

Abstract
We reviewed the phenotypic and molecular characteristics of MCF10DCIS.com and the SUM cell lines based on numerous studies performed over the years. The major signaling pathways that give rise to the phenotype of these cells may serve as a good resource of information when researchers in drug discovery and development use these cells to identify novel targets and biomarkers. Major signaling pathways and mutations affecting the coding sequence are also described providing important information when using these cells as a model in a variety of studies.
1. Introduction
Human tumor-derived cell lines grown in vitro and in vivo are important models to study cancer development, progression, and therapeutic response and resistance to anticancer drugs. However, for the last several years the relevance of cell lines in clinical cancer research has been criticized due to the properties of cell lines that differ when compared with primary tumor cells. New technologies such as next generation sequencing have enabled molecular characterization and identification of signaling pathways in specific cell lines. This characterization has facilitated the identification of cancer cell lines that are more clinically relevant for biological experiments as well as drug research and development. In addition, well characterized cells enable the identification of potential biomarkers for the development of companion diagnostics. Although there are still limitations with the relevance to the clinic, well characterized cancer cell lines will continue to be an important source for drug R&D and studying cancer biology.
In this paper, we review relevant information available in numerous studies that encompass the characterization of the different breast cancer cell lines available at Asterand US. This review will be a valuable resource for researchers in academia and industry for the use of the relevant cell type in their research. The MCF10DCIS cell line was licensed to Asterand US by Wayne State University and the SUM cell lines were licensed to Asterand US by the University of Michigan.
2. MCF10DCIS.com
MCF10DCIS.com is a clonal breast cancer cell line derived from a xenograft originating from premalignant MCF10AT cells that were injected into severe combined immune-deficient mice. The morphology of the MCF10DCIS cell line is shown in Figure 1. Injection of the MCF10DCIS cells into SCID mice resulted in rapidly growing lesions that are predominantly comedo ductal carcinoma in situ. Solid or comedo growth patterns are high grade ductal carcinoma in situ [1]. MCF10DCIS cells were shown to be reproducible from DCIS-like comedo lesions that spontaneously progress to IDC as xenografts in immunodefficent mice. Polizzotti and coworkers [2] used six (MCF10 series) in vitro cultures including MCF10DCIS to mimic the three grades of breast cancer along the metastatic cascade namely, nonmalignant, noninvasive carcinoma, and invasive carcinoma in vivo.
Figure 1.
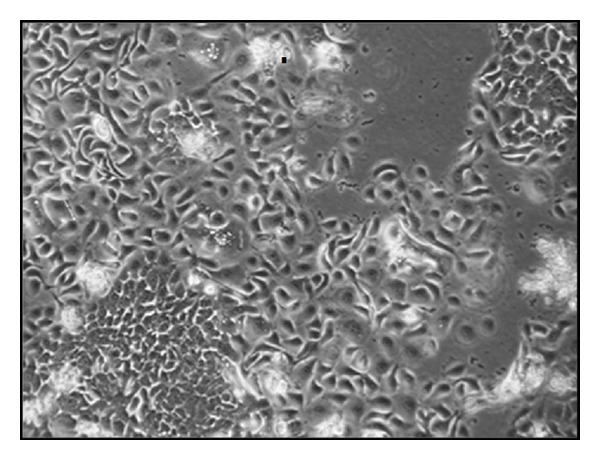
Morphology of MCF10DCIS and human breast cancer cell line in culture.
2.1. Key Signaling Proteins
CD44 is a cell-surface glycoprotein involved in cell-cell and cell extracellular matrix interactions including the migration and invasion of cancer cells and has been used as a key cancer stem cell-surface marker in various malignancies including breast cancer [3]. So et al. [4] showed growing malignant potential of MCF10 cell lines including MCF10DCIS with the decrease of expression of the standard 85-KDa isoform of CD44 and an increased expression of its 10–250 KDa variants, namely, CD44v, CD44v3, and CD44v6, and categorized them as markers of breast cancer progression. They have described a cascade of signaling proteins namely, Pak4, Stat3, pAkt, and pErk, in the MCF10 breast cancer model including two tumorigenic cell lines, MCF10DCIS and MCF10Ca1. High expression of CD44v can trigger the activities of several transmembrane receptor kinases [5]. Membrane receptors known to interact with CD44 and their downstream signals activate Stat3 leading to tumor progression and invasion [6]. Stat3 (signal transducer and activator of transcription 3) is part of a family known as the STAT genes. Normally, the Stat3 protein is switched on and off in response to signals that control cell growth and development. Abnormal Stat3 protein activation has been identified in many cancers that include breast, prostate, and pancreas, as well as cancers of blood-forming cells (leukemia and lymphoma). Excess Stat3 protein potentially contributes to the growth of cancers. Pak4 is a serine/threonine kinase and found to be highly expressed in breast cancer and associated with a signaling pathway leading to malignancy and formation of mammary tumors in nude mice [7]. It is highly expressed in MCF10DCIS compared to its precursor nontumorigenic cells lines [4]. Similarly they found high levels of phosphorylated forms of Erk and Akt namely pErk and pAkt were found only in the tumorigenic MCF10DCIS and MCF10CA1a cell lines indicating that overactivation of Erk and Akt is critical for developing malignant breast cancer. Erk and Akt are central protein kinases that mediate cellular responses to a diverse range of extracellular stimuli, including growth factors and cytokines, to regulate cell cycle progression and cell motility [8]. PAK (p21 activated kinase) belongs to a family of serine/threonine kinases that play a pivotal role in physiologic processes including motility, survival, invasion, and mitosis [9]. PAK's are widely expressed in a variety of tissues and are often upregulated or hyperactivated in a variety of human cancers including breast cancer [9, 10]. Upregulation of PAK1 and downregulation of HoxD10 were observed in MCF10DCIS cells [11]. HoxD10 is a homeobox transcription factor mediating gene responsible for the inhibition of invasiveness of cancer cells [12]. In luminal breast cancer, the expression and localization of PAK1 protein were assessed in primary tumors from 403 premenopausal patients that were randomized for two years of adjuvant tamoxifen or no treatment. Elevated expression and/or nuclear localization of PAK1 were associated with resistance to tamoxifen therapy [13].
2.2. PI3K Mutation
Courtney and coworkers [14] described the phosphatidylinositol 3-kinase (PI3K) signaling impact on cancer cell growth, survival, and metabolism as shown in Figure 2. There are three classes of PI3K based on structure and function. Class IA PI3K is the most clearly implicated in human cancer. The MCF10DCIS cell line has been shown to have a missense mutation H1047R that maps to the kinase domain of P13K [15–17]. This substitution generates the most potently oncogenic PI3K that is known to occur with high frequency in various cancers [16]. This gain of function mutation, H1047R, is one of the “hot spot” mutations in the catalytic domain p110a of the PIK3A gene [14]. The components of the PI3K pathway are being studied as drug targets in human cancer with PI3K itself being a target for therapeutic intervention [18]. As also described in Section 3.8. Chakrabarty et al. [19] showed that the H1047R PI3K mutant enhances HER2-mediated transformation via heregulin production and activation of HER3 thereby suggesting that the PI3K H1047R mutant enhances HER2-mediated transformation by amplifying the ligand-induced signaling output in the ERB network.
Figure 2.
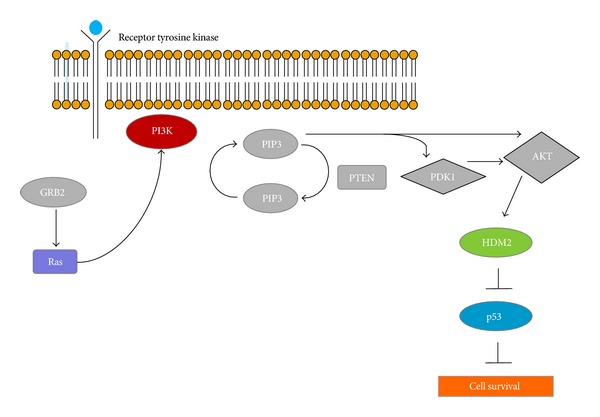
Signaling mechanism of PI3K.
2.3. Apoptosis
MCF10DCIS.com cells were found to undergo spontaneous apoptosis in vitro, in both monolayers and spheroids [20]. Shekhar and co-workers [20] evaluated both clinically derived specimens of comedo-DCIS and MCF10DCIS cells and showed that the apoptotic pathway was associated with increased mitochondrial membrane permeability and an increase in Bax/Bcl-2 ratio occurring via a caspase-9-dependent p53-independent pathway.
2.4. Metastasis
Malignant precursor cells with metastatic potential may already develop at early stages of tumorigenesis [21]. In addition, stromal cells in the microenvironment surrounding the primary tumor have been shown to be involved in facilitating metastasis [22]. Therefore, both tumor microenvironment and epithelial cells have to be considered in tumor invasion and metastasis. MCF10DCIS.com cells have been described to have a dynamic interplay of epithelium and stroma in the development of carcinoma in situ and subsequently in invasive ductal carcinoma (IDC) [23]. Stromal cell-derived factor-1 (SDF-1, also known as CXCL12) is a member of the CXC chemokine family [24]. SDF-1 has been identified as an estrogen-regulated gene in estrogen-receptor-(ER) positive ovarian and breast cancer cells [25]. Its effect is mediated by interaction with CXC chemokine receptor-4 (CXCR4) which is the only physiologic receptor for SDF-1 known to play a role in tumor metastasis, chemotaxis, and other metastasis components [26]. Tumors derived from the MCF10DCIS.com xenograft showed increased expression of SDF-1 in stromal cells, which is known to be highly induced by tumor-associated fibroblasts, with increased expression of CXCR4, in epithelial cancer cells during the DCIS to IDC transition [23]. This study showed that despite the phenotype of the epithelial cells being dependent upon the stroma, the malignant epithelium in these cells induced the development of the stroma which is necessary for their progression to the IDC.
