

Abstract
Excess generation of reactive oxygen species (ROS) and cytosolic calcium accumulation play major roles in the initiation of programmed cell death during acute myocardial infarction. Cell death may include necrosis, apoptosis and autophagy, and combinations thereof. During ischemia, calcium handling between the sarcoplasmic reticulum and myofilament is disrupted and calcium is diverted to the mitochondria causing swelling. Reperfusion, while essential for survival, reactivates energy transduction and contractility and causes the release of ROS and additional ionic imbalance. During acute ischemia–reperfusion, the principal death pathways are programmed necrosis and apoptosis through the intrinsic pathway, initiated by the opening of the mitochondrial permeability transition pore and outer mitochondrial membrane permeabilization, respectively. Despite intense investigation, the mechanisms of action and modes of regulation of mitochondrial membrane permeabilization are incompletely understood. Extrinsic apoptosis, necroptosis and autophagy may also contribute to ischemia–reperfusion injury. In this review, the roles of dysregulated calcium and ROS and the contributions of Bcl-2 proteins, as well as mitochondrial morphology in promoting mitochondrial membrane permeability change and the ensuing cell death during myocardial infarction are discussed.
Keywords: apoptosis, Bcl-2, caspase, ischemia–reperfusion, necrosis, oxidative stress
Overview of death pathways activated during acute myocardial infarction
Necrosis, apoptosis and autophagy are all implicated in myocardial cell loss during ischemic heart disease (reviewed in [1–7]). Apoptosis and autophagy play essential roles in eliminating unwanted cells during pre- and post-natal development and damaged or senescent cells in adult tissues [1]. These roles are physiological. Classical necrosis occurs during sustained, severe ischemia; it is an unregulated irreversible response to a fatal insult often involving energy failure. Reducing the magnitude of the insult may be the only way to prevent such death. Loss of myocardial cells by programmed death and autophagy after ischemia–reperfusion is at least partially pathological. In this case, the cells respond to environmental signals that they cannot distinguish from normal physiological cues, many of which have been conserved from fungi and worms over a billion years of evolution [8]. It may be possible to salvage cells undergoing pathological apoptosis, programmed necrosis and/or autophagy through interventions that block the signaling pathways (reviewed in [3]). Although each death pathway has distinguishing features, they are not mutually exclusive and tissue loss during ischemia of the heart and other organs may involve simultaneous or alternate contributions of multiple death programs, reviewed in [2–7,9].
Passive necrosis
Necrosis typically involves swelling of the mitochondria, whole-cell swelling and loss of plasma membrane integrity. Owing to the loss of membrane intergrity, toxic cellular contents are released causing additional collateral damage, triggering an inflammatory response that perpetuates the collateral injury. In the absence of reperfusion, necrosis develops rapidly as a consequence of energy failure; without oxygen and glucose, oxidative phosphorylation ceases and cells cannot generate the ATP needed to fuel the ionic pumps that maintain ionic gradients across the plasma membrane. Consequently, sodium and calcium accumulate in the cytoplasm and cause swelling, degeneration of organelles, loss of membrane integrity and dissolution of the cell [10,11].
Programmed necrosis
Necrosis continues after reperfusion but it may be through a different process. Until recently, reperfusion-induced necrosis was also considered to be passive, and was called coagulation necrosis; however, it is now clear that signaling pathways not dissimilar to apoptosis can also regulate necrotic cell death [2–5,12–17]. Such death termed ‘programmed necrosis’ is initiated by large amplitude swelling of the mitochondrial matrix, dispersion of the mitochondrial membrane potential, ATP loss and opening of the so-called mitochondrial permeability transition pore (MPTP). Caspase activation is not a feature of programmed necrosis per se, but it may occur in parallel if matrix swelling causes rupture of the outer membrane (OM) before ATP is depleted in a significant number of mitochondria. In this case, pro-apoptotic factors including cytochrome c are released and caspases may be activated during programmed necrosis [2,12,13]. As discussed in detail in Section ‘Death domains’, energy depletion, ROS, calcium, pH, inorganic phosphate, mitochondrial morphology as well as Bcl-2 proteins may contribute to the regulation of the MPTP [18–24]. The adenine nucleotide translocator (ANT) and voltage dependent anion channel (VDAC) may also contribute to the regulation of the MPTP although they do not appear to be essential [15,25–34]. On the other hand, cyclophilin D (CypD), an inner membrane peptidyl-prolyl cis-trans isomerase, regulates the sensitivity of the MPTP opening by calcium and ROS and is a potential target for reducing injury during acute myocardial infarction (AMI) [3,12,13,35]. Controversies over the structure and regulation of the MPTP are discussed in more detail in Section ‘Death domains’.
Extrinsic apoptosis
Apoptosis is characterized by membrane blebbing, cell shrinkage, nuclear fragmentation and chromatin condensation. Activation of a family of cysteine proteases called caspases is the distinguishing biochemical feature of apoptotic death. Caspases are activated by one of two major pathways (reviewed in [1–7]). The extrinsic pathway is a cellular response to inflammation. In this pathway, plasma membrane receptors are activated by proinflammatory ligands, including FAS, TNF-α and TRAIL. These ligands bind to death domain-containing receptors at the plasma membrane that become activated and recruit the adapter proteins FADD and TRADD to form a death-inducing signaling complex (DISC). DISC formation initiates the proteolytic cleavage of procaspases 8, 10 and 2, leading to the activation of caspases 7, 6 and 3. Cell death by the extrinsic pathway may require amplification by coactivation of the intrinsic pathway mediated by caspase-8-cleavage and activation of the pro-apoptotic Bcl-2 family protein, Bid, that can induce mitochondrial outer membrane permeability (MOMP) (reviewed in [36]). Activation of the extrinsic pathway requires a primary inflammatory response to tissue injury and may lag behind the intrinsic pathway that responds immediately to calcium and ROS. Evidence for the contribution of the extrinsic pathway to AMI includes observations that mice lacking Fas ligand are more resistant to infarction [37,38], and the presence of elevated Fas ligands in the plasma of patients with AMI [39].
