

Abstract
Since the discovery of IL-17 in 1995 as a T-cell cytokine, inducing IL-6 and IL-8 production by fibroblasts, and the report of a separate T-cell lineage producing IL-17(A), called Th17 cells, in 2005, the role of IL-17 has been studied in several inflammatory diseases. By inducing IL-8 production and subsequent neutrophil attraction towards the site of inflammation, IL-17A can link adaptive and innate immune responses. More specifically, its role in respiratory diseases has intensively been investigated. We here review its role in human respiratory diseases and try to unravel the question whether IL-17A only provides a link between the adaptive and innate respiratory immunity or whether this cytokine might also be locally produced by innate immune cells. We furthermore briefly discuss the possibility to reduce local IL-17A production as a treatment option for respiratory diseases.
1. Introduction
In 1995, Yao and coworkers discovered a cytokine, produced by human T cells, which induced IL-6 and IL-8 production and enhanced the surface expression of the intracellular adhesion molecule-1 (ICAM-1) by human fibroblasts [1]. Since then, several IL-17 family members have been described [2–4]. The family member, which was originally described, is referred to as IL-17A ever since. More recently, a separate T-cell lineage, called Th17 cells or inflammatory T cells, producing IL-17A, has been identified [5, 6]. Th17 cells might potentially play an important role in the pathophysiology of respiratory diseases such as asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), and lung transplant rejection. Human Th17 cells express retinoic acid receptor-related orphan receptor (ROR)C and produce, besides IL-17A, also IL-17F, IL-21, IL-22, and IL-26 [7, 8]. Human Th17 cells furthermore express CCR6 [8]. Patients with defective Th17 cells are known to suffer from severe infections by fungi and extracellular bacteria such as Candida albicans and Staphylococcus aureus [8]. Even Th17 cells might form a heterogeneous family: indeed, human hybrid Th17/Th1 cells (producing both IL-17A and IFN-γ as well as expressing both RORC and T-bet), human hybrid Th17/Treg cells (expressing CCR6, RORC and Foxp3), and human Th17/Tr1-like cells (producing both IL-17 and, for a limited period of 3 to 5 days after initial stimulation, also IL-10) have been described [8]. There recently has been a lot of interest in the local microbiota interacting with specific T-cell subsets, and hereby controlling recruitment and further differentiation of certain T-cell subsets. Candida albicans-specific Th17 cells are hybrids and produce IL-17 and IFN-γ, but no IL-10, whereas Staphylococcus aureus-specific Th17 cells produce IL-17 and also IL-10 upon restimulation [9]. IL-6, IL-23, and IL-1β were shown to contribute to Th17 differentiation induced by both pathogens. However, amongst these cytokines, IL-1β was essential in C. albicans-induced Th17 differentiation to counteract the inhibitory activity of IL-12 and to prime for IL-17/IFN-γ double-producing cells [9]. On the other hand, germination and biofilm formation of certain fungi can also be induced upon their interaction with “host” IL-17A [10]. Aspergillus fumigatus can increase its biofilm formation upon binding IL-17A and its filamentation can be promoted, inducing resistance to local antifungal defenses [10]. This interaction might be of utmost importance in CF patients, in whom the defective clearance of certain pathogens leads to chronic colonization and/or infection with certain pathogens, including Staphylococcus aureus and/or Aspergillus fumigatus.
It still remains enigmatic whether the cited respiratory diseases are specifically mediated by local Th17 cells, able to produce several other cytokines, or by the cytokine IL-17A itself, which might be secreted besides by Th17 cells, by several other cell types. Indeed, IL-17A can also be produced by γδT cells, NKT cells, and innate lymphoid cells, such as NK cells, lymphoid tissue inducer (LTi) cells, and innate lymphoid cells with specific IL-17A-secreting capacity, ILC17 [11, 12]. In humans, LTi cells and ILC17 cells might be overlapping cell populations [11]. Moreover, in murine models, IL-17A production in the lungs during invasive fungal infection was even shown to be mediated by CD11b(+) Ly-6G(+) neutrophils [13] and by alveolar macrophages upon allergen challenge [14]. Human neutrophils from cystic fibrosis patients were also shown to produce IL-17A [15].
Human IL-17A binds to its receptor IL-17RA, which, albeit weakly, also binds IL-17F [12]. IL-17RA binds with IL-17RC and can form heterodimers for optimal signalling [12]. IL-17RA is highly expressed in haematopoietic tissues and main responses to IL-17A are observed in epithelial cells, endothelial cells, fibroblasts, macrophages, and dendritic cells [12]. Several airway cells might therefore respond locally to IL-17A.
IL-17A is especially important for the recruitment of neutrophils [16]. The mechanism of neutrophil recruitment to the airways is still unclear, but several chemokines were suggested to play a role [17]. Among those, CXCL8 (IL-8), secreted by T lymphocytes, epithelial cells, smooth muscle cells, and macrophages might especially be important, as CXCL8 (IL-8) is also increased in the airways of patients with some respiratory diseases such as CF, COPD, and asthma [18–22]. Neutrophil recruitment is an utmost important defence mechanism against local pathogens, but persisted neutrophil recruitment might lead to chronic inflammation with local production of neutrophilic mediators, probably involved in the physiopathology of different respiratory diseases.
IL-17 (and IL-6) are furthermore major inducers of mucin genes in vitro, suggesting that IL-17 production might also be responsible for mucus secretion in IL-17-mediated respiratory diseases [23]. We here review the role of IL-17A and neutrophil recruitment in several human respiratory diseases: asthma, COPD, CF, and lung transplant rejection.
