

Abstract
In addition to inhibiting the cyclooxygenasemediated biosynthesis of prostanoids, various widely used non-steroidal anti-inflammatory drugs (NSAIDs) enhance endocannabinoid signaling by blocking the anandamidedegrading membrane enzyme, fatty acid amide hydrolase (FAAH). The X-ray structure of FAAH in complex with the NSAID carprofen, along with studies of site-directed mutagenesis, enzyme activity assays, and nuclear magnetic resonance, now reveal the molecular details of this interaction, providing information that may guide the design of dual FAAH-cyclooxygenase inhibitors with superior analgesic efficacy.
Non-steroidal anti-inflammatory drugs (NSAIDs), one of the most widely used classes of therapeutic agents, alleviate pain and inflammation1 by inhibiting the enzymes cyclooxygenase-1 (COX-1) and COX-2,2 which catalyze the conversion of membrane-derived arachidonic acid into the prostaglandin endoperoxides, PGG2 and PGH2.3 This reaction is the first committed step in the biosynthesis of the prostanoids,4 lipid messengers that cause pain and inflammation by engaging G protein–coupled receptors present on the surface of innate-immune and neural cells.5
Evidence indicates that the analgesic actions of the NSAIDs are enhanced in a synergistic manner by drugs that inhibit fatty acid amide hydrolase (FAAH),6 a serine enzyme responsible for the deactivation of the endogenous cannabinoid receptor agonist anandamide.7 By increasing anandamide levels, FAAH inhibitors8 heighten the ability of this compound to control emerging nociceptive signals9 – such as the prostanoids – resulting in a super-additive potentiation of NSAID-mediated analgesia. In addition to magnifying the analgesic actions of the NSAIDs, FAAH inhibitors reduce the frequency and severity of gastric side effects exerted by those compounds.10
These data suggest that dual inhibitors of FAAH and COX might provide superior efficacy and greater safety than current non-narcotic analgesics.11 This possibility is supported by recent studies that have implicated FAAH blockade in the analgesic properties of indomethacin and ibuprofen, two clinically important NSAIDs.12 Despite the therapeutic relevance of this hypothesis, the molecular mechanism through which NSAIDs inhibit FAAH remains unknown. To fill this knowledge gap, in the present study we solved the crystal structure of FAAH in complex with the ibuprofen analogue carprofen, and investigated this interaction using a combination of site-directed mutagenesis, enzyme activity assays, and nuclear magnetic resonance (NMR).
In a first set of experiments, we tested a representative set of commercially available NSAIDs for their ability to inhibit FAAH and identified one, carprofen [(RS)-2-(6-chloro-9H-carbazol-2-yl)propanoic acid], which reduced FAAH activity in rat brain homogenates with a median effective concentration (IC50) of 79±20 μM (mean±s.e.m., n=3; assays were conducted at pH 7.4, Supporting information, Methods). Carprofen was approximately as potent as indomethacin (IC50 = 68±4 μM) and more potent than ibuprofen (IC50 = 711±44 μM), two NSAIDs that have been previously shown to inhibit FAAH.13 As expected from studies with other NSAIDs, carprofen’s inhibition of FAAH activity was weaker at neutral than acidic pH conditions (IC50 at pH 6.0 = 15.5±0.1 μM; Supporting Figure 1 and Methods). To investigate the mechanism through which carprofen inhibits FAAH, we crystallized recombinant rat FAAH in complex with this drug and solved the structure at 2.25 Å resolution (Supporting information, Table 1). Diffracting crystals of the FAAH/carprofen complex could be obtained by pre-incubating FAAH with the O-arylcarbamate inhibitor URB597 ([3-(3-carbamoylphenyl)phenyl] N-cyclohexylcarbamate).14
The electron density map revealed that carprofen occupied a space located at the entrance of the membrane-access (MA) channel of FAAH (Figure 1a), an elongated cavity that allows substrates to enter the enzyme’s active site (Figure 1b). The propanoic acid group of carprofen remained partially exposed to the solvent, where its higher mobility produced a weaker and less defined electron density map (Figure 1a). This group, which is likely to be ionized at the pH used for crystallization (pH = 7.5), formed an H-bond with the side-chain nitrogen of Trp531 (Figure 1b). On the other hand, the carbazole ring and chloride atom of carprofen were positioned within the MA channel and were enshrouded by hydrophobic amino-acid residues, which formed a tight and well-modeled binding site (Figure 1b and Figure 2a) at ideal interaction distances (Figure 2b).