Intrinsic apoptosis
Caspase activation by the intrinsic pathway requires the release of mitochondrial death factors including cytochrome c, inhibitor of apoptosis (IAP) binding proteins, second mitochondria-derived activator of caspase (SMAC), Omi/HtrA2 apoptosis-inducing factor, and endonuclease G [40–43]. Release of these factors usually follows calcium-dependent swelling of mitochondria and OM permeabilization. The balance of pro- and anti-apoptotic Bcl-2 proteins in mitochondrial and endoplasmic reticulum (ER) membranes regulates MOMP in part by regulating calcium compartmentalization. Bcl-2, Bcl-XL and Mcl-1 prevent MOMP whereas pro-apoptotic members, Bax and Bak, activate MOMP (reviewed in [1–7]). The intracellular localization and activities of these Bcl-2 proteins are under the regulation of a third set of Bcl-2-related proteins, known as BH3-only proteins, that respond directly to apoptotic stimuli. In addition to regulating MOMP by interacting with mitochondrial membranes, Bcl-2 family proteins also differentially regulate ER calcium and its release by death-inducing stimuli [44–47]. Anti-apoptotic Bcl-2 proteins within the ER facilitate slow calcium leakage through an inositol trisphosphate receptor type 1 (IP3R-1), allowing partial calcium unloading and lower resting ER stores. Bax and Bak counter this by blocking calcium leakage by IP3R-1, increasing ER calcium stores, and magnifying the release of calcium by the ER and subsequent uptake by the mitochondria during the initiation of programmed death. The Bcl-2-regulated two-way cross-talk between the ER and mitochondria may be especially relevant to death pathway signaling within the ER junctional clefts that contain the so-called calcium ‘hot spots’ (see below). Both extrinsic and intrinsic pathways culminate in the activation of the executor caspase-3 that digests cellular proteins and macromolecules, and activates DNases (CAD) that together with endonuclease G and other DNases degrade proteins and DNA [48]. After the initiation of programmed death, phosphatidylserine is externalized on the plasma membrane and targets dying cells for phagocytosis by macrophages and elimination from the tissue (reviewed in [49]). The intrinsic pathway predominates over the extrinsic pathway during immediate phase of cell death (2–24 h) following ischemia–reperfusion [1–7].
Autophagy & ER stress
During normal autophagy, damaged organelles and macromolecules are packaged into double membrane structures called autophagosomes and are transferred to lysosomes for elimination from the cell. Autophagy is activated by ER stress through the unfolded protein response and provides a housekeeping function that favors cell survival in the face of multiple stresses caused, for example, by nutrient deprivation, hypoxia and elevated oxidative stress (reviewed in [50–53]). During persistent or extreme ER stress, such as occurs during severe ischemia–reperfusion, the cytoprotective functions of the unfolded protein response and autophagy can be switched to a cell-death response [51]. Neither the pathways of autophagic death nor the circumstances that determine the switch from cytoprotection to cell destruction during AMI are understood [52,53]. It is possible that hyperactive autophagy causes mitochondrial dysfunction and this provokes necrosis and/or apoptosis. Bnip3 is a hypoxia-induced atypical BH3-only member of the Bcl-2 family that has been linked with autophagy [52]. Controversy exists concerning the contributions of Bnip3 to myocardial cell death during infarction and it has been assigned various key roles in autophagy, apoptosis and programmed necrosis [5,7,54–56]. These will be discussed in more detail in Section ‘Regulation of infarction by mitochondrial permeability channels’. There is evidence for cross-talk between autophagy, apoptosis and ER stress, in the form of binding and regulation of key intermediates, such as beclin and CHOP by Bcl-2 (please refer to recent reviews for detailed discussion of autophagy, ER stress and mitochondrial remodeling in relation to ischemic cardiovascular disease [52,53,57–59]).
Owing to the dynamic nature of calcium movements in cardiac myocytes and the presence of high-affinity binding sites on multiple intracellular organelles, in particular the sarcoplasmic reticulum (SR), myofilament and mitochondria, precise calcium handling and compartmentalization are essential in order to maintain cardiac function. Calcium homeostasis is progressively lost during ischemia, caused in part by declining energy levels and partly by changes of other ions including decreased intracellular pH and increased inorganic phosphate (Pi). When ischemia is extended, calcium build-up contributes to classical necrosis and may also predispose cells to programmed death. Reperfusion causes additional dysregulation of calcium flux and compartmentalization, and excess ROS production that cause contractile arrhythmias (myocardial stunning) and predispose cells to programmed death. To understand how this occurs, it is important to document the movements of calcium during normal excitation–contraction coupling and the effect of ischemia–reperfusion.
Normal calcium handling by the plasma membrane, SR & myofilament
Defective calcium handling causes reversible, as well as irreversible myocardial injury and is a primary therapeutic target for cardioprotection during AMI and heart failure (reviewed in [60,61]). Under normal conditions, the sarcoplasmic calcium concentration is low with a gradient of several orders of magnitude between intra- and extracellular compartments. During an action potential, voltage-gated Na+ channels are activated, and the inward Na+ current induces a rapid depolarization of the sarcolemma that opens L-type Ca2+-channels. The Ca2+-influx triggers opening of ryanodine receptors (RyRs), through which large amounts of Ca2+ are released from the SR, a process termed Ca2+-induced Ca2+-release [62–64]. The wave of Ca2+ from the RyRs generates a rapid (ms) >500-fold increase in calcium concentration in the space between the SR and the sarcolemma, called the junctional cleft [65,66]. The junctional cleft calcium then rapidly diffuses into the cytosol [67,68]. The dynamic changes of calcium in different regions of the sarcoplasm are referred to as ‘Ca2+ microdomains’ [69]. During systole, Ca2+ binds to troponin C of the myofilament and induces contraction of the left ventricle. During diastole, the muscle relaxes, Ca2+ diffuses from the myofilaments back to the cytosol and is pumped back into the SR by the SR-calcium ATPase (SERCA channel), and out of the myocyte by a sarcolemmal calcium ATPase (Figures 1 & 2).