2. IL-17A in Asthma
The role of IL-17A and/or Th17 cells in asthma has been extensively studied in mouse models (reviewed in [24]). However, human data are scarce. Studies to unravel the role of IL-17A in asthma started in the late nineties by expression studies in airway cells. IL-17A was shown to be expressed in bronchial biopsies, bronchoalveolar lavage fluid and sputum of patients with asthma [22, 25–27]. We were the first to show that compared to healthy control individuals, sputum IL-17A and IL-8 mRNA levels were significantly higher in mild and moderate-to-severe asthmatics, whereas sputum IL-17A and IL-8 mRNA expression was similarly distributed in nonallergic and allergic patients [22]. Later on Doe and coworkers observed increased numbers of IL-17A+ cells in bronchial biopsies of mild asthmatics compared to healthy controls, whereas IL-17F+ cells were increased in mild and especially in moderate-to-severe asthmatics [28]. A recent study furthermore showed that upon bronchial house-dust-mite challenge systemic IL-17A levels increase in house-dust-mite allergic individuals [29]. Although this increase was clearly demonstrated, it remains unknown whether IL-17 (or Th17 cells?) is (are) increased in an attempt to protect the individual, or in contrary, might cause local harm. IL-17A is thought to be responsible for neutrophil recruitment, as a result of chemokine induction, such as IL-8. Neutrophils are especially prominent in acute, severe exacerbations of asthma [30, 31]. The subgroup of patients with neutrophilic asthma is characterized by poor response to corticosteroids [32]. Neutrophils potentially contribute to airway gland hypersecretion, bronchial hyperreactivity and to airway wall remodelling [33, 34] by producing matrix metalloproteinase-9 (MMP-9) observed in bronchoalveolar lavage fluid from moderate-to-severe asthma patients [35]. We found in our asthmatic study population that both sputum IL-17A and IL-8 mRNA levels correlate significantly with the sputum neutrophil count [22]. This correlation was also observed by other authors at the protein level [26]. A significant correlation between IL-8 mRNA and IL-17A mRNA on the one hand and with neutrophils on the other hand suggests (although it does not provide proof for) cause-effect relationship. On the other hand, Doe and coworkers observed that the number of IL-17A+ cells was correlated with FEV1% predicted (R s = 0.38; P = .04), and the sputum neutrophil count (R s = −0.43,P = .03) in asthma but was not associated with the number of neutrophils or eosinophils in the bronchial mucosa [28]. They furthermore reported a good correlation between the number of IL-17F+ cells and eosinophils in the bronchial submucosa [28]. Human bronchial epithelial cells have recently been shown to secrete IL-17F in response to IL-33 [36]. IL-33, secreted by epithelial cells or macrophages (innate “immune” cells), might therefore induce IL-17F production by epithelial cells, leading to eosinophil recruitment in initial asthma [37]. Later on IL-17A, produced by both innate and adaptive immune cells, can induce neutrophil recruitment mediated by IL-8 secretion, leading to persistent neutrophilic asthma.
3. IL-17A in COPD
COPD is characterized by airway obstruction as well as emphysematous destruction, secondary to chronic inflammation induced by cigarette smoke, involving neutrophil recruitment.
As for asthma, human studies on the potential involvement of IL-17 in COPD, started with expression studies in sputum or BAL from COPD patients [27, 28]. However, levels of IL-17 in sputum of COPD patients were significantly lower than in asthma (P = 0.004) and did not differ from levels in healthy controls [27]. Even decreased serum and sputum IL-17A levels in COPD patients compared to healthy controls have been reported [38]. More recently Doe and coworkers, on the other hand, were able to demonstrate that smoking was associated with increased numbers of IL-17A+ cells in bronchial biopsies, but numbers were not further increased in patients suffering from COPD [28]. Along the same line, increased RORγt expression in lung tissue was found in COPD patients and normal smokers compared to healthy control nonsmokers [39]. However, these authors also observed that increased RORγt levels paralleled the aggravation of the disease [39]. Smoking therefore certainly seems to upregulate IL-17A production in the airways, but it remains unclear whether COPD is associated with further increase in airway IL-17A expression. Differences in microbiota might furthermore be responsible for differences in airway IL-17A cytokine expression [40]. In a recent study, the authors demonstrated that some smokers had less diverse lung microbiota relative to smokers with normal lung function, indicating that alteration in lung microbiota can occur in subjects with no spirometric evidence of disease COPD, but this could be the first signature of disease progression. Although more study is warranted, at least, the results of the cited study provide a framework for characterizing the role of two important pathogens, Pseudomonas and Haemophilus, in the development, progression, and/or exacerbation of COPD [40]. The role of these two pathogens is certainly well studied in CF patients.