In addition, elevated levels of MMP-2, MMP-3, MMP-9, and MMP-11 were observed in the stroma and epithelia of solid DCIS lesions prior to conversion to comedo-DCIS. The MMPs are a large family of proteases which include the stromelysins, collagenases, gelatinases, elastases, and the membrane-type MMPs. Overexpression of MMPs associated with metastasis has been reported in several cancers including breast [27]. The role of MMPs in metastasis is not only through the basement membrane (BM) and extracellular matrix (ECM) degradation but also due to the release of growth factors, such as VEGF and fibroblast growth factor (FGF), which stimulate angiogenesis.
2.5. Galectin-3
Galectin-3 is a mammalian β-galactoside-binding protein that is expressed by various types of human cells and plays an important role in cancer cell growth, transformation, apoptosis, angiogenesis, adhesion, invasion, and metastasis [28]. The importance of Galectin-3 protein is manifested by its many effects on cancer cells. Based on a secretome study, MCF10DCIS cells were found to secrete high levels of metastatic marker Galectin-3-binding protein [11]. Amm and Buschsbaum [29] highlighted that Galectin-3 expression and genotype may be useful markers in predicting TRAIL or agonistic antibody sensitivity of breast cancer patients.
2.6. Osteopontin
Shevde and coworkers [30] found that when MCF10DCIS.com cells were grown in spheroids they secreted high levels of the oncogenic protein osteopontin (OPN). OPN was shown to contribute to tumorigenicity and is critical in the development of vascular-like structures in spheroids. OPN-targeting hsa-mir-299-5p was downregulated in the MCF10DCIS spheroids compared to the monolayer-derived cells. OPN is speculated to have a binding site in its 3′UTR in a region that is likely to be recognized by hsa-mir-299-5p.
3. SUM Breast Cancer Cell Lines
There are 11 SUM cell lines, ten of which are provided by Asterand US as shown in Table 1. Each cell line was derived from a separate individual and represents a different subtype of breast carcinoma.
Table 1.
Cell source and clinicopathological characteristics SUM cell lines provided by Asterand.
| Cell line | Cell source | Cellular morphology | Clinicopathology | |
|---|---|---|---|---|
| 1 | SUM102PT | Intraductal carcinoma/microinvasion | nd | Basal B |
| 2 | SUM1315MO2 | Skin metastasis of infiltration ductal carcinoma | Spindle | Mesenchymal |
| 3 | SUM149PT | Invasive ductal carcinoma (inflammatory) | Mixed | Basal B |
| 4 | SUM159PT | Anaplastic carcinoma | Spindle | Mesenchymal |
| 5 | SUM229PE | Pleural effusion | Spindle | Basal B |
| 6 | SUM185PE | Pleural effusion | nd | Luminal |
| 7 | SUM190PT | Invasive ductal carcinoma (inflammatory) | nd | Luminal |
| 8 | SUM225CWN | Chest wall recurrence of ductal carcinoma in situ | Epithelial | Luminal |
| 9 | SUM44PE | Pleural effusion | nd | Luminal |
| 10 | SUM52PE | Pleural effusion | nd | Luminal |
The isolation and culture of these cell lines designated SUM44PE, SUM52PE, and SUM102PT have been described in detail in several publications [31–33]. Forozan et al. [34] developed seven additional human breast cancer cell lines from primary tumors. These cell lines were designated SUM149PT, SUM159PT, SUM185PE, SUM190PT, SUM225CWN, and SUM229PE. SUM206 is not available at Asterand, Inc. One cell line designated SUM1315MO2 was developed from a highly invasive breast cancer specimen that was grown for two transplant generations in immune-deficient mice before being explanted into culture. Nine of the 11 patients had received chemotherapy prior to sampling. SUM149 breast cancer cells were isolated from a patient with triple negative, inflammatory breast cancer (IBC) whose disease progressed through chemotherapy. All 11 SUM cell lines are immortal in culture with an abnormal karyotype [36] and express luminal cytokeratins 8 and 18 with the exception of SUM102PT line that expresses keratin 19. This is consistent with its basal breast epithelial cell origin [18]. Three distinct morphology groups were present among each of the SUM breast cancer cell lines, namely, epithelial cells, rounded cells, and spindle cells [35]. Keller et al. [35] classified human breast tissue through the use of a three-marker strategy into Luminal 1 cells, characterized by the majority of cells having an EpCAMhiCD24+CD49f-profile; Luminal 2 cells, characterized by a majority of EpCAMhiCD24+CD49f+ cells; basal cells, characterized by EpCAM+/loCD24-CD49f+ cells, Luminal 3 and mesenchymal cells, characterized by EpCAM-CD24-CD49f+ cells (see Section 3.7). The morphology of the SUM cell lines are shown in Figure 3 and summarized in Table 1.
Figure 3.
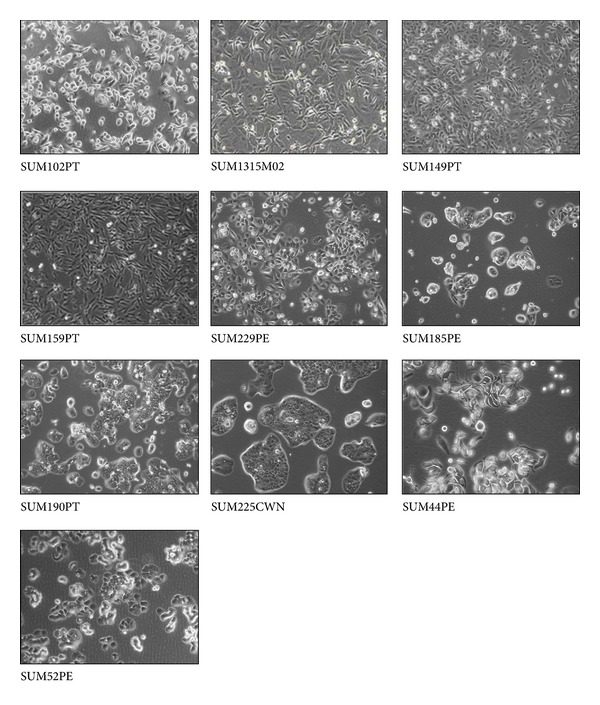
Morphology of SUM human breast cancer cell lines in culture.
3.1. Inflammatory Breast Cancer (IBC)
Inflammatory breast cancer (IBC) is among the most invasive, metastatic and lethal variant of human breast cancer [37]. IBC's have been reported to overexpress E-cadherin/α, β-catenin, and angiogenic features [38]. There have been very few advances in IBC-specific therapeutic targets and development of preclinical and clinical models of IBC that would enable the development of new therapeutic modalities to prolog survival of patients. The overall survival is currently 40% at three years [39]. Recently, in newly developed preclinical models of IBC and patient tumor tissues, E-cadherin, anaplastic lymphoma kinase (ALK), and HSP90 have been identified as potential targets for IBC. These targets are matched by therapeutics that are either currently in clinical trials or will be tested in clinical trials within the next year [40]. Molecular mechanisms have been implicated in IBC clinical aggressiveness and resistance to radiation [38, 40]. SUM149 and SUM190 are two cell lines established from primary IBC tumors. Both cell lines can form tumors in nude mice after mammary fat pad injection [41]. Forozan et al. [34] characterized these cell lines with respect to their ER, TP53, and other genes expression status. Laboratory investigations of SUM149 and SUM190 demonstrated radio resistance of these cell lines. Woodward et al. [40] used these cells as a clinical model to overcome radiation resistance to help guide clinical radiation trials against IBC. Charafe-Jauffret and coworkers [38] studied the role of cancer stem cells (CSC) in mediating metastasis in IBC and the association of these cells with patient outcome. This study suggested that the behavior of IBC could be mediated by the expression of aldehyde dehydrogenase-1 positive (ALDH1) in CSC. ALDH1 expression was the first independent prognostic marker that predicated metastasis and poor patient outcome in IBC. Victor et al. [42] investigated which proteases expressed by IBC cell lines SUM149 and SUM190 IBC cells are associated with Caveolin-1 (Cav-1) which is highly expressed in IBC patients. This protein participates in ECM degradation. Cav-1 is the major protein component of caveolac, expressed in IBC patients. Van Den Eynden and coworkers [43] showed that Cav-1 is hypomethylated and highly expressed on both IBC and SUM cell lines (SUM149 and SUM190) as opposed to the non-IBC cell line SUM 102PT. Yuan et al. [44] indicated that Cav-1 is critical for inflammatory responses regulating the STAT3/NF-κB pathway.