Figure 1. Calcium transients in sarcoplasmic reticulum and junctional cleft mitochondria.
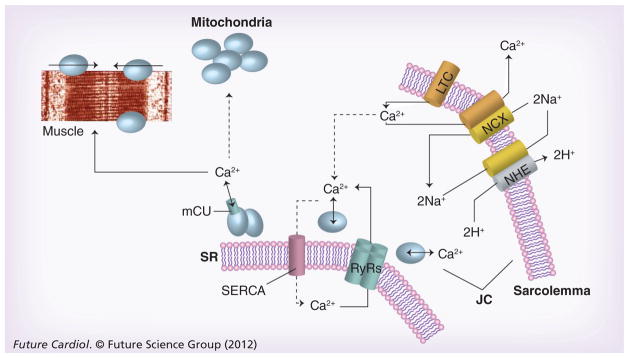
Calcium enters the cardiac myocyte at the beginning of the contraction cycle through LTC. This calcium diffuses through the myocyte to interact with the RyRs on the SR. The SR is a calcium storage organ and stimulation of the RyRs by calcium causes massive calcium release from the SR, which is channeled to muscle fibers where it initiates contraction. At the end of the contraction cycle, calcium is pumped back into the SR by a calcium pump called SERCA. The calcium that originally entered through the LTC is transported back out of the myocyte in exchange for sodium by the NCX exchanger in the sarcolemma. This causes net entry of sodium that can be removed in exchange for protons through the NHE exchanger. Calcium fluxes are highest in the JC between the SR and sarcolemmal membranes, known as a calcium microdomain. Mitochondria are also at high density within the JC and transport calcium in parallel with the calcium transient. Calcium is taken up by mitochondria through an mCU that normally operates in unison with a Na+/Ca2+ antiporter to maintain calcium homeostasis.
JC: Junctional cleft; LTC: L-type calcium channels; mCU: Electrogenic importer; NCX: Sodium calcium exchanger; NHE: Sodium-proton exchanger; RyRs: Ryanodine receptor; SERCA: Sarcoplasmic reticulum calcium ATPase; SR: Sarcoplasmic reticulum.
Figure 2. Calcium imbalance during ischemia–reperfusion causes mitochondrial swelling.

Glycolysis is activated by ischemia and produces excess intracellular acid. The protons are pumped out of the cell in exchange for sodium by the NHE. Sodium in turn is pumped out by reversal of the NCX causing net accumulation of calcium. Calcium is immediately taken up by the mCU. mCU activity is normally balanced by the mitochondrial NCX, but this balance fails during ischemia and calcium accumulates followed by phosphate and water causing mitochondrial swelling.
LTC: L-type calcium channels; mCU: Mitochondrial uniporter; NCX: Sodium calcium exchanger; NHE: Sodium-proton exchanger; SR: Sarcoplasmic reticulum.
Calcium handling during ischemia–reperfusion
Increased glycolytic activity during ischemia causes the intramyocyte pH to fall. The elevated proton concentration activates a Na+/H+ antiporter that expels H+ in an electroneutral exchange for Na+. This reduces acidosis but increases the intramyocyte sodium concentration. When the sodium level reaches a threshold, it forces the Na+/ Ca2+ antiporter to reverse so that sodium is expelled in exchange for calcium. The effect is a progressive increase of intracellular calcium that continues for the duration of ischemia. Calcium overload in the cytoplasm causes increased uptake by mitochondria and as the calcium-selective electrogenic importer (mCU) is nonsaturable, mitochondria accumulate calcium as long as an electrochemical potential exists across the inner membrane. During ischemia, glycolytic ATP is used to maintain the potential so that calcium flows into the mitochondria, as long as there is glycolytic ATP. The fall of intracellular pH during ischemia is also accompanied by build up of Pi; H+ and Pi both reduce the myofilament calcium sensitivity so that force generation is decreased despite increased calcium while ATP is conserved [70]. These changes favor calcium accumulation and swelling of the mitochondria, while the low pH suppresses mitochondrial permeability transition (MPT). This condition continues until ATP is depleted and myofilaments undergo contracture due to diastolic failure. Contracture is accompanied by progressive necrosis [70]. Paradoxically, calcium uptake and swelling may continue in viable cells after reperfusion due to a transient pH imbalance created by fresh extracellular blood. The imbalance occurs because the extracellular acidosis, built up during ischemia is immediately neutralized, but the intracellular compartment remains transiently acidotic. This causes a transient outside-to-inside proton gradient across the sarcolemma that stimulates proton extrusion through the Na+/H+ channel and an antiparallel uptake of calcium. This may contribute to increased hypercontracture, as well as to further calcium overload of the mitochondria (see below). Calcium overload contributes to the activation of protein kinases and opening of the death channels. It is a key inducer of both the MPTP and MOMP and as such the initiating stimulus for myocardial cell death and infarction during both phases of ischemia and reperfusion. Inhibition of the Na+/Ca2+ antiporter during ischemia–reperfusion can reduce mitochondrial Ca2+ overload, contracture and cell death, and improve bioenergetic profiles (Figure 2).