Figure 1.
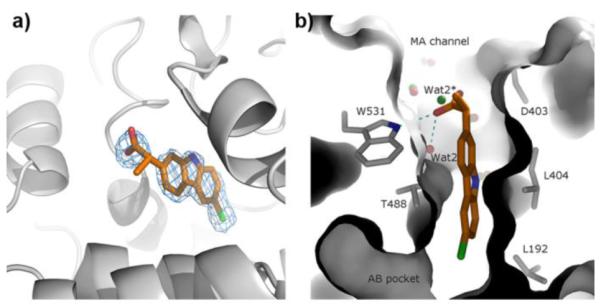
Structure of carprofen bound to FAAH. a) Position of carprofen (carbon atoms shown in orange) at the entrance of the active-site gorge of FAAH. The 2-arylpropionic acid group of carprofen protrudes from the enzyme active site cleft. The electron density map (2Fo-Fc) corresponding to the position of the drug (in sky-blue) is contoured at 1.0 σ. b) Binding of carprofen (carbon atoms shown in orange) in the membrane access channel of FAAH. MA, membrane access; AB, acyl binding. Water molecules are depicted as red spheres. H-bonds involving the carprofen carboxylate, Wat2, and W531 are represented as cyan dashed lines. Green spheres represent superimposed water molecules of the complex structure FAAH-URB597 (PDB code: 3LJ7) and show the different position of the corresponding Wat2 (highlighted by an asterisk) upon inhibitor binding. Single-letter abbreviations of amino acids have been used for clarity.
Figure 2.

Interaction between carprofen and FAAH. a) Stereoview showing the interaction between carprofen (carbon atoms shown in orange) and residues of the FAAH membrane access channel. Selected residues and water molecules facing carprofen are shown as sticks and red spheres, respectively, and their H-bonds represented as cyan dashed lines. Water molecules Wat1 and Wat2 form H-bonds involving the carbazole nitrogen and carboxylate of carprofen, respectively. Single-letter abbreviations of amino acids have been used for clarity. b) Interaction distances between carprofen and FAAH active site residues (Å).
An intense H-bond network of water molecules further stabilized the complex, filling the space between carprofen and the amino-acid residues flanking the MA cavity (Figure 2a). These water molecules occupied positions that were similar to those previously reported for the structure of the FAAH/URB597 complex.15 One notable exception was represented by water-2 (wat2), which in the FAAH/carprofen complex bridged through H-bonds the propionic acid moiety of carprofen with residues Thr488 and Gly485 (Figure 2a). Wat2 was located approximately 3.5 Å away from the site occupied by its counterpart in the FAAH/URB597 complex (Figure 1b). A comparison between the FAAH/carprofen and FAAH/URB597 complexes revealed that carprofen binding was associated with a marked structural rearrangement of the MA channel. For example, Phe432 and Met436 adopted side-chain conformations that were substantially different in the two complexes (Supporting information, Figure 2a). This restructuring of the MA cavity may contribute to maximize the interaction between the NSAID and FAAH.