3.2. BRCA and Hormonal Status
BRCA1 and BRCA2 are human genes that belong to a class of genes known as tumor suppressors. The names BRCA1 and BRCA2 stand for breast cancer susceptibility gene 1 and breast cancer susceptibility gene 2, respectively. BRCA1 has a central role in several pathways coordinating various cellular processes in response to DNA damage, including DNA repair and preservation of genomic integrity [45]. BRCA1 and BRCA2 are also involved in pathways that regulate cell cycle progression, ubiquitylation, and apoptosis. Cells deficient in BRCA1 are unable to repair double stranded breaks [46]. BRCA1 tumors of basal-like intrinsic subtype and more frequently of medullary histology are often ER negative [47]. All the SUM cell lines of basal origin are ER and PR negative (Table 2). Luminal breast cancers are more often ER and/or PR positive or have over expression of ERBB2 [36, 48]. The epidermal growth factor receptor gene (EGFR) was found to be overexpressed without amplification in SUM102, SUM149, and SUM229 cells. However, the fibroblast growth factor receptor 1 gene (FGFR1, at11q13) is amplified in SUM44 and SUM52 cells as well as the fibroblast growth factor receptor 2 gene (FGFR2, at 10q26) is amplified in SUM-52 and SUM190. SUM225 cells have an amplified ERBB2 (encoding Her-2/neu). SUM1315MO2 and SUM 149PT both of basal origin were found to have BRCA1 mutations and BRCA1 allelic loss [46]. In the SUM 149PT cell line, Elstrodt and coworkers [46] identified a shift in the BRCA1 reading frame with an insertion of 12 new amino-acids after codon 723, followed by a termination codon with a 2288delT. They also identified an AG nucleotide deletion in BRCA1 at position 185 in the SUM1315MO2 cell line [48]. The 185delAG is a well described pathogenic mutation in BRCA1 and is mainly found both in familial breast and ovarian cancer among Ashkenazi Jews, though it is also found in the general population [49]. Tumors with BRCA1 mutations have also been shown to have a higher frequency of p53 mutations than sporadic breast cancers (p53 will be discussed in Section 3.4).
Table 2.
ER, PR, and BRCA1 profile of SUM cell lines.
| Cell line | BRCA1 | ER protein | PR protein | ERBB2 | ||
|---|---|---|---|---|---|---|
| Allelic loss | Mutation | Protein effect | ||||
| SUM102PT | No loss | Wild type | NA | nd | nd | |
| SUM1315MO2 | Loss | 185del AG | truncation | − | − | |
| SUM149PT | Loss | 2288delT | truncation | − | − | |
| SUM159PT | No loss | Wild type | NA | − | − | |
| SUM229PE | No loss | Wild type | NA | − | − | |
| SUM185PE | Loss | Wild type | NA | − | − | |
| SUM190PT | Loss | Wild type | NA | − | − | +++ |
| SUM225CWN | Loss | Wild type | NA | − | − | +++ |
| SUM44PE | Loss | Wild type | NA | +++ | +++ | |
| SUM52PE | Loss | Wild type | NA | +++ | − | |
3.3. CHEK2 Mutation Analysis
The CHEK2 gene encodes a serine/threonine kinase. CHEK2 has been described as a tumor suppressor with proapoptotic, cell cycle checkpoint, and mitotic functions (Figure 4). ATM and CHEK2 have roles upstream of BRCA1. The ataxia telangiectasia mutated protein assists cells in recognizing damaged or broken DNA strands and phosphorylates CHEK2 following DNA damage by ionizing radiation, which prevents entry of the cell into mitosis. CHEK2 then associates with phosphorylates and activates functions of BRCA1 [50].
Figure 4.
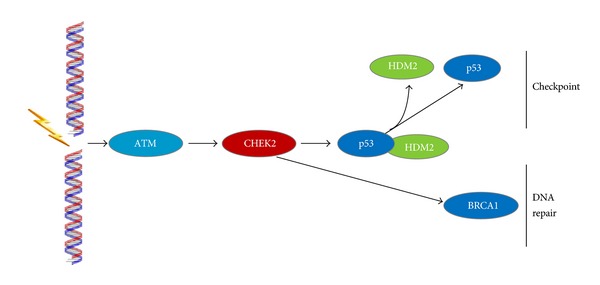
Signaling mechanism of CHEK2.
Inherited mutations in the CHEK2 gene have been identified in some cases of breast cancer. For example, a deletionmutation at nucleotide position 1100 is associated with an increased risk of breast cancer, particularly in the European population. Association studies estimated the risk of breast cancer carriers of 1100delC to be increased by 2.7-fold in women of Northern and Eastern European descent [51, 52]. The 1100delC mutation leads to the production of an abnormally short, nonfunctional version of the CHEK2 protein. Without this protein, cells are unable to regulate cell division properly. As a result, DNA damage accumulates and cells can divide without control or order. If cell division is not tightly controlled, cancers can develop. CHEK2 is located in the center of a pathway that transduces a DNA damage signal to cellular effectors that determine response to cellular damage [53]. Wasielewski et al. [52] showed that SUM 102PT has the CHEK2 1100delC germ line founder mutation and does not express any CHEK2 protein. Furthermore, Zoppoli et al. [54] studied a panel of 60 established cancer cell lines showing that the high heterogeneity of CHEK2 expression in cancer cells is primarily due to its inactivation (low gene expression, alternative splicing, point mutations, copy-number alterations, or premature truncation) or reduction of protein levels. CHEK2, phosphorylated at T68, is also commonly activated in cancers and precancerous lesions [54]. With the exception of SUM149PT, the majority of SUM cell lines overexpress CHEK2 protein. The latter has barely detectable CHEK2 protein expression and SUM 229PE has normal expression level. SUM 44PE was not studied.
3.4. Mutational Analysis and Regulation of p53 Pathway
p53 is a nuclear transcription factor and transactivates numerous target genes involved in the induction of cell cycle arrest and/or apoptosis [55]. Mutational inactivation is a major molecular mechanism of p53 dysfunction and over 50% of human cancers carry p53 mutations. Mutant p53 acts as a dominant-negative inhibitor of wild-type p53 and exhibits a longer half-life than wild-type p53 [56]. p53 truncating mutations were found in the SUM cell lines and were located throughout the p53 protein as described by Wasielewski et al. [57] and shown in Table 3. The SUM cells with truncating mutations had low transcript levels and low or no detectable protein expression levels. p53 was found to have variable transcripts and protein expression levels in SUM cell lines with p53 missense mutations (Table 3). These mutations were found in the sequence-specific DNA-binding region. This result is in concordance with the p53 missense mutations reported in clinical cancers [36].
Table 3.
p53 profile of the Asterand SUM cell lines.
| Cell line | 17p allelic loss | p53 gene sequence | p53 expression | p14ARF status | |||||
|---|---|---|---|---|---|---|---|---|---|
| Exon | Mutation | Amino acid change | Protein effect | Transcript size (kb) | Transcript expression | Gene | Transcript expression | ||
| SUM102PT | Wild type | nd | Deleted | − | |||||
| SUM1315MO2 | Loss | 5 | TGC>TTC | C135F | Missense | 1 | + | Deleted | − |
| SUM159PT | Loss | 5 | TCC>TCCTCC | S158insS | Missense | 0.7 | − | + | |
| SUM149PT | Loss | 7 | ATG>ATA | M237I | Missense | 1 | + | Deleted | − |
| SUM225CWN | Loss | 8 | CTG>CCG | L265P | Missense | 0.9 | ++ | nd | nd |
| SUM229PE | Loss | 8 | CGT>TGT | R273C | Missense | 0.7 | nd | Deleted | − |
| SUM44PE | Loss | 3 | GAA>CA | E28fsX16 | Truncating | 0.4 | − | wt | + |
| SUM185PE | Loss | 5 | CAG>TAG | Q144X | Truncating | 0 | − | wt | + |
| SUM52PE | Loss | 6 | CGA>TGA | R213X | Truncating | 0.3 | − | wt | + |
| SUM190PE | Loss | 9 | CAG>TAG | Q317X | Truncating | 0.4 | ± | nd | nd |
HDM2, a negative regulator of the p53 pathway, is amplified and the protein is overexpressed in the luminal subtype SUM52PE cells [48]. HDM2 was barely detectable in the remaining SUM cell lines. SUM102PT and SUM225CWN were not analyzed. Following DNA damage, phosphorylation of HDM2 leads to changes in protein function and stabilization of p53. This occurs when HDM2 functions as an ubiquitin E3 ligase, binds to the transcriptional activation domain of p53, blocking its function, and, via ubiquitination, targets p53 for proteosome-mediated enzymatic degradation [58].
