are best fit by a sequential two-site model (Table 3), indicating that both metal ions form a binuclear complex with H77A.
Table 3.
Fitting parameters from calorimetric titrations of MntR variants with Mn2+, Cd2+ and Co2+. Parameters allowed to vary during a fit are averages and standard deviations taken from three independent trials. The fitting parameters for the Co2+ titration of E11K are in italics to emphasize that background correction was performed before fitting.
| MntR Variant | Metal Ion | Binding Model | Number of subunits | Kd1 (μM)b | ΔH1 (kcal/mol) | Kd2 (μM)c | ΔH2 (kcal/mol) |
|---|---|---|---|---|---|---|---|
| E11K | Mn2+ | One site | 1.00 ± 0.16 | 22 ± 2 | 3.3 ± 0.1 | ||
| E11K | Cd2+ | Two independent sites with constraint that N1=2*N2 | 0.91 ± 0.22 | 6 ± 3 | −0.38 ± 0.22 | 150 ± 30 | −14.7 ± 1.9 |
| E11K | Co2+ | Two independent sites with constraint that N1=2*N2. Background adjusted. | 0.8 ±0.2 | 19 ± 7 | 2.6 ± 0.7 | 1.3 ± 1.1 | −2.4 ± 1.1 |
| H77A | Mn2+ | Sequential two site | 1 | 70 ± 20 | −2.4 ± 0.2 | 150 ± 60 | 0.4 ± 0.3 |
| H77A | Cd2+ | Sequential two site | 1 | 20 ± 20 d | −5.8 ± 1.6 | 48 ± 11 | −5.2 ± 1.6 |
| H77A | Co2+ | One site | 1.05 ± 0.19 | 40 ± 10 | −7.2 ± 0.4 |
N is the number of subunits determined to be present in the fitting process. For sequential models, the number of subunits is defined as unity.
Kd1 is the sole dissociation constant in the one-site model, the first dissociation constant in sequential fits and the subunit site in constrained fits to the two independent site model.
Kd2 is the second dissociation constant in a sequential binding model or the dissociation constant from the dimer site (at ½ stoichiometry) in constrained fits to the two independent site model.
The first dissociation constant in the formation of the H77A•Cd2+ complex varied from 7 to 100 μM in three trials.
Calorimetric titrations performed with cobalt indicate that E11K and H77A each bind one Co2+ at a subunit site. The titration of H77A is readily fit by a one-site model (Figure S6) with a dissociation constant of 40 ± 10 μM (Table 3), but the thermogram arising from the addition of Co2+ to E11K is more complex. A suitable fit for the data could not be found unless a background correction was performed to bring the baseline to zero at the end of the titration. That alteration permitted fitting of the data to the same two-site model employed in the Cd2+ titration of E11K (Figure S6), suggesting that E11K binds one Co2+ per subunit in addition to one cobalt ion to the dimer site formed by His104 residues.
The interactions of Co2+ with the E11K and H77A mutants were further investigated using visible spectroscopy. The titration of E11K with Co2+ gives a break point at 1.5 equivalents of added cobalt per subunit (Figure 7A), consistent with subunit and dimer site binding observed using ITC. The spectrum has a maximum absorbance at 560 nm with a molar absorptivity of 120 M−1cm−1 at 1.5 equivalents of added cobalt, suggesting pentacoordinate geometry as observed in the wild-type complex (36). Comparable titrations of H77A with Co2+ revealed a weakly absorbing species (molar absorptivity of 18 M−1cm−1 at 525 nm), which is typical of Co2+ in an octahedral coordination environment (36). The low intensity of the absorption spectrum prevented stoichiometric measurements of Co2+ binding to H77A. Despite the disruption of the A site, cobalt does bind to subunit sites within both mutant forms of MntR, though it does not form fully active complexes with either.
Figure 7.

(A) Titration of E11K with CoCl2, monitored by visible spectroscopy. Protein subunit concentrations were held at 300 μM, and spectra were collected after additions of 0.25 equivalents of Co2+ per subunit. The inset plot was prepared as in Figure 3. (B) The spectrum of 300 μM H77A•Co2+ at one equivalent of added cobalt per subunit of H77A.