4. IL-17A in Cystic Fibrosis
The major cause of morbidity and mortality in CF is lung damage characterised by bronchiectasis. This damage is the result of the vicious cycle of chronic infection and inflammation with production of harmful products, such as proteases and oxidants, secreted mainly by neutrophils. A major factor in the respiratory health of CF subjects is chronic Pseudomonas aeruginosa infection which is associated with a poor clinical outcome [41]. Chronic colonization with Staphylococcus aureus and/or Haemophilus influenzae also occur but seems to be more frequent in children suffering from CF compared to adults with CF [42]. McAllister and colleagues found elevated IL-17A (and IL-17A-inducing IL-23) protein levels in BAL-fluid and sputum of 8 CF patients during exacerbation [43]. We were the first to show increased sputum IL-17A (and IL-23 mRNA) levels in a group of stable adult CF patients when compared to healthy controls [44]. Chronic infection with P. aeruginosa (but not Staphylococcus aureus) was associated with higher mRNA expression of these cytokines in CF airways [44]. These results suggested a potential role for the IL-23/IL-17A axis in CF lung inflammation (and/or host response to P. aeruginosa) and suggested that the adaptive immune system might be involved in the pathophysiology of CF lung disease. However, the study did not reveal the source of this IL-17A protein expression. Recent data clearly demonstrated the presence of CD4+IL-17A+ adaptive T cells in airway walls of established CF as well as newly diagnosed CF patients [15]. On the other hand, these authors as well as Brodlie and their coworkers demonstrated that neutrophils might be a second source of IL-17A in CF patients, leading to a potential vicious cycle of neutrophilic inflammation in patients with CF [15, 45]. Consistent with this potential vicious cycle of IL-17A production after initial neutrophil recruitment to the airways, we observed higher sputum IL-17A mRNA levels in children with CF who had at least one exacerbation in the past 12 months, compared with those without exacerbation (Figure 1). We hypothesize that with each exacerbation neutrophils are attracted to the airways upon IL-8 production induced by local IL-17A and they have the potential to secrete IL-17A, leading to chronic neutrophil recruitment, explaining both the increased sputum neutrophil levels and the increased IL-17A levels in patients with stable CF. Moreover, recent data demonstrate that neutrophils from CF patients have deficient bacterial clearance capacity and slower rate of apoptosis compared to healthy subjects [46]. Neutrophils therefore remain in the airways of CF patients, while deficient in bacterial clearance. This in turn might lead to shift in the bacterial expression of virulence factors [46].
Figure 1.
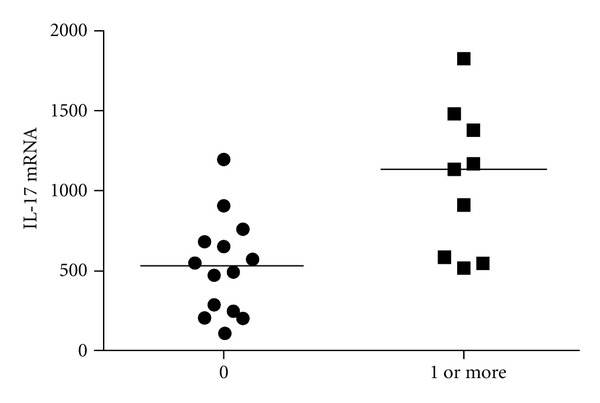
Sputum IL-17A mRNA levels in children with CF. Sputum mRNA was extracted from 23 children with CF aged 5–17 years, as described [47]. IL-17A mRNA levels were measured by real-time RT-PCR [44]. Sputum IL-17A mRNA levels in 9 CF children experiencing at least one exacerbation in the past 12 months (1 or more) were compared to those without exacerbation (n = 14). *P < 0.05 by Mann-Withney U test.
Furthermore, the chronic IL-17A expression might in turn instruct the local microbiota and lead to increased virulence of some pathogenic strains. Aspergillus fumigatus can increase its biofilm formation upon binding IL-17A and its filamentation can be promoted, inducing resistance to local antifungal defenses [10]. This might be of utmost relevance in a subgroup of CF patients, namely patients with allergic bronchopulmonary aspergillosis (ABPA). As indicated in Figure 2, and fitting with this hypothesis, we found increased sputum IL-17A mRNA levels in CF patients with APBA, compared to CF patients without ABPA. In these patients, the increased IL-17A levels might, although necessary for the defence against these fungi, in turn be responsible for the chronic aspergillosis. Amongst patients with ABPA, those patients receiving active antifungal treatment and corticosteroids (active APBA) had the highest sputum IL-17A mRNA levels (our own unpublished results).
Figure 2.
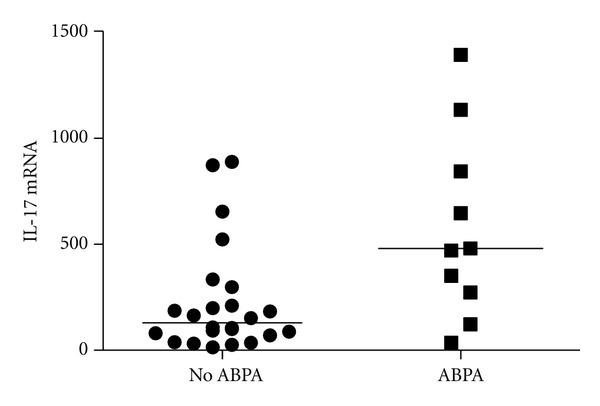
Sputum IL-17A mRNA levels in CF patients without and with ABPA. Sputum mRNA was extracted from adult CF patients without (n = 22) and with (n = 9) ABPA and IL-17A mRNA levels were measured by real-time RT-PCR [44]. ABPA was defined on the basis of (1) deteriorating lung function, (2) immediate Aspergillus fumigatus (af) skin test reactivity, (3) serum total IgE > 1000 IU/mL, (4) elevated af specific IgE and IgG antibodies, and (5) chest radiographic infiltrates. *P < 0.05 by Mann-Withney U test.