Another important partner of the p53 pathway is p14ARF with a role of keeping HDM2 localized in the nucleolus and preventing it from degrading p53 [59]. The induction of the p14ARF protein, the alternate reading frame product of the CDK (cyclin dependant kinase) locus, is also a mechanism that negatively regulates the p53-HDM2 interaction. p14ARF directly interacts with HDM2 and leads to upregulation of p53 transcriptional response. P14ARF therefore indirectly regulates the levels of p53. However, some evidence shows that p14ARF is able to bind p53 directly in the absence of Mdm2 [60]. The biological consequences of the p14ARF-p53 binding depend upon the cell cycle status when p53 is activated. G1 cell cycle arrest or apoptosis is observed when p53 is present and S-phase distortion is detected when p53 is inactive [61]. P14ARF is deleted in four of the five basal subtype SUM cell lines, SUM229PE, 1315MO2, SUM102PT, and SUM149PT. P14ARF showed normal transcript expression in SUM44PE, 185PE, 52PE, and 159PT (Table 3). The remaining two cell lines were not studied for this protein [48].
p21 is a potent cyclin-dependent kinase inhibitor and inhibits the activity of cyclin-CDK2 or -CDK1 complexes, and thus functions as a regulator of cell cycle progression in G1. The expression of this gene is tightly controlled by p53, through which this protein mediates the p53-dependent cell cycle G1 phase arrest in response to a variety of stress stimuli. p21 was barely detectable in 4 SUM cell lines (SUM 1315MO2, 149PT, 159 PT, and 52PE) and showed normal protein expression in the 4 SUM cell lines SUM 229PE, 185PE, 190PT, and 44PE. The remaining 2 cell lines were not analyzed [48].
3.5. Mutational Analysis of RB Pathway Genes
The Retinoblastoma protein (RB1) is a tumor suppressor that is a crucial regulator of appropriate cell cycle progression, including G1 to S and G2 to M phase transitions [62]. In cells entering the cell cycle, extracellular signals induce the expression of D-type cyclins, which bind to and activate cyclin-dependent kinases (CDK4 and CDK6) as shown in Figure 5. These complexes lead to the phosphorylation of RB1 which in turn transcriptionally activates genes required for S phase [63]. RB1 deregulation is frequently observed in multiple types of cancers [64]. RB1 is inactivated by CDK4-mediated phosphorylation, and the kinase activity of CDK4 is suppressed by p16INK4a (p16). Cyclin-dependent kinase inhibitor 2A, (CDKN2A, p16Ink4A) also known as tumor suppressor 1 (MTS-1), is a tumor suppressor protein, encoded by the CDKN2A gene and plays an important role in regulating the cell cycle. Mutations in p16 increase the risk of developing a variety of cancers [65]. Loss of functional p16 gives rise to unregulated CDK4 activity, leading to persistent Rb phosphorylation and uncontrolled cell proliferation [66]. Sørlie and co-workers [47] performed an extensive characterization of subtype-specific gene expression patterns at the protein and transcript level of the Rb pathway genes in SUM cells. The protein was overexpressed in SUM159PT and SUM44PE. SUM102PT and SUM44PE cells showed an overexpression of the cyclin D1 transcript and protein (Table 4).
Figure 5.
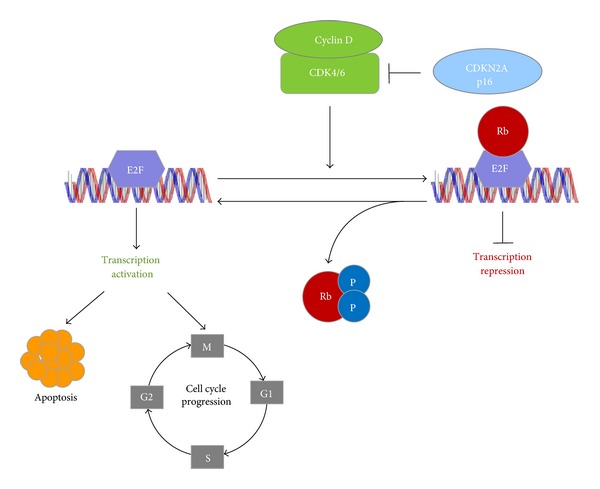
Signaling mechanism in Rb.
Table 4.
Mutational analysis Rb pathway genes for Asterand SUM cell lines.
| Cell line | RB1 protein expression | Cyclin D1 transcript expression | Cyclin D1 protein expression | p16 gene sequence |
p16 transcript expression |
p16 protein expression |
|---|---|---|---|---|---|---|
| SUM102PT | nd | ++ | nd | Deleted | + | nd |
| SUM1315MO2 | + | + | ± | Deleted | − | − |
| SUM149PT | + | + | ± | Deleted | − | − |
| SUM159PT | + + | + | + | Wild type | − | ± |
| SUM229PE | + | + | + | Deleted | − | − |
| SUM185PE | + + | + | + | Wild type | ± | ± |
| SUM190PT | nd | nd | nd | nd | nd | nd |
| SUM225CWN | nd | nd | nd | nd | nd | nd |
| SUM44PE | + | ++ | ++ | Methylated | ± | ± |
| SUM52PE | + | + | + | Mutant | + | + |
Source: [48]; nd: not determined, +: normal expression, ±: barely detectable, −: no detectable expression, ++: over expression.
The p16 gene was deleted in basal cell subtypes SUM102PT, SUM1315MO2, SUM149PT, and SUM229PE. The remaining three cell lines did not have detectable expression of p16 transcript or protein. No p16 deletions were found in the luminal cell subtypes. The p16 gene was found to be methylated in luminal subtype SUM 44PE and the transcript and protein levels were barely detectable (Table 4). Despite the gene being mutated in luminal subtype SUM52PE, the p16 transcripts and protein expression levels were found to be normal. Over expression or amplification of cyclin D1 (CCND1) is observed in as many as 50% of breast cancers, wherein it is believed to drive aberrant phosphorylation or inactivation of RB protein [67]. In breast cancer, the RB1 pathway is believed to be inactivated via several mutually exclusive mechanisms [68]. Hollestelle and coworkers [48] suggested a dichotomy in the genetic basis of human breast cancer, based on the observations whereby specific gene mutation profiles exist for the two major subtypes of breast cancer cell lines.
3.6. PIK3A and RAS Status (See Also Section 2.2)
Mutations that activated PIK3CA and RAS pathways in SUM cell lines have been identified [69] as shown in Table 5. PIK3CA was found to be constitutionally active in SUM102PT, SUM159PT, and SUM185PE, all of which possess the G12D mutation in KRAS. All the PIK3CA mutations were heterozygous with the exception of the H1047R mutation found in SUM185PE [69]. SUM159 had an H1047L mutation in P1K3CA (Table 5) and a G12D mutation in HRAS. SUM 229PE had wild-type P1K3CA. Since activating RAS isoforms have been reported for KRAS, HRAS and NRAS, Ras mutations have been identified in many tumor types and show tissue specificity [70].
Table 5.
PIK3CA, HRAS and KRAS mutation status for SUM cell lines.
| Cell line | PIK3CA status | HRAS status | KRAS status |
|||
|---|---|---|---|---|---|---|
| Gene sequence | Amino acid change | Gene sequence | Amino acid change | Gene sequence | Amino acid change | |
| SUM102PT | 3140A>G | H1047R | wt | None | wt | None |
| SUM1315MO2 | wt | None | wt | None | wt | None |
| SUM149PT | wt | None | wt | None | wt | None |
| SUM159PT | 3140A>T | H1047L | 35G>A | G12D | wt | None |
| SUM229PE | wt | None | wt | None | 35G>A | G12D |
| SUM185PE | 3140A>T | H1047R | wt | None | wt | None |
| SUM190PT | 3140A>G | H1047R | wt | None | wt | None |
| SUM225CWN | wt | None | wt | None | wt | None |
| SUM44PE | wt | None | wt | None | wt | None |
| SUM52PE | wt | None | wt | None | wt | None |
Source: [69]; +: normal expression; —: not detectable, nd: not determined, wt: wild type.
3.7. E-Cadherin/α-, β- and γ-Catenin Expression Pattern of Asterand SUM Cell Lines
The E-cadherin/catenin protein complex maintains the integrity of epithelial tissues through cell-cell adhesion [71, 72]. The E-cadherin/catenin protein complex consists of the cytoplasmic proteins α-catenin, β-catenin, γ-catenin, p120-catenin, and the transmembrane protein E-cadherin. Extracellular E-cadherin forms homodimers with E-cadherin proteins on adjacent epithelial cells in a calcium-dependent manner. Decreases in this adhesion ability of the cell have been linked to metastasis and tumor progression [73].
Hollestelle and colleagues [74] used human breast cancer cell lines that included all SUM cells to study E-cadherin inactivation. E-cadherin and α-, β-, and γ-catenin protein expressions were determined by immunohistochemistry. E-cadherin gene status was analyzed for gene mutations and promoter methylation status. All the spindle shaped, basal morphology SUM cell lines showed promoter hypermethylation indicating inactivation of E-cadherin. SUM1315MO2 had barely detectable γ-catenin protein expression and no E-cadherin expression. SUM159PT also did not show E-cadherin protein expression. SUM44PE with luminal subtype and rounded morphology had a protein truncating mutation in the E-cadherin gene with no detectable E-cadherin or α-catenin and barely detectable γ-catenin protein expression.