DISCUSSION
MntR is selectively activated by Mn2+ and Cd2+, due partly to the heptacoordinate geometry of interactions in the A site that position essential C-site residues. Smaller metal ions, such as Fe2+ and Co2+, fail to adopt the necessary coordination geometry. As a result, Mn2+ binding to the C site can be blocked by Co2+ binding in the A site (Figure 8A). By hindering C-site binding, non-cognate metal ions form complexes with MntR that are less structured and have lower affinity for DNA than the binuclear complexes with cognate metal ions (13, 14, 17). In addition, the C site is independently capable of selecting for Mn2+ and Cd2+ ions, as seen with the H77A and E11K variants of MntR. Both mutants are able to bind Mn2+ with native geometry in the C site, despite significant alterations to the A site, and adopt active DNA-binding conformations (Figure 8B,C). On the other hand, non-cognate metal ions fail to generate fully active complexes.
Figure 8.

Cartoon depictions of metal binding by (A) wild-type MntR, (B) E11K and (C) H77A. The center cartoon represents the metal-free form of each variant, while the Mn2+-bound form is shown to the left and the Co2+-bound form is shown to the right. Where evidence is available, the coordination number of each metal ion is represented by a polygon with an equivalent number of sides. The placement of Co2+ ions in E11K and H77A is speculative, and is chiefly intended to indicate the failure of each to bind Co2+ in a productive fashion. Ligands, including bound solvent molecules (Sol) are identified with connecting lines to the metal binding sites.
Interestingly, non-cognate metal ions do bind to H77A and E11K. Calorimetric titrations with cobalt indicate stoichiometric metal binding to the subunits of each mutant, and activity assays reveal weak activation by cobalt in vitro and iron in vivo. Non-cognate metal ions must partially organize the DNA-binding domains of the H77A and E11K subunits with respect to the dimerization domains in achieving even modest DNA-binding activity, but the mechanism is unclear. Absent crystallographic evidence, the visible spectra of cobalt complexes provide our best indication of the interactions taking place between a non-cognate metal ion and the H77A and E11K mutants.
The octahedral geometry of the H77A-bound Co2+ ion suggested by its low molar absorptivity is consistent with C-site binding. However, H77A•Co2+ has poor affinity for DNA, and C-site binding is otherwise a reliable feature of fully activated complexes of MntR (13, 14, 19). Instead, it is more likely that Co2+ binding occurs elsewhere at the interface of the DNA-binding and dimerization domains. The A′ site observed in the H77A•Mn2+ complex is a strong possibility (Figure 8). The coordinating side chains of Glu11 and Glu102 come from different domains, and solvent molecules could complete an octahedral coordination shell around Co2+ in the A′ site. The non-native geometry of the A′ site may produce an altered protein conformation in the Co2+-bound state, explaining the reduced anisotropy changes observed with H77A•Co2+ binding to fluoresceinated DNA.
Visible spectroscopy indicates that E11K binds Co2+ with pentacoordinate geometry, as observed in the A site of wild-type MntR•Co2+. Since Co2+ has to compete with the side chain of Lys11 for the remaining A-site residues in E11K, binding at that position likely precludes formation of even a partially active complex. Instead, the pentacoordinate geometry indicated for E11K-bound Co2+ likely results from non-native interactions of cobalt with the C site or with a mixture of residues from the A and C sites (Figure 8). Whatever the nature of its interaction with E11K, Co2+ fails to adopt the native C-site geometry observed in the fully active manganese and cadmium complexes.
The selectivity for manganese and cadmium in the C site of MntR is surprising, given the relative lack of specificity observed among related proteins. MntR belongs to the DtxR/MntR family of metalloregulators but is distinguished from other family members by its binuclear metal ion binding site with bridging ligands. Other structurally characterized proteins from this group bind two metal ions at sites composed of non-overlapping sets of residues, including residues from a third, C-terminal domain not found in MntR. Of particular interest is the so-called primary site found in the iron-dependent regulators DtxR of C. diphtheriae and IdeR of M. tuberculosis (37, 38), and in the manganese-dependent regulator ScaR of S. gordonii (39). Lying at the interface of the dimerization and DNA-binding domains, the primary site is essential for DNA binding activity (40, 41) and is homologous to the C site of MntR, comprising residues that align with Asp8, Glu99, Glu102 and His103 from MntR and adopting hexacoordinate geometry. Unlike the C site, the primary site accommodates a variety of metal ions. ScaR and its close homologs are activated by iron, cobalt, nickel, cadmium and manganese (39, 42–44), while DtxR is activated by manganese, iron, cobalt and nickel (19, 45, 46). The presence of sulfur ligands in the primary site of DtxR (Met10 and Cys102 substitute for Asp8 and Glu99 in MntR) may play a role in broadening selectivity, but the primary site of ScaR and the C site of MntR are composed of identical residues (39). Conceivably the C site, like the A site of MntR, favors larger cations, causing metal ions with smaller radii to bind with altered geometry or at other sites in the protein.