Mitochondrial transients in the junctional cleft
Mitochondria are sinks for calcium, and recent work has confirmed that mitochondria within the junctional cleft act as calcium buffers, rapidly taking up and extruding calcium in synchrony with the sarcoplasmic calcium transient [69]. Mitochondria occupy up to 30% of cardiac myocyte volume and are highly concentrated around the myofilament and SR, the so-called calcium hot spots. During the contraction cycle these mitochondria are cyclically exposed to high calcium levels and cycle calcium in parallel with the transient. The amount of calcium exchanged is related to the proximity of the mitochondria to the SR and myofilament, with the closest mitochondria cycling more calcium than those at a distance. These regions are called ‘mitochondrial calcium microdomains’ and the calcium cycling may modulate the filling state of the neighboring SR, as well as the dynamics of binding to troponin C and initiation of contraction. Such calcium cycling regulates mitochondrial respiration and ATP production, and is an integral component of energy coupling during muscle contraction. The effect on mitochondrial respiration and oxidative phosphorylation occurs through calcium-mediated activation of rate-limiting tricarboxylic acid cycle enzymes and the F1/F0-ATPase [71–73]. Calcium takes <100 ms to activate mitochondrial respiration, which is consistent with a synchronized contribution of microdomain mitochondria to energy transduction during excitation–contraction coupling (reviewed in [41]). As noted above, calcium enters the mitochondria down the inner membrane electrochemical gradient through a mCU. Patch clamping measurements of mitoplast membranes show that the mCU transports Ca2+ with a half-activation constant of approximately 20 mM and a Ca2+ carrying capacity (Vmax) of 5106 Ca2+ s−1 per single channel molecule [74,75]. These studies predict a channel density at the inner mitochondrial membrane of 10–40 channels per μm2, similar to the density of voltage-gated Ca2+ channels at the sarcolemmal membranes [74]. The current lacks Ca2+-dependent inactivation, is highly selective for Ca2+, and allows Ca2+ to accumulate in the mitochondria with minimal energy expenditure through a process that continues whenever there is a membrane potential [74]. The mCU normally functions in synchrony with a Na+/Ca2+ anti-porter, and calcium homeostasis is maintained by quantitative antiparallel extrusion of calcium in exchange for sodium. Some studies suggest that defective mitochondrial calcium domains associated with dysregulated calcium handling cause metabolic stress that contributes to heart failure [74–77]. It also seems likely that these microdomains are the primary sensors of dys-regulated calcium handling during ischemia and reperfusion. During the early stages of ischemia, before energy depletion, calcium transients can increase markedly, but force generation declines because a low pH and elevated Pi desensitize the myofilament to calcium [70]. Under this condition respiration is inhibited but glycolytic ATP maintains the mitochondrial membrane potential. Owing to the increased calcium transients, mitochondria within the calcium microdomains cycle more calcium, that may progress to calcium overload and swelling specifically in these regions as ischemia proceeds. This condition is predicted to predispose these mitochondria to MPT and MOMP and the release of prodeath factors for programmed necrosis and apoptosis during reperfusion. Responses of mitochondria in the calcium microdomains may also be transmitted throughout cardiac myocytes by fission and fusion as well as throughout the myofilament through gap junctions [78,79].
Oxidative stress of reperfusion
During ischemia, electron transport through the mitochondrial electron transport chain (ETC) ceases due to the lack of oxygen as an electron acceptor, and the ETC is rapidly reduced. An electron back-up primes the soluble ubiquinone components of the ETC to generate oxygen free radicals when oxygen returns. Additional ROS can also be generated from the NADPH oxidase pathways that are activated by ischemia–reperfusion. As illustrated in Figure 3, electrons are donated from intermediates of the citric acid cycle, through reduced nicotine adenine dinucleotide (NADH) and flavin adenine dinucleotide (FAD) to acceptor complexes of the ETC, and energy is released as the electrons move down the ETC, eventually reaching oxygen, the final and most electropositive electron acceptor. The electrical energy is used to pump protons (H+) across the mitochondrial membrane creating a proton motive force. The protons pass back through the proton-translocating ATPase of the mitochondrial inner membrane reversing ATPase and generating ATP. During normal function, it is estimated that 2–5% of the oxygen consumed by the mitochondria during respiration is converted to superoxide [80,81]. This is mostly neutralized by mitochondrial antioxidants, manganese superoxide dismutase, catalase, glutathione peroxidase, thioredoxin, peroxiredoxin and glutaredoxin (reviewed in [82]). Excess ROS activates second messenger pathways, including the protein kinases c-Jun N-terminal kinase (JNK), extracellular regulated kinase (ERK), P38, protein kinase C and PI3-kinase pathways[83–85]. ROS can also cause transitory loss of the mitochondrial membrane potential even during normal myocardial functions. Zorov et al. first described a pathway of ROS-induced ROS-release by mitochondria that could dissipate the membrane potential and even mediate MPTP opening [86]. Aon et al. extended these studies to demonstrate reversibility of the effects and proposed that ROS leakage from the ETC activates an inner mitochondrial anion channel (IMAC), distinct from the MPTP that releases ROS into the cytoplasm and simultaneously dissipates the membrane potential [87]. The signal is propagated to neighboring mitochondria also through the ROS-induced ROS mechanism, that sends a wave of oscillations across the cytoplasm. Under normal conditions the IMAC closes and the mitochondria recover. Under conditions of excess ROS generation during reperfusion and calcium overload, opening of the IMAC could induce MTP. Mitochondria in the calcium hot spots of the junctional cleft may be particularly sensitive to MPTP opening by this pathway. Reperfusion causes more than an order of magnitude increase in electron leakage by the mitochondria over the levels of normal function. The major ROS generators are the ubiquinone–ubiquinol mobile electron carriers, popularly known as coenzymes Q10 (Figure 3). Ubiquinone (coenzyme Q) acts as a sink to absorb and neutralize oxygen free radicals. Ubiquinone normally accepts electrons from complexes I and III of the ETC and transfers them to complex IV and cytochrome c. When oxygen is absent there is no final electron acceptor for complex IV. Consequently, the ubiquinol pool is highly reduced, increasing the level of ubisemiquinone, the reduced form of ubiquinone. Ubisemiquinone is a reactive free radical, and when oxygen returns to the mitochondria during reperfusion, ubisemiquinone donates electrons directly to oxygen generating superoxide. Superoxide is a diffusible free radical and it reacts rapidly with neighboring molecules. Lipid and protein components of the MPTP are present in the same membrane system as the ETC and may be immediate targets of the excess ROS generated during reperfusion. The combination of calcium accumulation in cytosolic and mitochondrial compartments, membrane swelling and high ROS levels caused by reperfusion are the initiating events that cause cellular injury, kinase activation, activation of autophagy and changes in BcL-2 protein compartmentalization that result in increased mitochondrial permeability (MPTP and MOMP) and cell death.