To determine whether the binding interaction identified by X ray crystallography might influence FAAH activity, we measured the inhibitory potencies of carprofen on wild-type FAAH and a FAAH mutant in which Thr488 – one of the residues most directly involved in the interaction with carprofen (Figure 1b and Figure 2a) – was replaced with alanine. In agreement with the X-ray complex outlined above, we found that carprofen was more potent at inhibiting purified recombinant wild-type FAAH (IC50 = 74±8 μM, assays conducted at pH 6) than the mutated protein Thr488Ala (IC50 = 165±7 μM) (Figure 3). A similar difference was observed when activity assays were carried out under neutral pH conditions (pH = 7.4; wild-type FAAH: IC50 = 381 μM, and Thr488Ala FAAH IC50 = 647 μM). In contrast with carprofen, the O-arylcarbamate derivative URB597 inhibited wild-type FAAH and Thr488Ala FAAH with similar potencies (3.1±0.96 nM and 3.4±1.04 nM at pH = 7.4, respectively), suggesting that the two compounds interact with distinct binding sites of FAAH. In agreement with this conclusion, previous15 and present X-ray crystallography studies showed that URB597 reacted with the catalytic nucleophile Ser241 to form a cyclohexyl carbamate adduct that was positioned at the inner extremity of the MA channel (Supporting information, Figure 2b).
Figure 3.
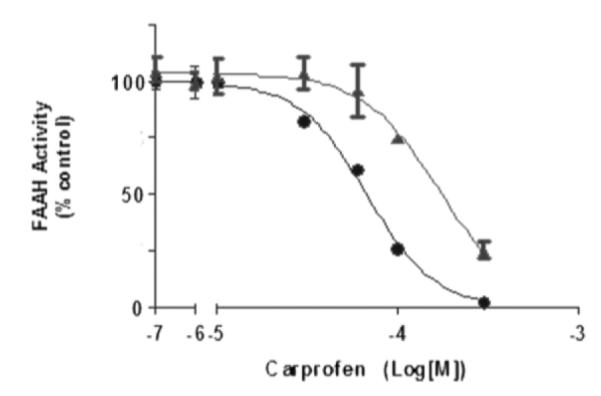
Carprofen inhibition profile on wild-type FAAH (•) and Thr488Ala FAAH (▲) using [H3]-anandamide hydrolysis assay. Data are expressed as percent of control activity, which was 81364.96 pmol/minute/mg protein for the wild-type, and 12605.33 pmol/minute/mg for the mutant. The results suggest that the interaction of carprofen with Thr488 is important for FAAH inhibition.
To assess whether this modification might impair the ability of carprofen to interact with FAAH, we measured the binding of this compound in the presence or absence of URB597, using the WaterLOGSY NMR spectroscopy method (Supporting information, BOX1). Consistent with the structural data, positive WaterLOGSY signals indicated that carprofen was able to bind FAAH, and that pre-incubating the enzyme with URB597 did not alter such binding (Supporting information, Figure 3).
Finally, we asked whether the structure of the carprofen complex reported here might provide insights on the interaction of FAAH with other NSAIDs. We used a highly sensitive NMR method (Fluorine Atoms for Biochemical Screening, n-FABS; Supporting information, BOX1) to monitor the FAAH-mediated hydrolysis of ARN1203 (Figure 4a and Supporting information, Methods) – a specifically designed fluorinated analogue of anandamide16 – in the presence of a representative set of commercially available NSAIDs (Figure 4a and Supporting information, Figure 4 and Table 2). Hydrolytic cleavage of ARN1203 by FAAH produced changes in the 19FNMR signal, which were used to measure enzyme inhibition. FABS analyses on twenty NSAIDs yielded the following rank order potency of FAAH inhibition: indomethacin ≈ carprofen > tenidap > flurbiprofen > ibuprofen. This was in line with the rank order potency obtained for carprofen, indomethacin and ibuprofen using a standard enzyme assay (above) and pointed to common pharmacophore functions in the NSAID class, which might be responsible for FAAH inhibition. Indeed, the three most active compounds (indomethacin, carprofen and tenidap) share a common planar bicyclic 5-6 fused system and a chlorine atom linked to one of the phenyl rings (Figure 4b). Possible interactions of these structural elements with the MA channel of the enzyme may be gleamed from the FAAH/carprofen complex (Figure 2).