5. IL-17A in Acute Rejection and/or BOS after Lung Transplantation
Lung transplantation is now a well-established treatment option for patients with end-stage pulmonary disease, end-stage cystic fibrosis in particular. Fifty to seventy percent of transplant patients experience one or more episodes of acute rejection within the first three months after transplantation [48, 49]. However, this acute rejection is associated with increased risk of chronic rejection or bronchiolitis obliterans syndrome (BOS), which causes transplant loss and bad clinical outcome [50]. In collaboration with our colleagues from the lung transplantation unit at our university, we were able to demonstrate that IL-17A protein is temporary upregulated in BAL fluid during acute rejection, associated with increased lymphocyte numbers as well as with increased neutrophil numbers (which correlated significantly with the IL-8 protein levels in BAL) [51]. Later on, we also showed that BOS patients have the highest BAL IL-17A (and IL-23) mRNA levels when compared to stable transplant patients and patients with acute rejection; levels were comparable to levels in infected patients [52]. Colonization with gram negatives, fungi, or multiple pathogens was on the contrary, only slightly more present in BOS (9, 2, and 1 (of whom 10 Pseudomonas+) on 36, resp.) than in stable transplant (6, 1, and 4 on 42, resp.) patients [52]. These data highlighted the involvement of the IL-23/IL17A axis in lung transplantation rejection. However, the source of the IL-17A production was not studied. Recently, Hodge and colleagues observed that IL-17+ NKT-like cells are increased in peripheral blood from lung transplant recipients compared to healthy control subjects [53]. Whether these cells are indeed also responsible for the local increase in IL-17A in BAL fluid from lung transplant patients is not known. Burlingham and colleagues suggested that while alloimmunity initiates lung transplant rejection, de novo autoimmunity mediated by collagen V (col(V))-specific Th17 cells as well as monocyte/macrophage accessory cells ultimately causes progressive airway obliteration [54]. Recent data also suggested that a shift in immunodominance of self-antigenic determinants of Col-V results in the induction of IFN-γ and IL-17A with loss of tolerance leading to autoimmunity to Col-V, which leads to chronic lung allograft rejection [55].
The impact of IL-17-producing cells in certain respiratory diseases leaded to booming research for new treatment options in those diseases.
6. Therapeutic Implications
Inhaled glucocorticosteroids, cornerstone in reducing airway eosinophils and gold standard in asthma treatment, are unfortunately less effective in attacking IL-17-dominated diseases. Glucocorticosteroids are successfully used in some patients with CF (mainly in patients with CF asthma) in whom they might reduce airway eosinophils. Fitting with this hypothesis, we observed a significantly higher sputum eosinophil percentage in CF patients with CF asthma (n = 13, mean age 24 years ±2.6), compared to patients without CF asthma (n = 21, mean age 27.1 years ±6.9) (median 1% versus 0.1%, P = 0.03). Asthma was defined in these CF patients based on airway FEV1% reversibility and the presence of symptoms: wheezing episodes and/or bronchial hyperreactivity (our own unpublished data). Almost 80% of these CF-asthmatics were daily treated with inhaled corticosteroids and reported clinical benefit of this treatment, whereas only 30% of the patients with CF without asthma used daily ICS treatment (unpublished data).
On the other hand, severe asthmatics (with neutrophil predominance), COPD patients, CF patients without asthma, and patients with acute rejection and/or BOS after lung transplantation are waiting for additional treatment options. Due to the IL-17 predominance in some of these disease phenotypes, researchers focused on the possibility to reduce local IL-17 production.
Indirectly, macrolides, and more specifically azithromycin, have been suggested to have anti-inflammatory capacities, besides their anti-infectious potentials, and their effects have been studied in several clinical trials. A recent study suggested no clinical benefit of 12-week azithromycin treatment (weekly dosed) compared to placebo in patients with asthma, although asthmatic patients who received the medication in open label reported improvement of quality of life, asthma symptoms, and asthma control [56]. This could indicate a placebo effect in the open-label-treated group. However, patients volunteered to take part in the placebo-controlled trial and received the active product if they refused to take part in the placebo-controlled trial. This could have induced selection bias towards more severe asthmatics in the open-label arm. These patients probably are more neutrophilic and might therefore be optimal candidates for azithromycin treatment. Open label patients had significantly more chronic sinusitis and less allergy than patients in the randomized trial, fitting with the hypothesis of selection bias [56]. The results of an earlier reported retrospective study also suggested that neomacrolides might be useful as an add-on therapy in patients with severe asthma and/or bronchiectasis. The treatment might be especially useful in older asthmatics with severe (most likely neutrophilic) asthma and/or children with neutrophilic asthma [57, 58]. While awaiting large prospective trials in severe asthmatics, older studies suggested no benefit in young asthmatics, known to be mostly allergic and/or eosinophilic asthmatics if persistent until adulthood [59] and also fitting with the hypothesis that preferentially older asthmatics might benefit from this therapy [60].
Azithromycin might offer new options to treat COPD patients [61]. Standard treatment should include smoking cessation, enrollment in a pulmonary rehabilitation program, and the use of evidence-based medications, including long-acting inhaled beta-agonists, long-acting inhaled anticholinergic agents, and inhaled glucocorticoids. Compliant patients with more than two exacerbations in the previous year can be treated with azithromycin [62]. Treatment should be for one year, after which a serious evaluation of benefit should be done, before continuing the treatment later on [62]. Resistance of certain pathogens due to chronic azithromycin use should certainly be studied.
Azithromycin treatment was also studied in CF patients. Since 2002, several studies showed improved clinical outcome of CF patients upon treatment with azithromycin [63–65]. Whether the effect of the treatment is primarily anti-infectious or primarily anti-inflammatory in patients with CF is difficult to disentangle. Long-term, low-dose treatment with azithromycin in CF patients certainly leads to reduced prevalence of S. aureus, S. pneumoniae, and H. influenzae, but might increase macrolide resistance in S. aureus in these patients [65]. In a prospective study approved by our local ethical committee, we prospectively recruited 13 adult CF patients (>16 years) in the outpatient clinic of the University Hospitals Gasthuisberg, after obtaining informed consent in 2011. Patients received oral azithromycin (Zitromax, Pfizer, Elsene, Belgium) (250 mg, 3x/week) during 3 months. Lung function parameters and sputum induction were performed as described [44], before and after three months of azithromycin treatment. Paired FEV1% predicted (P = 0.42) and FVC in liters (P = 0.86) remained unaltered (our own unpublished data). Sputum neutrophil percentages and sputum IL-17A mRNA levels also remained stable (our own unpublished data). However, sputum macrophage percentages (median 6% before treatment versus median 3% after treatment, P = 0.08) and sputum TNF mRNA levels (median TNF/β actin 200 ∗ 104 before treatment versus median TNF/β actin 160 ∗ 104 after treatment, P = 0.07) tended to decrease after azithromycin treatment (our own unpublished results). These data are in line with the data recently published by Saint-Criq and coworkers showing no effect of azithromycin on in vitro IL-8 production by epithelial cell lines, either from cystic fibrosis patients or from healthy subjects [66]. These data are however also in line with the reported ±30% reduction of in vitro TNF-alpha mRNA and protein levels produced by cystic fibrosis airway epithelial cell lines treated with azithromycin [67]. Along the same line, TNF production by monocytic THP-1 cells stimulated by lps as well as by murine M1 alveolar macrophages from CF and wild type mice, was also reduced upon in vitro treatment with azithromycin [68, 69]. The positive effect of azithromycin in CF patients could be the elegant combination of the anti-infectious and anti-inflammatory properties of the drug, which perhaps could also be attributed to other members of the macrolide family, reducing the risk to develop resistance.