The structure and function of E11K points to an evolutionary intermediate between MntR and its distant homologs DtxR, IdeR and ScaR. Replacement of Glu11 with lysine in E11K brings the structure of the C site even closer to that of the primary site. The residue at position 11 of ScaR is lysine, and it is arginine in DtxR and IdeR (residue 13 in those proteins). Structural alignment of the C site of E11K with the primary site of IdeR (Figure 9) illustrates that structural similarity, as well as the role of the basic residue at position 11 (MntR numbering), in positioning metal-binding residues at position 99 and 102 (MntR numbering) in E11K and IdeR, respectively. The substitution of Glu11 with lysine in E11K recapitulates a step in the evolutionary path between DtxR/MntR family members that are activated by a binuclear complex, like MntR, and those that only bind one metal ion at the interface of the dimerization and DNA-binding domains, such as ScaR, DtxR and IdeR.
Figure 9.

Structural alignment of the primary site residues of IdeR•Co2+ (PDB ID 1FX7; carbons and Co2+ ion colored cyan) with the C site residues of E11K•Mn2+ (carbons and Mn2+ ion colored violet). Metal ligand interactions taking place between IdeR residues and bound Co2+ are shown as green dashed lines. Hydrogen bonding interactions taking place between Arg13 and primary site residues are shown as dotted cyan lines, while hydrogen bonds between Lys11 of E11K and C site residues are shown as dotted violet lines.
The work described here and previously (15) indicates that MntR relies on a two-part mechanism to achieve selectivity. First, a large metal cation is needed at the A site to correctly orient and position the C-site ligands, while non-cognate metal ions will bind at the A site but disrupt the C site. Second, binding at the C site, which is needed to lock the repressor in the active, DNA-binding conformation, also favors manganese and cadmium over other metals. The two levels of discrimination achieved by MntR have generated remarkable selectivity for manganese, a metal ion that otherwise competes poorly for protein binding sites.
Supplementary Material
Acknowledgments
Funding was provided by NIH grant R15 GM0699644 (A. G.) and GM059323 (J. D. H.)
We thank Dr. Jay Nix of the Molecular Biology Consortium at the Advanced Light Source of Lawrence Berkeley Laboratory for his invaluable assistance in crystallographic data collection and processing. Also thanks to Professor Michael Chapman and Dr. Thomas Lerch of Oregon Health and Sciences University for generously providing access to x-ray diffraction instrumentation.
Abbreviations
- MntR
manganese transport regulator
- DtxR
diphtheria toxin repressor
- IdeR
iron dependent regulator
- ScaR
streptococcal cell adhesion regulator
- rmsd
root mean square deviation
- ITC
isothermal titration calorimetry
Footnotes
SUPPORTING INFORMATION AVAILABLE
Additional text related to crystallographic results, Tables S1–S4, and Figures S1–S6. This material is available free of charge via the Internet at http://pubs.acs.org.
BIBLIOGRAPHY
- 1.Giedroc DP, Arunkumar AI. Metal sensor proteins: nature’s metalloregulated allosteric switches. Dalton Trans. 2007:3107–3120. doi: 10.1039/b706769k. [DOI] [PubMed] [Google Scholar]
- 2.Pennella MA, Giedroc DP. Structural Determinants of Metal Selectivity in Prokaryotic Metal-responsive Transcriptional Regulators. Biometals. 2005;18:413–428. doi: 10.1007/s10534-005-3716-8. [DOI] [PubMed] [Google Scholar]
- 3.Chen P, He C. Selective recognition of metal ions by metalloregulatory proteins. Current Opinion in Chemical Biology. 2008;12:214–221. doi: 10.1016/j.cbpa.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 4.Waldron KJ, Rutherford JC, Ford D, Robinson NJ. Metalloproteins and metal sensing. Nature. 2009;460:823–830. doi: 10.1038/nature08300. [DOI] [PubMed] [Google Scholar]
- 5.Irving H, Williams RJP. The stability of transition-metal complexes. J Chem Soc. 1953;637:3192–3210. [Google Scholar]
- 6.Tottey S, Waldron KJ, Firbank SJ, Reale B, Bessant C, Sato K, Cheek TR, Gray J, Banfield MJ, Dennison C, Robinson NJ. Protein-folding location can regulate manganese-binding versus copper- or zinc-binding. Nature. 2008;455:1138–1142. doi: 10.1038/nature07340. [DOI] [PubMed] [Google Scholar]