Figure 4.
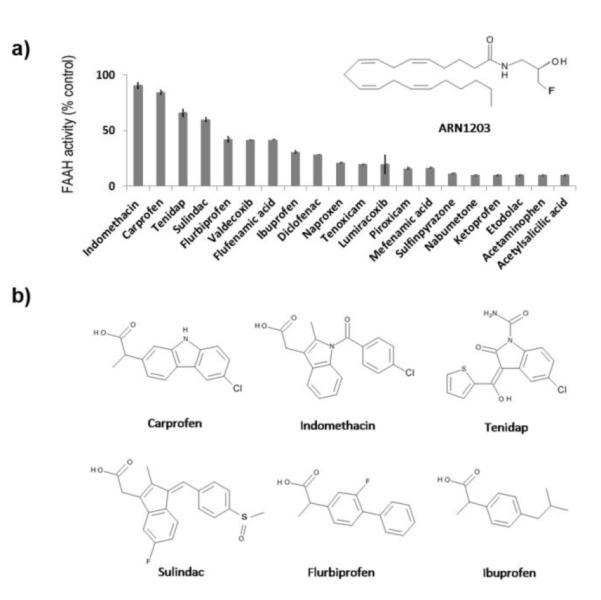
NSAIDs that inhibit FAAH activity share a common chemical signature. a) FAAH inhibition (% control) of representative NSAIDs assessed using the NMR-based FABS method. Compounds were tested at 200 μM. Inset: Chemical structure of the fluorinated substrate. b) Chemical structures of the NSAIDs that were most active at inhibiting FAAH. Note the presence of a planar bicyclic 5-6 fused system, a chlorine atom linked to one of phenyl ring, and a carboxylate group. The FAAH/carprofen complex (Figure 2) points to possible interactions of these chemical groups with FAAH.
Carprofen is currently utilized in veterinary medicine owing to its high analgesic effectiveness and relatively lack of gastric side effects.17 The present study shows that this NSAID binds to a set of amino acid residues located at the entrance of FAAH’s active site, with the carboxylate group of the molecule protruding at the exterior of the enzyme. This interaction (a) appears to influence FAAH function, because removal of one of its key components, Thr488, markedly weakens the ability of carprofen to inhibit FAAH activity; and (b) distinguishes carprofen from other known FAAH inhibitors, which occupy either the core of the substratebinding cavity (URB597, OL-135 and PF-3845)18 (Supporting information, Figure 5a) or, less frequently, the entirety of the MA channel (ketobenzimidazoles)19 (Supporting information, Figure 5b). Furthermore, our n-FABS experiments reveal several chemical commonalities that might underpin the ability of clinically important NSAIDs, such as indomethacin, to inhibit FAAH activity. This structural information provides novel insights into the mode of action of the NSAIDs and may help to design superior analgesic agents that act by simultaneously targeting FAAH and COX.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. Carlo Patrono, Dr Glauco Tarozzo, Dr. Rosalia Bertorelli, Dr Stefania Girotto, Dr Claudio Dalvit, and Dr Tiziano Bandiera for helpful discussions, and the ESRF BM14 beamline staff for help with data collection. We thank the National Institute on Drug Abuse (grant DA012413 to D.P.) and the Marie Curie Action IRG for financial support (FP7-PEOPLE-2010-RG, contract n. PIRG07-GA-2010-268385 to G. G.).
Footnotes
Drug Discovery and Development, Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genoa, Italy.
Supporting Information Placeholder
ASSOCIATED CONTENT Supporting Information Additional figures and experimental procedures for construct generation, mutagenesis, protein expression and purification, nuclear magnetic resonance, in vitro assays, structure determination, table of crystallographic analysis and refinement statistics, are included in the supporting information. This material is available free of charge via the internet at http://pubs.acs.org. The atomic coordinates and the crystallographic structure factors of FAAH complexed with carprofen have been deposited in the Protein Data Bank database with accession code 4DO3. Figures of the protein structure were prepared using PyMOL (http://www.pymol.org).