The observation that azithromycin could potentially reduce BOS by reducing neutrophilia as well as IL-8 production [70] is sustained by in vitro work, showing inhibition of IL-17-induced IL-8 release from human airway smooth muscle cells when treated with azithromycin [71]. Further study should now further unravel at what moment azithromycin treatment should best be started in lung transplant patients.
The anti-inflammatory properties of the drug in the cited respiratory diseases could furthermore be related to their potential to improve the phagocytic (and efferocytic) function of lung macrophages [72], which are reported to be defective in some of the cited diseases, such as in COPD [73] and BOS [74]. Several studies suggested that azithromycin could change the macrophage to a more M2 phenotype, which are characterized by specific markers and have increased IL-10 production [75–77]. If its anti-inflammatory effect would mainly be due to the induction of IL-10 gene expression and/or expression and release of this cytokine and specific membrane markers, one could try to develop a component of azithromycin/macrolides with similar function but without its anti-infectious properties. This would reduce its risk of bacterial resistance.
In all the cited diseases, we are awaiting the results of large clinical trials with anti-IL-17A or anti-IL-17R treatment, which will be available in the near future. These biologicals exert their function independent of the source of the local IL-17A overproduction in respiratory IL-17-mediated diseases. Innate or adaptive IL-17? Most likely both pathways are important, but depending on the respiratory disease type, the innate or the adaptive immunity might have a different weight.
References
- 1.Yao Z, Painter SL, Fanslow WC, et al. Human IL-17: a novel cytokine derived from T cells. Journal of Immunology. 1995;155(12):5483–5486. [PubMed] [Google Scholar]
- 2.Li H, Chen J, Huang A, et al. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(2):773–778. doi: 10.1073/pnas.97.2.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee J, Ho WH, Maruoka M, et al. IL-17E, a novel proinflammatory ligand for the IL-17 receptor homolog IL-17Rh1. Journal of Biological Chemistry. 2001;276(2):1660–1664. doi: 10.1074/jbc.M008289200. [DOI] [PubMed] [Google Scholar]
- 4.Starnes T, Robertson MJ, Sledge G, et al. Cutting edge: IL-17F, a novel cytokine selectively expressed in activated T cells and monocytes, regulates angiogenesis and endothelial cell cytokine production. Journal of Immunology. 2001;167(8):4137–4140. doi: 10.4049/jimmunol.167.8.4137. [DOI] [PubMed] [Google Scholar]
- 5.Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nature Immunology. 2005;6(11):1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nature Immunology. 2005;6(11):1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 7.Acosta-Rodriguez EV, Rivino L, Geginat J, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nature Immunology. 2007;8(6):639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 8.Sallusto F, Zielinski CE, Lanzavecchia A. Human Th17 subsets. Mini-review. European Journal of Immunology. 2012;49:2215–2220. doi: 10.1002/eji.201242741. [DOI] [PubMed] [Google Scholar]
- 9.Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β . Nature. 2012;484:514–518. doi: 10.1038/nature10957. [DOI] [PubMed] [Google Scholar]
- 10.Zelante T, Iannitti RG, De Luca A, Arroyo J, Blanco N, Servillo G. Sensing of mammalian IL-17A regulates fungal adaptation and virulence. Nature Communications. 2012;3, article 683 doi: 10.1038/ncomms1685. [DOI] [PubMed] [Google Scholar]
- 11.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nature Immunology. 2011;12(1):21–27. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- 12.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nature Reviews Immunology. 2009;9(8):556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Werner JL, Gessner MA, Lilly LM, Nelson MP, Metz AE, Horn D. Neutrophils produce interleukin 17A (IL-17A) in a dectin-1- and IL-23-dependent manner during invasive fungal infection. Infection and Immunity. 2011;76:3966–3977. doi: 10.1128/IAI.05493-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song C, Luo L, Lei Z, et al. IL-17-producing alveolar macrophages mediate allergic lung inflammation related to asthma. Journal of Immunology. 2008;181(9):6117–6124. doi: 10.4049/jimmunol.181.9.6117. [DOI] [PubMed] [Google Scholar]
- 15.Tan HL, Regamey N, Brown S, Bush A, Lloyd CM. The Th17 pathway in cystic fibrosis lung disease. American Journal of Respiratory and Critical Care Medicine. 2011;184:252–258. doi: 10.1164/rccm.201102-0236OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. 2004;21(4):467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 17.Lindén A. Role of interleukin-17 and the neutrophil in asthma. International Archives of Allergy and Immunology. 2001;126:179–184. doi: 10.1159/000049511. [DOI] [PubMed] [Google Scholar]
- 18.Folkard SG, Westwick J, Millar AB. Production of interleukin-8, RANTES and MCP-1 in intrinsic and extrinsic asthmatics. European Respiratory Journal. 1997;10(9):2097–2104. doi: 10.1183/09031936.97.10092097. [DOI] [PubMed] [Google Scholar]