- 7.Outten CE, Tobin DA, Penner-Hahn JE, O’Halloran TV. Characterization of the Metal Receptor Sites in Escherichia coli Zur, an Ultrasensitive Zinc(II) Metalloregulatory Protein. Biochemistry. 2001;40:10417–10423. doi: 10.1021/bi0155448. [DOI] [PubMed] [Google Scholar]
- 8.Que Q, Helmann JD. Manganese homeostasis in Bacillus subtilis is regulated by MntR, a bifunctional regulator related to the diphtheria toxin repressor family of proteins. Mol Microbiol. 2000;35:1454–1468. doi: 10.1046/j.1365-2958.2000.01811.x. [DOI] [PubMed] [Google Scholar]
- 9.Guedon E, Moore CM, Que Q, Wang T, Ye RW, Helmann JD. The global transcriptional response of Bacillus subtilis to manganese involves the MntR, Fur, TnrA and sigma B regulons. Mol Microbiol. 2003;49:1477–1491. doi: 10.1046/j.1365-2958.2003.03648.x. [DOI] [PubMed] [Google Scholar]
- 10.Papp-Wallace KM, Maguire ME. Manganese transport and the role of manganese in virulence. Annu Rev Microbiol. 2006;60:187–209. doi: 10.1146/annurev.micro.60.080805.142149. [DOI] [PubMed] [Google Scholar]
- 11.Lieser SA, Davis TC, Helmann JD, Cohen SM. DNA-binding and oligomerization studies of the manganese(II) metalloregulatory protein MntR from Bacillus subtilis. Biochemistry. 2003;42:12634–12642. doi: 10.1021/bi0350248. [DOI] [PubMed] [Google Scholar]
- 12.Golynskiy MV, Davis TC, Helmann JD, Cohen SM. Metal-induced structural organization and stabilization of the metalloregulatory protein MntR. Biochemistry. 2005;44:3380–3389. doi: 10.1021/bi0480741. [DOI] [PubMed] [Google Scholar]
- 13.Golynskiy MV, Gunderson WA, Hendrich MP, Cohen SM. Metal binding studies and EPR spectroscopy of the manganese transport regulator MntR. Biochemistry. 2006;45:15359–15372. doi: 10.1021/bi0607406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glasfeld A, Guedon E, Helmann JD, Brennan RG. Structure of the manganese-bound manganese transport regulator of Bacillus subtilis. Nat Struct Biol. 2003;10:652–657. doi: 10.1038/nsb951. [DOI] [PubMed] [Google Scholar]
- 15.Kliegman JI, Griner SL, Helmann JD, Brennan RG, Glasfeld A. Structural basis for the metal-selective activation of the manganese transport regulator of Bacillus subtilis. Biochemistry. 2006;45:3493–3505. doi: 10.1021/bi0524215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeWitt MA, Kliegman JI, Helmann JD, Brennan RG, Farrens DL, Glasfeld A. The conformations of the manganese transport regulator of Bacillus subtilis in its metal-free state. J Mol Biol. 2007;365:1257–1265. doi: 10.1016/j.jmb.2006.10.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Golynskiy M, Li S, Woods VL, Cohen SM. Conformational studies of the manganese transport regulator (MntR) from Bacillus subtilis using deuterium exchange mass spectrometry. J Biol Inorg Chem. 2007;12:699–709. doi: 10.1007/s00775-007-0216-z. [DOI] [PubMed] [Google Scholar]
- 18.Sen KI, Sienkiewicz A, Love JF, vanderSpek JC, Fajer PG, Logan TM. Mn(II) binding by the anthracis repressor from Bacillus anthracis. Biochemistry. 2006;45:4295–4303. doi: 10.1021/bi052288g. [DOI] [PubMed] [Google Scholar]
- 19.Guedon E, Helmann JD. Origins of metal ion selectivity in the DtxR/MntR family of metalloregulators. Mol Microbiol. 2003;48:495–506. doi: 10.1046/j.1365-2958.2003.03445.x. [DOI] [PubMed] [Google Scholar]
- 20.Allen KN, Lavie A, Glasfeld A, Tanada TN, Gerrity DP, Carlson SC, Farber GK, Petsko GA, Ringe D. Role of the divalent metal ion in sugar binding, ring opening, and isomerization by D-xylose isomerase: replacement of a catalytic metal by an amino acid. Biochemistry. 1994;33:1488–1494. doi: 10.1021/bi00172a027. [DOI] [PubMed] [Google Scholar]
- 21.Fisher CL, Pei GK. Modification of a PCR-based site-directed mutagenesis method. BioTechniques. 1997;23:570–574. doi: 10.2144/97234bm01. [DOI] [PubMed] [Google Scholar]
- 22.Teng TY. Mounting of crystals for macromolecular crystallography in a free-standing thin film. J Appl Crystallogr. 1990;23:387–391. [Google Scholar]
- 23.Leslie A. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4+ ESF-EAMCB Newsletter on Protein Crystallography. 1992;26:27–33. [Google Scholar]