Figure 3. Generation of superoxide by the electron transport chain.

Ubiquinone is a lipid-soluble electron carrier that can accept electrons from complexes I–III generating QH2 that diffuses in the membrane and can donate electrons to cytochrome c or oxygen. The ubiquinone–QH2 pool is the major source of oxygen free radicals generated by the electron transport chain. Under ischemic or hypoxic conditions electrons cannot exit the electron transport chain and there is an electron back-up that causes the pool of QH2 to increase. When oxygen returns during reperfusion, electrons leak from QH2 to oxygen causing massive production of superoxide. It has been proposed that superoxide activates an IMAC through which superoxide radicals can exit the mitochondria. Reactive oxygen species may also collapse the membrane potential and open the mitochondrial permeability transition pore.
IMAC: Inner membrane anion channel.
Hypercontracture & cell death during AMI
One of the earliest histological features of infarction is rupture of the sarcolemmal and extensive areas of hypercontracted muscle, recognizable within minutes of reperfusion, and referred to as contraction-band necrosis (reviewed in [88]). When cardiac myocytes are re-energized during reperfusion the excess calcium available to the sarcomere causes exaggerated contraction, called hypercontracture. Calcium overload and contracture coincide with cell death and infarction, elegantly illustrated by studies that have demonstrated protection against infarction by temporary inhibition of contraction during reperfusion to allow recovery of calcium regulation [89–94]. The links between contracture and cell death are not fully defined, but include oxidative stress and ATP depletion as well as calcium overload (discussed in Sections ‘Normal calcium & ROS handling & dysregulation by ischemia–reperfusion’ and ‘Regulation of infarction by mitochondrial permeability channels’). Hypercontracture is propagated between myocytes within the reperfusion zone by sodium exchange through gap junctions [93]. Such propagation may explain the continuous distribution of dead cardiac myocytes within areas of contracture [94]. The link between defective calcium handling, elevated ROS and contracture is compelling, as is the link with defective mitochondrial function, membrane permeabilization (MPTP and MOMP) and programmed death.
Death channels
MPT structure & mechanism
Electrophysiologically, the MPTP acts as a non-specific, voltage-independent megachannel with a molecular cutoff of approximately 1.5 kDa and a diameter of approximately 2.3 nm that traverses both mitochondrial membranes [14]. These characteristics have been confirmed by diffusion kinetics, reconstitution and patch clamp studies of isolated mitochondria (reviewed in [2–4,14]). Opening of the MPTP results in mitochondrial swelling, dissipation of the mitochondrial membrane potential and loss of oxidative phosphorylation. By abolishing mitochondrial energy production and causing energy collapse, the MPTP initiates necrotic cell death during ischemia and reperfusion. No structural component has been unequivocally identified as essential for activity of the MPTP, thus the molecular mechanism of action of the MPTP is unknown. Mitochondrial channels formerly linked to MPTP function include the OM VDAC, the inner membrane ANT and the inner membrane phosphate ion channel (Figure 4) [14]. Gene ablation studies revealed that neither ANT, nor VDAC, are essential for MPTP activity [15,95]. Hepatocytes from mice with liver-specific ablation of both mouse ANT genes retained MPTP function, although with decreased sensitivity to both calcium and oxidative stress. Similarly, cells from mice with ablation or combined ablation and knockdown of each of the three VDAC isoforms retained normal MPTP function as well as responses to necrotic and apoptotic stimuli. ANT and VDAC normally transport ATP and ADP across mitochondrial membranes. In the gene ablation studies it was not reported whether these functions were retained. If they were, then it seems possible that substitute transport systems for adenine nucleotides could also substitute for ANT or VDAC in promoting MPTP function. Furthermore, the effects of combined ablation or knockdown of VDAC and ANT on MPTP have not been reported. Therefore, it is still possible that there is functional redundancy of ANT and VDAC (and/or phosphate ion channel and IMAC) in the normal operation of the MPTP. Interestingly, the IMAC may be regulated by the mitochondrial benzodiazepine receptor (mBzR), formerly thought to be a component of the MPTP. Another possibility is that denatured/ oxidized proteins play a role in forming the MPTP [96]. MPTP accessory proteins include CypD, a prolyl isomerase present on the matrix surface of the inner membrane [12,23], hexokinase-II (Hex-II), a glycolytic pathway enzyme that interacts with VDAC2 and may suppress the MPTP under some conditions [97–99], and the peripheral-type benzodiazepine receptor that has been reported to complex with VDAC and ANT and induce or inhibit the MPTP depending on ligand binding. A regulator role for CypD in MPT and programmed necrosis during AMI is unequivocal, confirmed biochemically and genetically [12–14]. CypD determines the sensitivity of MPT to calcium and ROS in vitro and in vivo. Preliminary clinical studies indicate a small but significant protection from infarction when patients were infused with cyclosporine A (CsA), a selective CypD inhibitor [100].
Figure 4. Mitochondrial death channels.