3.8. Expression of CK8/18/19 and CD49f after CD146/EpCAM Enrichment of SUM Cell Lines
Epithelial cell adhesion molecule (EpCAM) also known as CD326 is a type I transmembrane 39–42 kDa glycoprotein that functions as a homophilic, epithelial-specific intercellular cell-adhesion molecule. The 314 amino acid-long EpCAM protein comprises a large extracellular domain with an epidermal growth-factor- (EGF-) like domain and a putative thyroglobulin (TY) domain, a single transmembrane region, and a short (26 amino acids) cytoplasm tail [75]. Recent data suggest that EpCAM's role is not limited to cell adhesion, but it is also involved in cellular signaling, cell migration, proliferation, and differentiation [76]. EpCAM's association with proliferation, adhesiveness, tissue stabilization, promotion of tumor growth, and metastasis suggests that EpCAM is a pleiotropic molecule that potentially offers therapeutic applications in cancer treatment [53]. Mostert and coworkers [77] undertook a clinical study in primary breast cancer patients in which both EpCAM and CD146-positive circulating tumor cells (CTCs) were studied to establish the clinical relevance of cell lines including the SUM cell lines. CD146 was highly expressed in SUM1315MO2 and SUM159PT with epithelial-mesenchymal transition (EMT) features (as shown in Table 1). In addition to examining the added value of selecting cancer cells with a broader array of cytokeratins (CKs), they tested CD49f (ITGA6; integrin, alpha 6) as an alternative selection marker for CTC. CD49f is also an integral cell-surface protein involved in cell adhesion which has also been described as a stem cell marker in breast cancer [78], making it a candidate to select EMT-like breast cancer cells with stem cell-like features. Mostert and colleagues [77] set out to identify an additional new marker to be used after capturing breast cancer cells with combined anti-CD146/anti-EpCAM, to detect those cancer cells that lack CK8/18/19 expression. Table 6 summarizes their work in which SUM159PT was the only SUM cell line negative for these markers.
Table 6.
Protein expression of cytokeratins and CD49f/EPCAM in SUM cells.
| Cell lines | CK5 (KRT5) | CK14 (KRT14) | CK8-18 | CK19 (KRT19) | CD49f (ITGA6) | CD146 (MCAM) | EpCAM (TACSTD1) |
|---|---|---|---|---|---|---|---|
| IHC | IHC | IHC | IHC | IHC | FACS (s/n)a | FACS (s/n)a | |
| Normal like | |||||||
|
| |||||||
| SUM1315MO2 | + | − | nd | nd | + | 20–200 | <5 |
| SUM159PT | − | − | − | − | +++ | 20–200 | <5 |
| SUM102PT | nd | − | nd | nd | nd | 520 | <5 |
|
| |||||||
| Basal | |||||||
|
| |||||||
| SUM149PT | ++++ | − | ++++ | ++ | + | 20–200 | 20–200 |
| SUM229PE | +++ | − | ++++ | ++ | + | 20–200 | 20–200 |
|
| |||||||
| Luminal | |||||||
|
| |||||||
| SUM44PE | − | − | nd | +++ | ++++ | nd | nd |
| SUM185PE | − | − | ++++ | +++ | ++++ | nd | nd |
Interestingly, some SUM cell lines express markers of some of cancer stem cell (CSC) populations namely, ALDH, CD44+, and CD24− [35, 84]. ALDH is a detoxifying enzyme responsible for the oxidation of intracellular aldehydes and is thought to play a role in stem cell differentiation via metabolism of retinol to retinoic acid [78]. Breast carcinomas have been reported to contain a subpopulation of CD44+/CD24− tumor cells with stem cell-like properties. The subpopulation of breast cancer cells with CD44+/CD24− has been shown to exhibit enhanced invasive phenotype as an early step to metastasis [35, 85].
The phenomenon of heterogeneity has also been well documented in breast cancer [86]. This has been attributed to the presence of cancer stem cells (CSC's) that have the properties of differentiating along divergent pathways. The percentage of cancer cells that express CD44+/CD24− phenotype correlate with spindle/mesenchymal physical features [87]. CD44, a multifunctional class I transmembrane glycoprotein, generally acts as a specific receptor for hyaluronic acid, promoting migration in normal cells and is highly expressed in almost every cancer cell [88]. CD44 is mainly associated with proteins that monitor the extracellular changes and plays a critical role in regulating cell adhesion, proliferation, growth, survival, motility, migration, angiogenesis, and differentiation [89–91]. CD24 is a small cell-surface protein molecule anchored by glycosyl-phosphotidyl inositol in a wide variety of cancer cells. It is heavily glycosylated and functions in cell-cell and cell-matrix interactions [91]. CD24 is another important marker whose prognostic value and significance remains controversial [92, 93]. CD24 is highly expressed in ovarian, breast, prostate, bladder, renal, non-small cell carcinomas, and other human cancers and is involved in cell adhesion and metastasis [92]. This indicates that CD24 could be a significant marker in tumor prognosis and diagnosis. The metastatic association of CD24 increases its importance as a prognostic factor and a CSC marker [90]. Marotta and coworkers [94] targeted the JAK2/Stat3 signaling pathway for specific breast cancer therapies by highlighting the difference between distinct breast cancer cell types. They found that the IL-6/JAK2/Stat3 pathway was preferentially active in CD44+ CD24− breast cancer cells compared to other tumor cell types.
Subpopulations of SUM149PT and SUM159PT cells, selected for a given phenotypic state both luminal and basal cells, were shown to have the ability to give rise to stem cells [95]. SUM149PT and SUM159PT cells were treated with either paclitaxel or 5-fluorouracil in vitro and showed an increased number of stem cells in both cell lines. The authors used the Markov mathematical model in which cells transition stochastically between states to predict the conversions that were most likely to have occurred to achieve this outcome.
3.9. Clinical Relevance of Markers and Targets Aberrantly Expressed in MCF10DCIS and SUM Cell Lines
Mutations affecting the coding sequence in these cell lines are reported in the COSMIC database and other resources are shown in Table 7. Proia and coworkers [96, 97] reported that basal breast epithelial cells derived from patients and cell lines SUM1315 MO2 and harboring deleterious mutations in BRCA1 (BRCA1mut/+) give rise to tumors with increased basal differentiation relative to cells from BRCA1+/+ patients.
Table 7.
Summary of mutations affecting the coding sequence of Asterand breast cancer cell lines.
| Coding mutations listed in COSMIC database | ||||||
|---|---|---|---|---|---|---|
| Cell line | Gene | AA mutation | CDS mutation | Mutation | Zygosity | Reference |
| MCF10DCIS | PI3KCA | c.3140A>G | p.H1047R | Missense | Unknown | Kalaany and Sabatini (2009) [17] |
| SUM102PT | CDKN2A | 471 del 471 | None | Whole gene deletion | Homozygous | Hollestelle et al. (2010) [48] |
| PIK3CA | c.3140A>G | p.H1047R | Missense | Heterozygous | ||
| SUM1315MO2 | BRCA1 | 66_67 del AG | p.E23fs*17 | Frameshift | Homozygous | Hollestelle et al. (2010) [48] |
| CDKN2A | 471 del 471 | None | Whole gene deletion | Homozygous | ||
| SUM149PT | FBXW7 | c.1644 1645 ins 416 | F549Fs*6 | Frameshift | Homozygous | Strohmaier et al. (2001) [79] |
| BRCA1 | c.2169 del T | p.P724fs*12 | Frameshift | Homozygous | Hollestelle et al. (2010) [48] | |
| CDKN2A | 471 del 471 | None | Whole gene deletion | Homozygous | ||
| EP300 | c.4025+(28)del3 | unknown | Unknown | Heterozygous | Gayther et al. (2000) [80] | |
| SUM159PT | HRAS | c.35G>A | p.G12D | Missense | Heterozygous | Hollestelle et al. (2007) [69] |
| PIK3CA | c.3140A>T | p.H1047L | Missense | Heterozygous | ||
| SUM229PE | KRAS | c.35G>A | p.G12D | Missense | Heterozygous | Hollestelle et al. (2010) [48] |
| CDKN2A | 471 del 471 | None | Whole gene deletion | Homozygous | ||
| SUM185PE | PIK3CA | 3140A>G | p.H1047R | Missense | Heterozygous | Saal et al. (2005) [81] |
| SUM190PT | PIK3CA | 3140A>G | p.H1047R | Missense | Heterozygous | Hollestelle et al. (2007) [69] |
| SUM225CWN | None | |||||
| SUM44PE | CDH1 | 1269delT | F423fs*8 | Frameshift | Homozygous | Van de Wetering et al. (2001) [82] |
| SUM52PE | CDKN2A | 203C>T | A68V | Missense | Homozygous | Hollestelle et al. (2010) [48] |
Source: [83].
The PI3K pathway is one of the most important pathways in cancer metabolism and growth. Mutations in the PI3K pathway are frequent in breast cancer and result in resistance to HER2-targeted agents hormonal agents [98]. The PI3K inhibitors XL147 and XL765 developed by Exelixis and Sanofi-aventis have been shown to effectively block the PI3K and ERK pathways in Phase I studies [98]. Phase II clinical trials with XL147 in hormone-refractory disease and also in patients who have failed trastuzumab (trade name Herceptin), are currently in progress [99].
Hormone receptor-positive breast cancer therapies have been the main drug targets for several decades. HER2 became an accepted therapeutic target in standard breast cancer practice. Trastuzumab, a monoclonal antibody that interferes with the HER2/neu receptor, has been developed by Genentech. In addition to the treatment with trastuzumab other approaches have been developed [100, 101]. One approach has been the development of HER2-directed antibody-drug conjugates. Trastuzumab-DM1 (T-DM1) is an anti-HER2 antibody-drug conjugate consisting of trastuzumab covalently bound via a linker to DM1, a derivative of the antimicrotubule chemotherapy maytansine. Patient cohorts with HER2-positive breast cancer that progressed during trastuzumab therapy and developedresistance to the treatment were assignedto the clinical trials with T-DM1. A significantclinical benefit among patients treated at the recommended dose of T-DM1 was observed in Phase I studies [102]. This single-agent activity was subsequently confirmed in a Phase II study in another cohort of trastuzumab-resistant patients [103].