- 19.Gibson PG, Simpson JL, Saltos N. Heterogeneity of airway inflammation in persistent asthma: evidence of neutrophilic inflammation and increased sputum interleukin-8. Chest. 2001;119(5):1329–1336. doi: 10.1378/chest.119.5.1329. [DOI] [PubMed] [Google Scholar]
- 20.Xiao W, Hsu YP, Ishizaka A, Kirikae T, Moss RB. Sputum cathelicidin, urokinase plasminogen activation system components, and cytokines discriminate cystic fibrosis, COPD, and asthma inflammation. Chest. 2005;128(4):2316–2326. doi: 10.1378/chest.128.4.2316. [DOI] [PubMed] [Google Scholar]
- 21.Ordoñez CL, Shaughnessy TE, Matthay MA, Fahy JV. Increased neutrophil numbers and IL-8 levels in airway secretions in acute severe asthma: clinical and biologic significance. American Journal of Respiratory and Critical Care Medicine. 2000;161(4):1185–1190. doi: 10.1164/ajrccm.161.4.9812061. [DOI] [PubMed] [Google Scholar]
- 22.Bullens DMA, Truyen E, Coteur L, et al. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respiratory Research. 2006;7, article 135 doi: 10.1186/1465-9921-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. Journal of Biological Chemistry. 2003;278(19):17036–17043. doi: 10.1074/jbc.M210429200. [DOI] [PubMed] [Google Scholar]
- 24.Cosmi L, Liotta F, Maggi E, Romagnani S, Annunziato F. Th17 cells: new players in asthma pathogenesis. Allergy. 2011;66(8):989–998. doi: 10.1111/j.1398-9995.2011.02576.x. [DOI] [PubMed] [Google Scholar]
- 25.Molet S, Hamid Q, Davoine F, et al. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. Journal of Allergy and Clinical Immunology. 2001;108(3):430–438. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 26.Sun YC, Zhou QT, Yao WZ. Sputum interleukin-17 is increased and associated with airway neutrophilia in patients with severe asthma. Chinese Medical Journal. 2005;118(11):953–956. [PubMed] [Google Scholar]
- 27.Barczyk A, Pierzcha W, Sozañska E. Interleukin-17 in sputum correlates with airway hyperresponsiveness to methacholine. Respiratory Medicine. 2003;97(6):726–733. doi: 10.1053/rmed.2003.1507. [DOI] [PubMed] [Google Scholar]
- 28.Doe C, Bafadhel M, Siddiqui S, et al. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest. 2010;138(5):1140–1147. doi: 10.1378/chest.09-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bajoriuniene I, Malakauskas K, Lavinskiene S, Jeroch J, Gasiuniene E, Vitkauskiene A. Response of peripheral blood Th17 cells to inhaled dermatophagoides pteronyssinus in patients with allergic rhinitis and asthma. Lung. 2012;190:487–495. doi: 10.1007/s00408-012-9411-y. [DOI] [PubMed] [Google Scholar]
- 30.Jatakanon A, Uasuf C, Maziak W, Lim S, Chung KF, Barnes PJ. Neutrophilic inflammation in severe persistent asthma. American Journal of Respiratory and Critical Care Medicine. 1999;160(5 I):1532–1539. doi: 10.1164/ajrccm.160.5.9806170. [DOI] [PubMed] [Google Scholar]
- 31.Tillie-Leblond I, Gosset P, Tonnel AB. Inflammatory events in severe acute asthma. Allergy. 2005;60(1):23–29. doi: 10.1111/j.1398-9995.2005.00632.x. [DOI] [PubMed] [Google Scholar]
- 32.Green RH, Brightling CE, Woltmann G, Parker D, Wardlaw AJ, Pavord ID. Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax. 2002;57(10):875–879. doi: 10.1136/thorax.57.10.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woodruff PG, Khashayar R, Lazarus SC, et al. Relationship between airway inflammation, hyperresponsiveness, and obstruction in asthma. Journal of Allergy and Clinical Immunology. 2001;108(5):753–758. doi: 10.1067/mai.2001.119411. [DOI] [PubMed] [Google Scholar]
- 34.Barnes PJ. Pharmacology of airway smooth muscle. American Journal of Respiratory and Critical Care Medicine. 1998;158(5):S123–S132. doi: 10.1164/ajrccm.158.supplement_2.13tac800. [DOI] [PubMed] [Google Scholar]
- 35.Cundall M, Sun Y, Miranda C, Trudeau JB, Barnes S, Wenzel SE. Neutrophil-derived matrix metalloproteinase-9 is increased in severe asthma and poorly inhibited by glucocorticoids. Journal of Allergy and Clinical Immunology. 2003;112(6):1064–1071. doi: 10.1016/j.jaci.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Fujita J, Kawaguchi M, Kokubu F, Ohara G, Ota K, Huang SK. Interleukin-33 induces interleukin-17F in bronchial epithelial cells. Allergy. 2012;67:744–750. doi: 10.1111/j.1398-9995.2012.02825.x. [DOI] [PubMed] [Google Scholar]
- 37.Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. Journal of Allergy and Clinical Immunology. 2012;129:216–227. doi: 10.1016/j.jaci.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, Zheng H, Zhang H, Ma W, Wang F, Liu C. Increased interleukin (IL)-8 and decreased IL-17 production in chronic obstructive pulmonary disease (COPD) provoked by cigarette smoke. Cytokine. 2011;56:717–725. doi: 10.1016/j.cyto.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 39.Chu S, Zhong X, Zhang J, Lao Q, He Z, Bai J. The expression of Foxp3 and ROR gamma t in lung tissues from normal smokers and chronic obstructive pulmonary disease patients. International Immunopharmacology. 2011;11:1780–1788. doi: 10.1016/j.intimp.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 40.Erb-Downward JR, Thompson DL, Han MK, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS ONE. 2011;6(2) doi: 10.1371/journal.pone.0016384.e16384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Høiby N, Koch C. Pseudomonas aeruginosa infection in cystic fibrosis and its management. Thorax. 1990;45(11):881–884. doi: 10.1136/thx.45.11.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Navarro J, Rainisio M, Harms HK, et al. Factors associated with poor pulmonary function: cross-sectional analysis of data from the ERCF. European Respiratory Journal. 2001;18(2):298–305. doi: 10.1183/09031936.01.00068901. [DOI] [PubMed] [Google Scholar]