- 24.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 25.Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 26.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 27.Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr D Biol Crystallogr. 1999;55:484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- 28.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Meth Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 29.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Crystallogr. 2006;39:109–111. [Google Scholar]
- 31.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gabriel SE, Helmann JD. Contributions of Zur-controlled ribosomal proteins to growth under zinc starvation conditions. J Bacteriol. 2009;191:6116–6122. doi: 10.1128/JB.00802-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller J. Experiments in molecular genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 34.Cotton FA, Wilkinson G, Murillo CA, Bochmann M. Advanced Inorganic Chemistry. 6. John Wiley & Sons, Inc; New York: 1999. [Google Scholar]
- 35.Ma Z, Faulkner MJ, Helmann JD. Origins of specificity and cross-talk in metal ion sensing by Bacillus subtilis Fur. Mol Microbiol. 2012;86:1144–1155. doi: 10.1111/mmi.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bertini I, Luchinat C. High spin cobalt(II) as a probe for the investigation of metalloproteins. Adv Inorg Biochem. 1984;6:71–111. [PubMed] [Google Scholar]
- 37.Ringe D, White A, Chen S, Murphy JR. Diphtheria Toxin Repressor: Metal Ion Mediated Control of Transcription. In: Messerschmidt A, Huber R, Poulas T, Wieghardt K, Cygler M, Bode W, editors. Handbook of Metalloproteins. John Wiley & Sons, Ltd; Chichester: 2006. pp. 929–938. [Google Scholar]
- 38.Feese MD, Pohl E, Holmes RK, Hol WG. Iron-Dependent Regulators. In: Messerschmidt A, Huber R, Poulas T, Wieghardt K, Cygler M, Bode W, editors. Handbook of Metalloproteins. John Wiley & Sons, Ltd; Chichester: 2006. pp. 850–863. [Google Scholar]
- 39.Stoll KE, Draper WE, Kliegman JI, Golynskiy MV, Brew-Appiah RAT, Phillips RK, Brown HK, Breyer WA, Jakubovics NS, Jenkinson HF, Brennan RG, Cohen SM, Glasfeld A. Characterization and Structure of the Manganese-Responsive Transcriptional Regulator ScaR. Biochemistry. 2009;48:10308–10320. doi: 10.1021/bi900980g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spiering MM, Ringe D, Murphy JR, Marletta MA. Metal stoichiometry and functional studies of the diphtheria toxin repressor. Proc Natl Acad Sci USA. 2003;100:3808–3813. doi: 10.1073/pnas.0737977100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goranson-Siekierke J, Pohl E, Hol WG, Holmes RK. Anion-coordinating residues at binding site 1 are essential for the biological activity of the diphtheria toxin repressor. Infect Immun. 1999;67:1806–1811. doi: 10.1128/iai.67.4.1806-1811.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bates CS, Toukoki C, Neely MN, Eichenbaum Z. Characterization of MtsR, a new metal regulator in group A streptococcus, involved in iron acquisition and virulence. Infect Immun. 2005;73:5743–5753. doi: 10.1128/IAI.73.9.5743-5753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jakubovics NS, Smith AW, Jenkinson HF. Expression of the virulence-related Sca (Mn2+) permease in Streptococcus gordonii is regulated by a diphtheria toxin metallorepressor-like protein ScaR. Mol Microbiol. 2000;38:140–153. doi: 10.1046/j.1365-2958.2000.02122.x. [DOI] [PubMed] [Google Scholar]
- 44.Spatafora G, Moore M, Landgren S, Stonehouse E, Michalek S. Expression of Streptococcus mutans fimA is iron-responsive and regulated by a DtxR homologue. Microbiology. 2001;147:1599–1610. doi: 10.1099/00221287-147-6-1599. [DOI] [PubMed] [Google Scholar]
- 45.Tao X, Murphy JR. Binding of the metalloregulatory protein DtxR to the diphtheria tox operator requires a divalent heavy metal ion and protects the palindromic sequence from DNase I digestion. J Biol Chem. 1992;267:21761–21764. [PubMed] [Google Scholar]
- 46.Qiu X, Verlinde CL, Zhang S, Schmitt MP, Holmes RK, Hol WG. Three-dimensional structure of the diphtheria toxin repressor in complex with divalent cation co-repressors. Structure. 1995;3:87–100. doi: 10.1016/s0969-2126(01)00137-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.