Recently, HER3 and its physiologic ligand heregulin (HRG) have been implicated in the development of resistance to antiestrogen therapies [102]. Presently, a humanized anti-HER3 monoclonal antibody MM-121 is available. This antibody binds to HER3 and prevents phosphorylation of HER3 and also effectively inhibits the HER2/HER3 heterodimer [104]. This compound is in Phase II studies, in combination with the nonsteroidal AI exemestane, in patients with advanced breast cancer that had previously progressed on endocrine therapies [100].
Triple-negative breast cancer (TNBC) is an aggressive disease with about 70% of breast tumors falling within the basal-like group of breast cancers [105]. A large majority of women with germline BRCA1 mutations have the triple-negative phenotype clustered within the basal group [101]. In 2009, Iglehart and Sliver [106] showed that inhibiting the remaining DNA repair machinery within BRCA-deficient cancer cells using (PARP) poly ADP ribose polymerase inhibition results in synthetic lethality. In 2011, AstraZeneca performed a proof of principal clinical trial on 54 subjects with BRCA1 or BRCA2 mutations with advanced breast cancer that were treated with PARP inhibitor olaparib [107]. Oral olaparib was administered to patients without significant toxicity. In addition, survival improvement was observed when Sanofi performed a randomized Phase II trial among patients with TNBC using another PARP inhibitor, iniparib (BSI-201) combined with carboplatin and gemcitabine [108]. The Hoosier Oncology group has currently an active Phase II [109] ongoing with a PARP inhibitor to evaluate 2-year disease-free survival in patient population treated with single-agent Cisplatin and patients treated with Cisplatin in combination with Rucaparib, another PARP inhibitor following preoperative chemotherapy treatment. The Lee Moffitt Cancer Center and Research Institute also has currently an active phase I/II clinical trial in progress [110] studying the Ad.p53 DC Vaccine and 1-MT in metastatic invasive breast cancer. Based on a report published by Bioseeker group (BSG) in October 2012 there are over 350 companies developing over 479 drugs targeting breast cancer. They identified drugs that are linked to more than 247 different targets which are divided into 61 classifications of molecular function.
Recently the cancer genome network (TCGA) reported a comprehensive molecular analysis of breast cancer tumors with a catalogue of likely genomic drivers of the most common breast cancer subtypes [111]. They categorized breast cancer into four main phenotypic cancer classes to explain the phenotypic heterogeneity observed within defined breast cancer subtypes. They identified two novel protein-expression-defined subgroups by identifying specific signaling pathways dominant in each molecular subtype including a HER2/phosphorylated HER2/EGFR/phosphorylated EGFR signature within the HER2-enriched expression subtype. Anti-EGFR (epidermal growth factor receptor) therapies, including tyrosine kinase inhibitors (TKIs) and monoclonal antibodies, are available and could potentially be used in breast cancer therapy.
4. Conclusions
This is a summary of several studies performed over the years that reveal the signaling pathways governing the MCF10DCIS.com and SUM cell lines. Breast tumors can be classified by their intrinsic subtypes. These subtypes have been associated with differences in survival with the basal-like and HER2-positive subtypes having the shortest survival times [112]. There are major gene pathways that give rise to the nature of MCF10DCIS.com and the SUM cells. This paper will serve as a valuable resource for future research in the identification of biomarkers and drug discovery, in breast cancer. Another important resource summarized in Table 7 is a list of mutation affecting the coding sequence in these cell lines as reported in the COSMIC database [83].
As we complete this paper, there are 538 clinical trials focused on breast cancer currently in progress. The breast cancer clinical trials are categorized either by type of breast cancer such as inflammatory or triple negative breast cancer or by response and resistance to treatments [113]. Most of these trials are not stratified by the analysis of individual tumor molecular marker profiles or mechanistic hypothesis. Although a signature specific to a specific subtype may not predict overall outcome, such a signature would have both biological and clinical utility. The utilization of highly characterized cells in the R&D will aid in the development of highly specific therapy and patient stratification.
Acknowledgments
The authors thank Dr. Victoria Blanc (Asterand US) and Dr. Evelyn Barrack (Henry Ford Health System, Detroit, MI) for a critical review of the paper and Mr. Justin Farkas (Asterand US) for his help in designing the figures and images.
References
- 1.Miller FR, Santner SJ, Tait L, Dawson PJ. MCF10DCIS.com xenograft model of human comedo ductal carcinoma in situ. Journal of the National Cancer Institute. 2000;92(14):1185–1186. doi: 10.1093/jnci/92.14.1185a. [DOI] [PubMed] [Google Scholar]
- 2.Polizzotti LM, Oztan B, Bjornsson CS, et al. Novel image analysis approach quantifies morphological characteristics of 3D breast culture acini with varying metastatic potentials. Journal of Biomedicine and Biotechnology. 2012;2012:16 pages. doi: 10.1155/2012/102036.102036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nature Reviews Molecular Cell Biology. 2003;4(1):33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 4.So JY, Lee HJ, Kramata P, et al. Differential expression of key signaling proteins in MCF10 cell lines, a human breast cancer progression model. Molecular and Cellular Pharmacology. 2012;4(1):31–40. [PMC free article] [PubMed] [Google Scholar]
- 5.Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nature Reviews Cancer. 2011;11(4):254–267. doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
- 6.Orian-Rousseau V, Morrison H, Matzke A, et al. Hepatocyte growth factor-induced Ras activation requires ERM proteins linked to both CD44v6 and F-actin. Molecular Biology of the Cell. 2007;18(1):76–83. doi: 10.1091/mbc.E06-08-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Chen N, Cui X, et al. The protein kinase Pak4 disrupts mammary acinar architecture and promotes mammary tumorigenesis. Oncogene. 2010;29(44):5883–5894. doi: 10.1038/onc.2010.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nature Reviews Cancer. 2004;4(12):937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 9.Kumar R, Gururaj AE, Barnes CJ. p21-activated kinases in cancer. Nature Reviews Cancer. 2006;6(6):459–471. doi: 10.1038/nrc1892. [DOI] [PubMed] [Google Scholar]
- 10.Bokoch GM. Biology of the p21-activated kinases. Annual Review of Biochemistry. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- 11.Mbeunkui F, Metge BJ, Shevde LA, Pannell LK. Identification of differentially secreted biomarkers using LC-MS/MS in isogenic cell lines representing a progression of breast cancer. Journal of Proteome Research. 2007;6(8):2993–3002. doi: 10.1021/pr060629m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449(7163):682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 13.Ong CC, Jubb AM, Haverty PM, et al. Targeting p21-activated kinase 1 (PAK1) to induce apoptosis of tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(17):7177–7182. doi: 10.1073/pnas.1103350108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. Journal of Clinical Oncology. 2010;28(6):1075–1083. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.So JY, Lee HJ, Smolarek AK, et al. A novel Gemini vitamin D analog represses the expression of a stem cell marker CD44 in breast cancer. Molecular Pharmacology. 2011;79:360–367. doi: 10.1124/mol.110.068403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogt PK, Hart JR, Gymnopolous M, et al. Phosphatidylinositol 3-Kinase: the oncoprotein. Current Topics in Microbiology and Immunology. 2011;347:79–104. doi: 10.1007/82_2010_80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalaany NY, Sabatini DM. Tumours with PI3K activation are resistant to dietary restriction. Nature. 2009;458(7239):725–731. doi: 10.1038/nature07782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janku F, Tsimberidou AM, Garrido-Laguna I, et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Molecular Cancer Therapeutics. 2011;10(3):558–565. doi: 10.1158/1535-7163.MCT-10-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakrabarty A, Rexer BN, Wang SE, Cook RS, Engelman JA, Arteaga CL. H1047R phosphatidylinositol 3-kinase mutant enhances HER2-mediated transformation by heregulin production and activation of HER3. Oncogene. 2010;29(37):5193–5203. doi: 10.1038/onc.2010.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shekhar MPV, Tait L, Pauley RJ, et al. Comedo-ductal carcinoma in situ: a paradoxical role for programmed cell death. Cancer Biology and Therapy. 2008;7(11):1774–1782. doi: 10.4161/cbt.7.11.6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Espina V, Mariani BD, Gallagher RI, et al. Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PLoS ONE. 2010;5(4) doi: 10.1371/journal.pone.0010240.e10240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nature Reviews Cancer. 2009;9(4):239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tait LR, Pauley RJ, Santner SJ, et al. Dynamic stromal-epithelial interactions during progression of MCF10DCIS.com xenografts. International Journal of Cancer. 2007;120(10):2127–2134. doi: 10.1002/ijc.22572. [DOI] [PubMed] [Google Scholar]