- 43.McAllister F, Henry A, Kreindler JL, et al. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-α and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. Journal of Immunology. 2005;175(1):404–412. doi: 10.4049/jimmunol.175.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Decraene A, Willems-Widyastuti A, Kasran A, De Boeck K, Bullens DM, Dupont LJ. Elevated expression of both mRNA and protein levels of IL-17A in sputum of stable Cystic Fibrosis patients. Respiratory Research. 2010;11, article 177 doi: 10.1186/1465-9921-11-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brodlie M, McKean MC, Johnson GE, et al. Raised interleukin-17 is immunolocalised to neutrophils in cystic fibrosis lung disease. European Respiratory Journal. 2011;37(6):1378–1385. doi: 10.1183/09031936.00067110. [DOI] [PubMed] [Google Scholar]
- 46.Cohen TS, Prince A. Cystic fibrosis: a mucosal immunodeficiency syndrome. Nature Medicine. 2012;18:509–519. doi: 10.1038/nm.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bullens DMA, Decraene A, Dilissen E, et al. Type III IFN-λ mRNA expression in sputum of adult and school-aged asthmatics. Clinical and Experimental Allergy. 2008;38(9):1459–1467. doi: 10.1111/j.1365-2222.2008.03045.x. [DOI] [PubMed] [Google Scholar]
- 48.Aurora P, Boucek MM, Christie J, et al. Registry of the International Society for Heart and Lung Transplantation: 10th Official Pediatric Lung and Heart/Lung Transplantation Report-2007. Journal of Heart and Lung Transplantation. 2007;26(12):1223–1228. doi: 10.1016/j.healun.2007.07.035. [DOI] [PubMed] [Google Scholar]
- 49.Trulock EP, Christie JD, Edwards LB, et al. Registry of the International Society for Heart and Lung Transplantation: 24th Official Adult Lung and Heart-Lung Transplantation Report-2007. Journal of Heart and Lung Transplantation. 2007;26(8):782–795. doi: 10.1016/j.healun.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 50.Boehler A, Kesten S, Weder W, Speich R. Bronchiolitis obliterans after lung transplantation: a review. Chest. 1998;114(5):1411–1426. doi: 10.1378/chest.114.5.1411. [DOI] [PubMed] [Google Scholar]
- 51.Vanaudenaerde BM, Dupont LJ, Wuyts WA, et al. The role of interleukin-17 during acute rejection after lung transplantation. European Respiratory Journal. 2006;27(4):779–787. doi: 10.1183/09031936.06.00019405. [DOI] [PubMed] [Google Scholar]
- 52.Vanaudenaerde BM, De Vleeschauwer SI, Vos R, et al. The role of the IL23/IL17 axis in bronchiolitis obliterans syndrome after lung transplantation. American Journal of Transplantation. 2008;8(9):1911–1920. doi: 10.1111/j.1600-6143.2008.02321.x. [DOI] [PubMed] [Google Scholar]
- 53.Hodge G, Hodge S, Li-Liew C, Reynolds PN, Holmes M. Increased natural killer T-like cells are a major source of pro-inflammatory cytokines and granzymes in lung transplant recipients. Respirology. 2012;17:155–163. doi: 10.1111/j.1440-1843.2011.02075.x. [DOI] [PubMed] [Google Scholar]
- 54.Burlingham WJ, Love RB, Jankowska-Gan E, et al. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. Journal of Clinical Investigation. 2007;117(11):3498–3506. doi: 10.1172/JCI28031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tiriveedhi V, Angaswamy N, Brand D, Weber J, Gelman AG, Hachem R. A shift in the collagen V antigenic epitope leads to T helper phenotype switch and immune response to self-antigen leading to chronic lung allograft rejection. Clinical & Experimental Immunology. 2012;167:158–168. doi: 10.1111/j.1365-2249.2011.04486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hahn DL, Grasmick M, Hetzel S, Yale S. Azithromycin for bronchial asthma in adults: an effectiveness trial. Journal of the American Board of Family Medicine. 2012;25:442–459. doi: 10.3122/jabfm.2012.04.110309. [DOI] [PubMed] [Google Scholar]
- 57.Coeman M, van Durme Y, Bauters F, Deschepper E, Demedts I, Smeets P. Neomacrolides in the treatment of patients with severe asthma and/or bronchiectasis: a retrospective observational study. Therapeutic Advances in Respiratory Disease. 2011;5:377–386. doi: 10.1177/1753465811406771. [DOI] [PubMed] [Google Scholar]
- 58.Piacentini GL, Peroni DG, Bodini A, et al. Azithromycin reduces bronchial hyperresponsiveness and neutrophilic airway inflammation in asthmatic children: a preliminary report. Allergy and Asthma Proceedings. 2007;28(2):194–198. doi: 10.2500/aap.2007.28.2958. [DOI] [PubMed] [Google Scholar]