Proteins associated with the MPTP include VDAC, ANT and CypD. The IMAC and phosphate ion channels have also been implicated. Structural elements of MOMP has not been defined structurally, but channel activity is regulated by interactions of Bax, Bak and tBid; some studies implicate VDAC as a component of MOMP while others suggest that Bax not only regulates MOMP, but may also be a structural component. Mitochondrial permeability transition and MOMP are induced by calcium and oxidative stress and Bcl-2 proteins may regulate both. Some reports suggest that VDAC and ANT bridge the IM and OM by interacting at contact points. Other reports suggest that neither ANT nor VDAC are required for MPTP function but both ANT and CypD regulate MPTP opening. The MPTP opens under conditions of ischemia–reperfusion when matrix calcium and reactive oxygen levels increase. Cytochrome c release from the intermembrane space and ICS triggers apoptosis by activating the apoptosome, followed by caspase and DNAse activation. Approximately 15% of total cytochrome c is in the IMS and the rest is in the ICS; membrane fission and fusion events regulated in part by Bax and tBid are required for efficient release of cytochrome c. Low matrix pH inhibits the opening of the MPTP when ΔTM is dissipated but a low pH induces MPTP opening in energized mitochondria.
ΔTM: Membrane potential; ANT: Adenine nucleotide translocase; BDR: Benzodiazepine receptor; CypD: Cyclophilin D; ICS: Intercristal space; IM: Inner membrane; IMAC: Inner membrane anion channel; IMS: Inter-membrane space; MAC: Membrane anion channel; mCU: Mitochondrial uniporter; MOMP: Mitochondrial outer membrane permeability; MPTP: Mitochondrial permeability transition pore; OM: Outer membrane; PiC: Phosphate ion channel; ROS: Reactive oxygen species; VDAC: Voltage-dependent anion channel.
The role of the VDAC is perhaps the most controversial because VDAC has been linked with both MPTP and cell death regulated by Bcl-2 family proteins [26–30]. VDACs are related to bacterial porins, β-barrel proteins, that traverse cellular membranes and act as pores for the exchange of small molecules by diffusion. Mammalian mitochondria contain three VDAC isoforms of similar size (28–36 kDa) and are the most abundant proteins in the outer mitochondrial membrane (OMM). VDACs allow the exchange of metabolic substrates, NADH and ATP, between the intermembrane space and cytosol [101]. At first glance, VDACs are unlikely candidates for the megachannel activity of MPTP owing to their small molecule conductance and selectivity. However, porins in general and VDACs in particular are unusually sticky, interacting with and possibly modulating the functions of other proteins at both membrane surfaces. Interactions between VDACs and Hex-II, ANT, and Bcl-2 proteins, tBid, Bcl-xL and Bak, have been described [14,31–34,102,103]. Mathupal et al. showed that Hex-II binds to and enhances the functions of VDAC in cancer cells, possibly by inducing a conformational change of VDAC [78]. The Hex-II–VDAC interaction protected cells against cytotoxic stimuli including oxidative stress, a property that they attributed to blocking of the MPTP and/or preventing the binding of pro-apoptotic Bcl-2 proteins. These effects were prevented when VDAC was phosphorylated by GSK3β that disrupts the binding of Hex-II, and were reversed by Akt-mediated inhibition of GSK3β[104,105]. These results suggest that VDAC/Hex-II controls an MPTP-dependent death pathway, responsive to oxidative stress that is sensitive to GSK3β and Akt, both known to regulate the MPTP [106].
Multiple studies have reported infarct protection by anti-apoptotic Bcl-2 proteins [107,108]. Perfusion of hearts with a cell-permeant (TAT)-peptide containing the protective BH4 domain of Bcl-xL reduces infarction up to 50% by blocking apoptosis and/or necrosis [109,110]. In support of a role for the MPTP in this response, Juhaszova et al. demonstrated that treatment of isolated cardiac myocytes with a TAT–BH4 peptide enhanced the threshold ROS required to open the MPTP by approximately 40%, confirming that the BH4 peptide blocked MPTP opening [110]. In further studies they used HA14-1, a small cell-permeable selective inhibitor of Bcl-2. HA14-1 treatment blocked the protection afforded by TAT–BH4, as well as that of several other protective pathways including an NHE-inhibitor, Akt, or direct inhibition of GSK-3β with lithium. The authors concluded that Bcl-2 is downstream of GSK3β and regulates the MPTP. If this is true, then Bcl-2 family members exert regulation of both mitochondrial death channels and may control the pathways of necrosis and apoptosis during ischemia–reperfusion. VDAC is the only MPTP-associated protein known to bind Bcl-2 family proteins. Therefore, it seems possible that VDAC is also a component of this activity; one or more of the VDAC isoforms may confer regulation of the MPTP by Bcl-2. In the VDAC deletion studies reported by Baines et al., the effects of Bcl-2 on MPTP-dependent necrotic death was not investigated [15]. Therefore, these results do not preclude a role for VDAC in the regulation of MPTP by Bcl-2.