Notes The authors declare no competing financial interests.
REFERENCES
- (1).Scholz J, Woolf CJ. Nat. Neurosci. 2002;5:1062–1067. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- (2) (a).Vane JR. Nature. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]; (b) Nuki G. Br. Med. J. 1983;287:39–43. doi: 10.1136/bmj.287.6384.39. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Meade EA, Smith WL, DeWitt DL. J Biol Chem. 1993;268:6610–6614. [PubMed] [Google Scholar]
- (3) (a).Bergström S, Danielsson H, Klenberg D, Samuelsson B. J. Biol. Chem. 1964;239:4006–4008. [PubMed] [Google Scholar]; (b) Marnett LJ, Rowlinson SW, Goodwin DC, Kalgutkar AS, Lanzo CA. J. Biol. Chem. 1999;274:22903–22906. doi: 10.1074/jbc.274.33.22903. [DOI] [PubMed] [Google Scholar]; (c) Malkowsky MG, Ginell SL, Garavito RM. Science. 2000;289:1933–1937. doi: 10.1126/science.289.5486.1933. [DOI] [PubMed] [Google Scholar]; (d) Funk CD. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- (4) (a).Kiefer JR, Pawlitz JL, Moreland KT, Stegeman RA, Hood WF, Gierse JK, Stevens AM, Goodwin DC, Rowlinson SW, Marnett LJ, Stallings WC, Kurumbail RG. Nature. 2000;405:97–101. doi: 10.1038/35011103. [DOI] [PubMed] [Google Scholar]; (b) Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Annu. Rev. Pharmacol. Toxicol. 2001:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- (5).Pertwee RG, Howlett AC, Abood ME, Alexander SPH, Di Marzo V, Elphick MR, Greasley PJ, Hnasen HS, Kunos G, Mackie K, Mechoulam R, Ross RA. Pharmacol. Rev. 2010;62:588–632. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6) (a).Holt S, Comelli F, Costa B, Fowler C. J. Br. J. Pharm. 2005;146:467–476. doi: 10.1038/sj.bjp.0706348. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Naidu PS, Booker L, Cravatt B, Lichtman AH. J. Pharmacol. Exp. Ther. 2009;329:48–56. doi: 10.1124/jpet.108.143487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]; (b) Bracey MH, Hanson MA, Msuda KR, Stevens RC, Cravatt B. J. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]; (c) Cravatt BF, Lichtman AH. Curr. Opin. Chem. Biol. 2003;7:469. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]
- (8) (a).Piomelli D. Nature Rev. Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]; (b) Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier A. J. Br. J. Pharmacol. 2006;148:102. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Palmer JA, Higuera ES, Chang L, Chaplan SR. Neuroscience. 2008;154:1554. doi: 10.1016/j.neuroscience.2008.04.047. [DOI] [PubMed] [Google Scholar]; (d) Kinsey SG, Long JZ, O’Neal ST, Abdulla RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. J. Pharmacol. Exp. Ther. 2009;330:902. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BF. J. Pharmacol. Exp. Ther. 2004;311:441. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]; (e) Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. J. Med. Chem. 2004;47:4998. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- (9) (a).Calignano A, La Rana G., Giuffrida A, Piomelli D. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]; (b) Cravatt BF, Demarest K, Patricelli MP, Bracey HM, Giang DK, Martin BR, Lichtman AH. Proc. Nat. Acad. Sci. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Clapper JR, Moreno Sanz G., Russo R, Guijarro A, Vacondio F, Durati A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, Hohmann AG, Calignano A, Mor M, Tarzia G, Piomelli D. Nat. Neurosci. 