- 24.Tsutsumi A, Okada H, Nakamoto T, Okamoto R, Yasuda K, Kanzaki H. Estrogen induces stromal cell-derived factor 1 (SDF-1/CXCL12) production in human endometrial stromal cells: a possible role of endometrial epithelial cell growth. Fertility and Sterility. 2011;95(1):444–447. doi: 10.1016/j.fertnstert.2010.08.037. [DOI] [PubMed] [Google Scholar]
- 25.Hall JM, Korach KS. Stromal cell-derived factor 1, a novel target of estrogen receptor action, mediates the mitogenic effects of estradiol in ovarian and breast cancer cells. Molecular Endocrinology. 2003;17(5):792–803. doi: 10.1210/me.2002-0438. [DOI] [PubMed] [Google Scholar]
- 26.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107(5):1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 27.Brooks SA, Lomax-Browne HJ, Carter TM, Kinch CE, Hall DMS. Molecular interactions in cancer cell metastasis. Acta Histochemica. 2010;112(1):3–25. doi: 10.1016/j.acthis.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 28.Anna U, Newlaczyl AU, Yu L. Galectin-3—a jack-of-all-trades in cancer playing an important role in cancer cell growth, transformation. Cancer Letters. 2011;313(2):123–128. doi: 10.1016/j.canlet.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 29.Amm HM, Buschsbaum DJ. Relationship between galectin-3 expression and TRAIL sensitivity in breast cancer. Expert Review of Anticancer Therapy. 2011;11(8):1193–1196. doi: 10.1586/era.11.108. [DOI] [PubMed] [Google Scholar]
- 30.Shevde LA, Metge BJ, Mitra A, et al. Spheroid-forming subpopulation of breast cancer cells demonstrates vasculogenic mimicry via hsa-miR-299-5p regulated de novo expression of osteopontin. Journal of Cellular and Molecular Medicine. 2010;14(6):1693–1706. doi: 10.1111/j.1582-4934.2009.00821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ethier SP, Mahacek ML, Gullick WJ, et al. Differential isolation of normal luminal mammary epithelial cells and breast cancer cells from primary and metastatic sites using selective media. Cancer Research. 1993;53:627–635. [PubMed] [Google Scholar]
- 32.Ethier SP. Human breast cancer cell lines as models of growth regulation and disease progression. Journal of Mammary Gland Biology and Neoplasia. 1996;1(1):111–121. doi: 10.1007/BF02096306. [DOI] [PubMed] [Google Scholar]
- 33.Sartor CI, Dziubinski ML, Yu CL, Jove R, Ethier SP. Role of epidermal growth factor receptor and STAT-3 activation in autonomous proliferation of SUM-102PT human breast cancer cells. Cancer Research. 1997;57(5):978–987. [PubMed] [Google Scholar]
- 34.Forozan F, Veldman R, Ammerman CA, et al. Molecular cytogenetic analysis of 11 new breast cancer cell lines. British Journal of Cancer. 1999;81(8):1328–1334. doi: 10.1038/sj.bjc.6695007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keller PJ, Lin AF, Arendt LM, et al. Mapping the cellular and molecular heterogeneity of normal and malignant breast tissues and cultured cell lines. Breast Cancer Research. 2010;12(5, article R87) doi: 10.1186/bcr2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10(6):515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dawood S, Merajver SD, Viens P, et al. International expert panel on inflammatory breast cancer: consensus statement for standardized diagnosis and treatment. Annals of Oncology. 2011;22(3):515–523. doi: 10.1093/annonc/mdq345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charafe-Jauffret E, Ginestier C, Iovino F, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clinical Cancer Research. 2010;16(1):45–55. doi: 10.1158/1078-0432.CCR-09-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robertson FM, Cristofinilli M. New therapeutic targets in Inflammatory breast cancer. Current breast cancer reports. 2012;4(4):264–270. [Google Scholar]
- 40.Woodward WA, Debeb BG, Xu W, Buchholz TA. Overcoming radiation resistance in inflammatory breast cancer. Cancer. 2010;116(supplement 11):2840–2845. doi: 10.1002/cncr.25173. [DOI] [PubMed] [Google Scholar]
- 41.Van Den Eynden GG, Van Laere SJ, Van der Auwera I, et al. Overexpression of caveolin-1 and -2 in cell lines and in human samples of inflammatory breast cancer. Breast Cancer Research and Treatment. 2006;95(3):219–228. doi: 10.1007/s10549-005-9002-1. [DOI] [PubMed] [Google Scholar]
- 42.Victor BC, Anbalagan A, Mohamed MM, et al. Inhibition of cathesin B activity attenuates extracellulaer matrix degradation and inflammatory breast cancer invasion. Breast Cancer Research. 2011;13, article R115 doi: 10.1186/bcr3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Den Eynden GG, Van Laere SJ, Van der Auwera I, et al. Overexpression of calveolin-1 and -2 in cell lines and in human samples of human inflammatory breast cancer. Breast cancer Research and Treatment. 2006;95(3):219–228. doi: 10.1007/s10549-005-9002-1. [DOI] [PubMed] [Google Scholar]
- 44.Yuan K, Huang C, Fox J, et al. Elevated inflammatory response in caveolin-1-deficient mice with Pseudomonas aeruginosa infection is mediated by STAT3 protein and Nuclear Factor κB (NF-κB) Journal of Biological Chemistry. 2011;286(24):21814–21825. doi: 10.1074/jbc.M111.237628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25(43):5864–5874. doi: 10.1038/sj.onc.1209874. [DOI] [PubMed] [Google Scholar]
- 46.Elstrodt F, Hollestelle A, Nagel JHA, et al. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Research. 2006;66(1):41–45. doi: 10.1158/0008-5472.CAN-05-2853. [DOI] [PubMed] [Google Scholar]
- 47.Sørlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hollestelle A, Nagel JHA, Smid M, et al. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Research and Treatment. 2010;121(1):53–64. doi: 10.1007/s10549-009-0460-8. [DOI] [PubMed] [Google Scholar]
- 49.Bar-Sade RB, Kruglikova A, Modan B, et al. The 185delAG BRCA1 mutation originated before the dispersion of Jews in the Diaspora and is not limited to Ashkenazim. Human Molecular Genetics. 1998;7(5):801–805. doi: 10.1093/hmg/7.5.801. [DOI] [PubMed] [Google Scholar]
- 50.Smith J, Mun Tho L, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Advances in Cancer Research. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 51.Domagala P, Wokolorczyk D, Cybulski C, Huzarski T, Lubinski J, Domagala W. Different CHEK2 germline mutations are associated with distinct immunophenotypic molecular subtypes of breast cancer. Breast Cancer Research and Treatment. 2011;7(5):801–805. doi: 10.1007/s10549-011-1635-7. [DOI] [PubMed] [Google Scholar]
- 52.Wasielewski M, Hanifi-Moghaddam P, Hollestelle A, et al. Deleterious CHEK2 1100delC and L303X mutants identified among 38 human breast cancer cell lines. Breast Cancer Research and Treatment. 2009;113(2):285–291. doi: 10.1007/s10549-008-9942-3. [DOI] [PubMed] [Google Scholar]
- 53.Tung N, Silver DP. Chek2 DNA Damage Response Pathway and Inherited Breast Cancer Risk. Journal of Clinical Oncology. 2011;29(28):3813–3815. doi: 10.1200/JCO.2011.37.1476. [DOI] [PubMed] [Google Scholar]
- 54.Zoppoli G, Solier S, Reinhold WC, et al. CHEK2 genomic and proteomic analyses reveal genetic inactivation or endogenous activation across the 60 cell lines of the US National Cancer Institute. Oncogene. 2012;31:403–418. doi: 10.1038/onc.2011.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nature Reviews Cancer. 2002;2(8):594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 56.Ozaki T, Nakagawara A. Role of p53 in cell death and human cancers. Cancers. 2011;3(1):994–1013. doi: 10.3390/cancers3010994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wasielewski M, Elstrodt F, Klijn JGM, Berns EMJJ, Schutte M. Thirteen new p53 gene mutants identified among 41 human breast cancer cell lines. Breast Cancer Research and Treatment. 2006;99(1):97–101. doi: 10.1007/s10549-006-9186-z. [DOI] [PubMed] [Google Scholar]
- 58.Polsky D, Bastian BC, Hazan C, et al. HDM2 protein overexpression, but not gene amplification, is related to tumorigenesis of cutaneous melanoma. Cancer Research. 2001;61(20):7642–7646. [PubMed] [Google Scholar]
- 59.Agrawal A, Yang J, Murphy RF, Agrawal DK. Regulation of the p14ARF-Mdm2-p53 pathway: an overview in breast cancer. Experimental and Molecular Pathology. 2006;81(2):115–122. doi: 10.1016/j.yexmp.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 60.Lin AW, Lowe SW. Oncogenic ras activates the ARF-p53 pathway to suppress epithelial cell transformation. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(9):5025–5030. doi: 10.1073/pnas.091100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yarbrough WG, Bessho M, Zanation A, Bisi JE, Xiong Y. Human tumor suppressor ARF impedes S-phase progression independent of p53. Cancer Research. 2002;62(4):1171–1177. [PubMed] [Google Scholar]
- 62.Hernando E, Nahlé Z, Juan G, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430(7001):797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- 63.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81(3):323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 64.Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nature Reviews Cancer. 2008;8(9):714–724. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368(6473):753–756. doi: 10.1038/368753a0. [DOI] [PubMed] [Google Scholar]