- 59.Wenzel SE. Asthma: defining of the persistent adult phenotypes. Lancet. 2006;368(9537):804–813. doi: 10.1016/S0140-6736(06)69290-8. [DOI] [PubMed] [Google Scholar]
- 60.Strunk RC, Bacharier LB, Phillips BR, et al. Azithromycin or montelukast as inhaled corticosteroid-sparing agents in moderate-to-severe childhood asthma study. Journal of Allergy and Clinical Immunology. 2008;122(6):1138–1144. doi: 10.1016/j.jaci.2008.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Albert RK, Connett J, Bailey WC, Casaburi R, Cooper JAJ, Criner GJ. Azithromycin for prevention of exacerbations of COPD. New England Journal of Medicine. 2011;365:689–698. doi: 10.1056/NEJMoa1104623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wenzel RP, Fowler AAR, Edmond MB. Antibiotic prevention of acute exacerbations of COPD. New England Journal of Medicine. 2012;367:340–347. doi: 10.1056/NEJMct1115170. [DOI] [PubMed] [Google Scholar]
- 63.Wolter J, Seeney S, Bell S, Bowler S, Masel P, McCormack J. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax. 2002;57(3):212–216. doi: 10.1136/thorax.57.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steinkamp G, Schmitt-Grohe S, Döring G, et al. Once-weekly azithromycin in cystic fibrosis with chronic Pseudomonas aeruginosa infection. Respiratory Medicine. 2008;102(11):1643–1653. doi: 10.1016/j.rmed.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 65.Hansen CR, Pressler T, Hoiby N, Johansen HK. Long-term, low-dose azithromycin treatment reduces the incidence but increases macrolide resistance in Staphylococcus aureus in Danish CF patients. Journal of Cystic Fibrosis. 2009;8(1):58–62. doi: 10.1016/j.jcf.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 66.Saint-Criq V, Ruffin M, Rebeyrol C, Guillot L, Jacquot J, Clement A. Azithromycin fails to reduce inflammation in cystic fibrosis airway epithelial cells. European Journal of Pharmacology. 2012;674:1–6. doi: 10.1016/j.ejphar.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 67.Cigana C, Assael BM, Melotti P. Azithromycin selectively reduces tumor necrosis factor alpha levels in cystic fibrosis airway epithelial cells. Antimicrobial Agents and Chemotherapy. 2007;51(3):975–981. doi: 10.1128/AAC.01142-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ikegaya S, Inai K, Iwasaki H, Naiki H, Ueda T. Azithromycin reduces tumor necrosis factor-alpha production in lipopolysaccharide-stimulated THP-1 monocytic cells by modification of stress response and p38 MAPK pathway. Journal of Chemotherapy. 2009;21(4):396–402. doi: 10.1179/joc.2009.21.4.396. [DOI] [PubMed] [Google Scholar]
- 69.Meyer M, Huaux F, Gavilanes X, et al. Azithromycin reduces exaggerated cytokine production by M1 alveolar macrophages in cystic fibrosis. American Journal of Respiratory Cell and Molecular Biology. 2009;41(5):590–602. doi: 10.1165/rcmb.2008-0155OC. [DOI] [PubMed] [Google Scholar]
- 70.Verleden GM, Vanaudenaerde BM, Dupont LJ, Van Raemdonck DE. Azithromycin reduces airway neutrophilia and interleukin-8 in patients with bronchiolitis obliterans syndrome. American Journal of Respiratory and Critical Care Medicine. 2006;174(5):566–570. doi: 10.1164/rccm.200601-071OC. [DOI] [PubMed] [Google Scholar]
- 71.Vanaudenaerde BM, Wuyts WA, Geudens N, et al. Macrolides inhibit IL17-induced IL8 and 8-isoprostane release from human airway smooth muscle cells. American Journal of Transplantation. 2007;7(1):76–82. doi: 10.1111/j.1600-6143.2006.01586.x. [DOI] [PubMed] [Google Scholar]
- 72.Hodge S, Reynolds PN. Low-dose azithromycin improves phagocytosis of bacteria by both alveolar and monocyte-derived macrophages in chronic obstructive pulmonary disease subjects. Respirology. 2012;17:802–807. doi: 10.1111/j.1440-1843.2012.02135.x. [DOI] [PubMed] [Google Scholar]
- 73.Hodge S, Hodge G, Jersmann H, et al. Azithromycin improves macrophage phagocytic function and expression of mannose receptor in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2008;178(2):139–148. doi: 10.1164/rccm.200711-1666OC. [DOI] [PubMed] [Google Scholar]
- 74.Hodge S, Dean M, Hodge G, Holmes M, Reynolds PN. Decreased efferocytosis and mannose binding lectin in the airway in bronchiolitis obliterans syndrome. Journal of Heart and Lung Transplantation. 2011;30(5):589–595. doi: 10.1016/j.healun.2011.01.710. [DOI] [PubMed] [Google Scholar]
- 75.Murphy BS, Sundareshan V, Cory TJ, Hayes D, Anstead MI, Feola DJ. Azithromycin alters macrophage phenotype. Journal of Antimicrobial Chemotherapy. 2008;61(3):554–560. doi: 10.1093/jac/dkn007. [DOI] [PubMed] [Google Scholar]
- 76.Feola DJ, Garvy BA, Cory TJ, et al. Azithromycin alters macrophage phenotype and pulmonary compartmentalization during lung infection with Pseudomonas. Antimicrobial Agents and Chemotherapy. 2010;54(6):2437–2447. doi: 10.1128/AAC.01424-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vrančić M, Banjanac M, Nujić K, Bosnar M, Murati T, Munić V. Azithromycin distinctively modulates classical activation of human monocytes in vitro. British Journal of Pharmacology. 2012;165:1348–1360. doi: 10.1111/j.1476-5381.2011.01576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]