2010;13:1265–1270. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sasso O, Bertorelli R, Bandiera T, Scarpelli R, Colombano G, Armirotti A, Moreno-Sanso G, Reggiani A, Piomelli D. Pharmacol. Res. 2012;65:553–563. doi: 10.1016/j.phrs.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Fowler CJ, Naidu PS, Lichtman A, Onnis V. Br. J. Pharmacol. 2009;156:412–419. doi: 10.1111/j.1476-5381.2008.00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gühring H, Hamza M, Sergejeva M, Ates M, Kotalla CE, Ledent C, Brune K. Eur. J. Pharmacol. 2002;454:153–163. doi: 10.1016/s0014-2999(02)02485-8. [DOI] [PubMed] [Google Scholar]
- (13) (a).Holt S, Paylor B, Boldrup L, Alajakku K, Vandevoorde S, Sundstrom A, Cocco MT, Onnis V, Fowler C. J. Eu. J Pharmacol. 2007;565:26–36. doi: 10.1016/j.ejphar.2007.02.051. [DOI] [PubMed] [Google Scholar]; (b) Holt S, Nilsson J, Omeir R, Tiger G, Fowler C. J. Br. J. Pharmacol. 2001;133:513–520. doi: 10.1038/sj.bjp.0704113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Piomelli D, Tarzia G, Durati A, Tontini A, Mor M, Compton TR, Dasse O, Monaghan EP, Parrott JA, Putman D. CNS Drug Reviews. 2006;12:21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mileni M, Kamtekar S, Wood DC, Benson TE, Cravatt BF, Stevens RC. J. Mol. Biol. 2010;400:743–754. doi: 10.1016/j.jmb.2010.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lambruschini C, et al. in preparation.
- (17) (a).Fox SM, Johnston SA. J. Am. Vet. Med. Assoc. 1997;210:1493–1498. [PubMed] [Google Scholar]; (b) Radi ZR, Nasir KK. Exp. Toxicol. Pathol. 2006;58:163–173. doi: 10.1016/j.etp.2006.06.004. [DOI] [PubMed] [Google Scholar]
- (18) (a).Mileni M, Johnson DS, Wang Z, Everdeen DS, Liimatta M, Pabst B, Bhattacharya K, Nugent RA, Kamtekar S, Cravatt BF, Ahn K, Stevens RC. Proc. Natl. Acad. Sci. U S A. 2008;105:12820–12824. doi: 10.1073/pnas.0806121105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mileni M, Garfunkle J, DeMartino JK, Cravatt BF, Boger DL, Stevens RC. J. Am. Chem. Soc. 2009;131:10497–10506. doi: 10.1021/ja902694n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF. Chem Biol. 2009;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Min X, Thibault ST, Porter AC, Gustin DJ, Carlson TJ, Xu H, Lindstrom M, Xu G, Uyed C, Ma Z, Yihong L, Kayser F, Walker NPC, Wang Z. Proc. Natl. Ac. Sci. USA. 2011;108:7379–7384. doi: 10.1073/pnas.1016167108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ezzili C, Mileni M, McGlinchey N, Long JZ, Kinsey SG, Hochstatter DG, Stevens RC, Lichtman AH, Cravatt BF, Bilsky EJ, Boger DL. J. Med. Chem. 2011;54:2805–2822. doi: 10.1021/jm101597x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Mileni M, Garfunkle J, Ezzili C, Cravatt BF, Stevens RC, Boger DL. J. Am. Chem. Soc. 2011;133:4092–40100. doi: 10.1021/ja110877y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Otrubova K, Ezzili C, Boger D. Bioorg. Med. Chem. Lett. 2011;16:4674–4685. doi: 10.1016/j.bmcl.2011.06.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Min X, Thibault ST, Porter AC, Gustin DJ, Carlson TJ, Xu H, Lindstrom M, Xu G, Uyed C, Ma Z, Yihong L, Kayser F, Walker NPC, Wang Z. Proc. Natl. Ac. Sci. 2011;108:7379–7384. doi: 10.1073/pnas.1016167108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.