MPTP opening by calcium & ROS
The combination of increased calcium and oxidative stress promotes opening of the MPTP during ischemia–reperfusion, and the extent of MPTP opening correlates closely with the extent of infarction. In isolated mitochondria, calcium alone causes MPTP opening and calcium chelators close the pore (reviewed in [14]). Positive and negative modulators of pore opening by calcium have been identified. Agents that cause oxidative stress, adenine nucleotide depletion, elevated phosphate concentration and mitochondrial depolarization sensitize the MPTP to calcium. MPTP is inhibited by Mg2+, protons (pH <7.0), ATP or ADP, and ligands of the ANT. Carboxyatractyloside, an ANT inhibitor that locks the ANT in a cytosol-directed conformation, increases the sensitivity of pore opening to Ca2+ while another inhibitor, bongkrekic acid, locks the ANT in the opposite orientation, and blocks opening [14]. Some studies suggest that oxidative stress caused by reperfusion alone, in the presence of resting calcium, is sufficient to open the MPTP in vivo confirming equivalent roles of calcium and ROS [111]. The physiological role of MPTP may include a rapid reversible opening that releases excess calcium; this may be particularly important in the mitochondrial calcium domains of the junctional cleft. Ichas et al. first reported a transitory low conductance mode of MPTP opening that mediated a Ca2+-induced, CypA-sensitive release of Ca2+ from mitochondria [112]. Invitro this activity was shown to propagate from one mitochondrion to another, generating traveling depolarization and Ca2+ waves. By using calcein fluorescence imaging of mitochondria, Petronilli et al. reported that MPTP opening is reversible and flickers on and off within a millisecond range [113]. The authors also found that as many as 50% of the mitochondria that undergo permeability transition during early reperfusion subsequently close the pore. Hence the MPTP may be able to release apoptotic inducers while the cell retains sufficient mitochondrial function to complete apoptosis. Petronilli et al. also demonstrated CypA-sensitive transients and long-lasting openings of the MPTP in intact cells [113]. These results suggested that the MPTP fluctuates rapidly between open and closed states in intact cells mediating a fast Ca2+, and perhaps H+, release process.
Mitochondrial OM permeabilization
Irreversible tissue injury during myocardial ischemia–reperfusion involves necrosis and Bcl-2/caspase-dependent apoptosis. In mammalian cells, the latter is activated by release of cytochrome c and other pro-apoptotic factors from the mitochondrial inter-membrane space and cristae, and requires MOMP. Whereas matrix swelling due to MPT may cause OM rupture and release of pro-apoptotic factors including cytochrome c [12], this is probably not the major pathway as apoptosis requires energy for completion, and typically proceeds without OM rupture or loss of the inner membrane potential, at least during the early stages [114]. Translocation of pro-apoptotic proteins, Bax and Bid, from sites within the cytoplasm to the mitochondrial OM, and conformational changes of Bak within the OM are classical steps that lead to MOMP under most circumstances of intrinsic apoptosis [1–8,114]. Severe impairment of MOMP and apoptosis by double but not single deletions of Bax/ Bak genes in mice confirmed essential but partially redundant roles for Bax and Bak [115,116]. Interestingly, neither Bax/Bak, MOMP nor cytoplasmic cytochrome c are required for Bcl-2-dependent apoptosis in Caenorhabditis elegans or Drosophila melanogaster, perhaps indicating a later acquisition of this step for apoptosis. The precise molecular mechanisms of Bax/Bak/Bid-mediated MOMP are unclear and multiple alternatives have been proposed that may operate simultaneously. Homo- or heterodimerization of Bax, Bak and possibly Bid through BH3 domains may be a prerequisite for MOMP (reviewed in [2,3]). Oligomerized Bax and/or Bak may induce MPOP by forming structural OM channels [117,118] by causing lipid destabilization [3,118,119], or by associating with components of the MPTP including ANT and VDAC. Alternatively or in parallel, activated Bax may cause the dysregulation of mitochondrial fusion and fission, and ER stress (see below).
Mitochondrial fusion and fission occur continuously in eukaryotic cells and are required for normal cell function and survival (reviewed in [120–122]). The process involves morphological changes of the mitochondria, remodeling of the cristae and fusing together of four lipid bilayers. Mitochondrial fusion in mammals requires GTPases, mitofusin1 and mitofusin2 (Mfn1 and Mfn2), that are anchored to the OMM, and Optic Atrophy 1 (OPA1), an inner membrane dynamin family GTPase. Mitochondria fission requires dynamin-like protein 1 (DLP1/Drp1), a cytosolic GTPase, and fission protein 1 (Fis1) also anchored to the OMM. Keys steps of fusion and fission are regulated by calcium-dependent kinases [120]. Irregularities of fusion and fission mediated by Bax, Bak and Bid may contribute to apoptosis by promoting MOMP (reviewed in [121]). Fis1, Mfn1 and -2, and Drp1 are recruited during Bax-induced apoptosis. Cells lacking Fis1 are defective in staurosporine-induced translocation of Bax to the mitochondria, whereas cells lacking Drp1 translocate Bax but are defective in cytochrome c release. Drp1 and Mfn2 colocalize with Bax and Bak to the large foci on the surface of the OMM, and knockdown of Drp1 by siRNA or overexpression of a dominant negative Drp1 inhibits mitochondrial release of cytochrome c and apoptosis by Bax. BH3-proteins, tBid and Bik, also induce remodeling of mitochondria that includes fusion of the cristae, opening of the intermembrane space and mobilization of cytochrome c. tBid appears to work independently, whereas Bik localizes to the ER during apoptosis where it promotes calcium release and activation of Drp1 to induce mitochondrial remodeling. These proteins are also activated during ischemia–reperfusion and contribute to mitochondrial dysfunction and autophagy. It was recently reported that Bax targeting of OPA1 promoted cytochrome c release and apoptosis independently of MOMP [122]. Another important study demonstrated that fused mitochondria are more susceptible to MPTP opening, suggesting that fission/fusion may regulate programmed necrosis, as well as Bax-dependent apoptosis [123]. All of the components for mitochondrial fission and fusion are present in cardiac myocytes [120] and while it has been demonstrated that these processes are not essential for apoptosis to take place, it seems likely that they play roles in modulating and in some cases amplifying the responses.
Purified Bax is able to form channels in artificial membranes [117,118]. This combined with the description of a Bax-regulated, high-conductance, voltage-independent channel activity in the OMM that exports cytochrome c lead some to suggest that Bax alone constituted a MOMP channel [118,124]. While this is still a possibility, it is technically difficult to confirm even in isolated mitochondria, and the concept remains speculative.