- 66.Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Research. 2008;10(5, article R75) doi: 10.1186/bcr2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arnold A, Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. Journal of Clinical Oncology. 2005;23(18):4215–4224. doi: 10.1200/JCO.2005.05.064. [DOI] [PubMed] [Google Scholar]
- 68.Ertel A, Dean JL, Rui H, et al. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle. 2010;9(20):4153–4163. doi: 10.4161/cc.9.20.13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hollestelle A, Elstrodt F, Nagel JHA, Kallemeijn WW, Schutte M. Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Molecular Cancer Research. 2007;5(2):195–201. doi: 10.1158/1541-7786.MCR-06-0263. [DOI] [PubMed] [Google Scholar]
- 70.Pylayeva-Gupta Y, Grabockaand E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nature Reviews Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nature Reviews Molecular Cell Biology. 2005;6(8):622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 72.Van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cellular and Molecular Life Sciences. 2008;65(23):3756–3788. doi: 10.1007/s00018-008-8281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tanaka T, Lino M, Goto K. Knockdown of Sec6 improves cell-cell adhesion by increasing á-E-catenin in oral cancer cells. FEBS Letters. 2012;586(6):924–933. doi: 10.1016/j.febslet.2012.02.026. [DOI] [PubMed] [Google Scholar]
- 74.Hollestelle A, Elstrodt F, Timmermans M, et al. Four human breast cancer cell lines with biallelic inactivating α-catenin gene mutations. Breast Cancer Research and Treatment. 2010;122(1):125–133. doi: 10.1007/s10549-009-0545-4. [DOI] [PubMed] [Google Scholar]
- 75.Patriarca, Macchi RM, Marschner AK, Mellested H. Epithelial cell adhesion molecule expression (CD326) in cancer: a short review. Cancer Treatment Reviews. 2012;38(1):68–75. doi: 10.1016/j.ctrv.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 76.Maetzel D, Denzel S, Mack B, et al. Nuclear signalling by tumour-associated antigen EpCAM. Nature Cell Biology. 2009;11(2):162–171. doi: 10.1038/ncb1824. [DOI] [PubMed] [Google Scholar]
- 77.Mostert B, Kraan J J, Sieuwerts AM, van der Spoel P, et al. CD49f-based selection of circulating tumor cells (CTCs) improves detection across breast cancer subtypes. Cancer Letters. 2012;319:49–55. doi: 10.1016/j.canlet.2011.12.031. [DOI] [PubMed] [Google Scholar]
- 78.Lim E, Vaillant F, Wu D, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nature Medicine. 2009;15(8):907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 79.Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413(6853):316–322. doi: 10.1038/35095076. [DOI] [PubMed] [Google Scholar]
- 80.Gayther SA, Bailey SJ, Linger L, et al. Mutations truncating the EP300 acetylase in human cancer. Nature Genetics. 2000;32:300–203. doi: 10.1038/73536. [DOI] [PubMed] [Google Scholar]
- 81.Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Research. 2005;65(7):2554–2559. doi: 10.1158/0008-5472-CAN-04-3913. [DOI] [PubMed] [Google Scholar]
- 82.van de Wetering M, Barker N, Harkes IC, et al. Mutant E-cadherin breast cancer cells do not display constitutive Wnt signaling. Cancer Research. 2001;61(1):278–284. [PubMed] [Google Scholar]
- 83.COSMIC Database. http://www.sanger.ac.uk/genetics/CGP/cosmic.
- 84.Charafe-Jauffret E, Ginestier C, Iovino F, et al. Breast cancer cell lines contain functional cancer stem sells with metastatic capacity and a distinct molecular signature. Cancer Research. 2009;69(4):1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Park SY, Lee HE, Li H, Shipitsin M, Gelman R, Polyak K. Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Clinical Cancer Research. 2010;16(3):876–887. doi: 10.1158/1078-0432.CCR-09-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Polyak K. Heterogeneity in breast cancer. Journal of Clinical Investigation. 2011;121(10):2786–3788. doi: 10.1172/JCI60534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nakshatri H, Srour EF, Badve S. Breast cancer stem cells and intrinsic subtypes: controversies rage on. Current Stem Cell Research and Therapy. 2009;4(1):50–60. doi: 10.2174/157488809787169110. [DOI] [PubMed] [Google Scholar]
- 88.Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nature Reviews Molecular Cell Biology. 2003;4(1):33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 89.Giatromanolaki A, Sivridis E, Fiska A, Koukourakis MI. The CD44+/CD24− phenotype relates to “triple-negative” state and unfavorable prognosis in breast cancer patients. Medical Oncology. 2011;28(3):745–752. doi: 10.1007/s12032-010-9530-3. [DOI] [PubMed] [Google Scholar]
- 90.Lee HJ, Choe G, Jheon S, Sung SW, Lee CT, Chung JH. CD24, a novel cancer biomarker, predicting disease-free survival of non-small cell lung carcinomas: a retrospective study of prognostic factor analysis from the viewpoint of forthcoming (Seventh) New TNM classification. Journal of Thoracic Oncology. 2010;5(5):649–657. doi: 10.1097/JTO.0b013e3181d5e554. [DOI] [PubMed] [Google Scholar]
- 91.Zhou L, Jiang Y, Yan T, et al. The prognostic role of cancer stem cells in breast cancer: a meta-analysis of published literatures. Breast Cancer Research and Treatment. 2010;122(3):795–801. doi: 10.1007/s10549-010-0999-4. [DOI] [PubMed] [Google Scholar]
- 92.Du H, Wang L, He J, et al. CD44 is of functional importance for colorectal cancer stem cells. Clinical Cancer Research. 2008;14:6751–6760. doi: 10.1158/1078-0432.CCR-08-1034. [DOI] [PubMed] [Google Scholar]
- 93.Ricardo S, Vieira AF, Gerhard R, D R, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. Journal of Clinical Pathology. 2011;64:937–946. doi: 10.1136/jcp.2011.090456. [DOI] [PubMed] [Google Scholar]
- 94.Marotta LLC, Almendro V, Marusyk A, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24− stem cell-like breast cancer cells in human tumors. Journal of Clinical Investigation. 2011;121(7):2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gupta PB, Fillmore CM, Jiang G, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 96.Proia TA, Keller PJ, Gupta PB, et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cancer Stem Cell. 2011;8:149–163. doi: 10.1016/j.stem.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schoeberl B, Faber AC, Li D, et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Research. 2010;70(6):2485–2494. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.LoRusso P. A phase I dose-escalation study of safety, pharmacokinetics and pharmacodynamics of XL765, a p13K/TORC1/TORC1 inhibitor administered orally to patients) with advanced solid tumors. Journal of Clinical Oncology. 2009;25(supplement 15):p. 3502. [Google Scholar]
- 99.Baselga J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. The Oncologist. 2011;16:12–19. doi: 10.1634/theoncologist.2011-S1-12. [DOI] [PubMed] [Google Scholar]
- 100.Merrimack. Trial of exemestane +/- MM-121 in postmenopausal women with locally advanced or metastatic estrogen receptor positive and/or progesterone receptor positive her2 negative breast cancer. NIH Web site, 2011, http://clinicaltrials.gov/ct2/show/results/NCT01151046.
- 101.Higgins MJ, Baselga J. Targeted therapies for breast cancer. The Journal of Clinical Investigation. 2011;121(10):3797–3803. doi: 10.1172/JCI57152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krop IE, Beeram M, Modi S, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. Journal of Clinical Oncology. 2010;28(16):2698–2704. doi: 10.1200/JCO.2009.26.2071. [DOI] [PubMed] [Google Scholar]
- 103.Burris HA, Rugo HS, Vukelja SJ, et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2) -positive breast cancer after prior HER2-directed therapy. Journal of Clinical Oncology. 2011;29(4):398–405. doi: 10.1200/JCO.2010.29.5865. [DOI] [PubMed] [Google Scholar]
- 104.Schaefer G, Haber L, Crocker LM, et al. A Two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cell. 2011;20(4):472–486. doi: 10.1016/j.ccr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 105.Irvin WJ, Jr., Carey LA. What is triple-negative breast cancer? European Journal of Cancer. 2008;44(18):2799–2805. doi: 10.1016/j.ejca.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 106.Iglehart JD, Silver DP. Synthetic lethality—a new direction in cancer-drug development. The New England Journal of Medicine. 2009;361(2):189–191. doi: 10.1056/NEJMe0903044. [DOI] [PubMed] [Google Scholar]
- 107.Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Breast Diseases. 2011;22(2):180–181. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 108.O'Shaughnessy J, Osborne C, Pippen JE, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. The New England Journal of Medicine. 2011;364(3):205–214. doi: 10.1056/NEJMoa1011418. [DOI] [PubMed] [Google Scholar]
- 109.Hoosier Oncology Group. PARP inhibition for triple negative breast cancer (ER-/PR-/HER2-) with BRCA1/2 mutations. NIH Web site, 2012, http://clinicaltrials.gov/ct2/show/NCT01074970.
- 110.Lee Moffitt H, Cancer Center and Research Institute Study of Ad.p53 DC Vaccine and 1-MT in Metastatic Invasive Breast Cancer. 2012, http://clinicaltrials.gov/ct2/show/NCT01042535.
- 111.The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kennecke H, Yerushalmi R, Woods R, et al. Metastatic behavior of breast cancer subtypes. Journal of Clinical Oncology. 2010;28(20):3271–3277. doi: 10.1200/JCO.2009.25.9820. [DOI] [PubMed] [Google Scholar]
- 113.Breast Cancer Trials. https://www.breastcancertrials.org.