Regulation of infarction by mitochondrial permeability channels
Relative contributions of necrosis & apoptosis during AMI
An extensive body of evidence supports the contributions of apoptotic as well as necrotic cell death to infarction, with the relative proportions reflecting at least in part the severity of ischemia and degree of energy deprivation of the affected cells. Apoptosis is known to contribute to infarct expansion at the border zones of the infarct and in remote regions after AMI, but it is unclear how much the apoptosis pathway contributes to acute infarction [125,126]. Acute death during reperfusion is thought to be mainly by necrosis regulated by the MPTP. Despite these distinctions, caspase inhibitors or reagents that interfere with intermediates of apoptosis confer protection against infarction that is at least equivalent to inhibitors of the MPTP. For example, reduction of acute infarction by 50% or more has been demonstrated for pan-caspase inhibitors [127], inhibitors of PKC-δ [128,129], overexpression of the apoptosis repressor with caspase recruitment domains [130,131], inhibition of the pro-apoptotic mitochondrial serine protease Omi/HtrA2 [132,133], over expression of IAPs [134], expression of a dominant negative form of death-associated protein (Daxx) [135] and genetic deficiency of Bax or enhanced Bcl-2 [136,137]. There are several possible interpretations: apoptosis contributes to 50% or more of acute infarction; early apoptotic signals are required to initiate MPTP-mediated necrosis; and apoptosis and programmed necrosis occur simultaneously in the same cells. Our group recently reported that infarction of mouse hearts was significantly regulated by the activity of JNK-1 and apoptosis correlated closely with infarct size [138]. It would be interesting to determine the effects of combined apoptotic (MOMP) inhibitors and MPTP desensitizers (CsA) on acute infarction.
Apoptosis requires energy, therefore energy-depleted cells in the center of an infarct become necrotic, whereas less energy-compromised cells in the border zone are more likely to die by apoptosis. Apoptosis may be initiated during ischemia, when the energy state is low, but completed after reperfusion when the energy level is restored [139]. Conversely, early stress signals may induce apoptosis that subsequently converts to necrosis if the energy level drops [140]. Levels of apoptosis are quantified by staining and visualizing cells or tissues for TUNEL, annexin-V, nuclear condensation and activated caspases. Nonapoptotic death is most frequently identified by the loss of plasma membrane integrity, swelling and leakage of enzymes. Using these techniques, Scarabelli et al. estimated maximal apoptosis rates for endothelial cells and cardiac myocytes respectively of 40 and 8% after 35 min of ischemia and 60–120-min reperfusion of rat hearts [141]. Using an in vivo annexin-V fluorescent imaging technique, Dumont et al. reported that 20% of cardiac myocytes were undergoing apoptosis after 30 min of ischemia and 90 min reperfusion of mouse hearts [142,143]. A modification of this technique in which annexin-V was labeled with Tc99-m was consistent with the presence of active apoptosis in the myocardial infarcts of patients after angioplasty [144]. These studies support multiple previous reports that have described the simultaneous presence of apoptotic and necrotic cell death during infarction in animal models and in humans. It is clearly important to determine how necrotic and apoptotic death pathways are activated and regulated during reperfusion. Bax/Bak-mediated apoptosis and MPTP-mediated cell death respond to different levels of the same stress. For example, low doses of arsenic trioxide promote apoptotic death that is dependent on Bax/Bak whereas higher doses cause MPTP-dependent necrosis that is independent of Bax or Bak [145]. Arsenic stimulates ROS production that can lower the threshold of the MPTP for calcium [146]. Low doses of H2O2 or calcium ionophore stimulate Bax/Bak-dependent apoptosis, whereas high doses induce MPTP opening and necrosis [12,147,148]. During myocardial ischemia–reperfusion cardiac myocytes in the area at risk are subjected to a graded level of oxidative stress determined in part by their distance from capillaries; myocytes closer to the capillaries receiving the highest stress [141]. It seems possible that such a graded stress may contribute to the independent initiation of necrotic and apoptotic pathways during reperfusion.
Regulation by Bcl-2 proteins
Bax/Bak have been labeled the ‘gateway to mitochondrial dysfunction and death’ [149]. The tight relationship between Bcl-2 proteins and reperfusion injury is supported by many observations: Bax translocates to the mitochondria during ischemia and reperfusion [150,151]; Bax−/− mice are resistant to ischemia–reperfusion injury displaying decreased necrosis, apoptosis and reduced infarction [128]; Bcl-xL overexpression blocks Bax translocation and reduces infarction [152]; defective MPTP opening in Bax−/− cells can be rescued by reintroducing Bax, coincident with enhanced calcium mobilization [153,154]; and the MPT requires a higher calcium threshold in Bax/Bak double knockout cells [155]. In addition to the possible role of Bax in directly regulating the MPTP, Bcl-2 proteins may modulate pore opening indirectly through the mobilization of intracellular calcium and loading of the SR [44–48]. Although Bax channels have not been described in the heart, a recent study demonstrated a similar activity in ischemia-reperfused neurons by using Bax-channel-specific inhibitors [156]. The inhibitors, named Bci1 and Bci2, were identified by their property to block the activity of oligomeric Bax channels reconstituted into liposomes. These inhibitors prevented cytochrome c release from neurons and protected the brain against global ischemia when delivered by intraperitoneal injections during reperfusion.
The Bcl-2 family of programmed death regulators
At least 22 Bcl-2 family members that regulate programmed cell death pathways in response to a wide variety of stimuli have been described. These proteins are usually associated with cell membranes, particularly of the mitochondria, ER and nuclear envelope where they are anchored by a COOH-terminal domain. Bcl-2 proteins contain regions of amino-acid sequence similarity known as Bcl-2 homology (BH) domains; these domains promote oligomerization between self and other Bcl-2 family members. Individual Bcl-2 family members may remain in the cytosol or be loosely membrane-bound and translocate into the membrane after a death signal is received (reviewed in [1–7]). The physical location and activity of each Bcl-2 family protein is partly determined by its binding to other Bcl-2-family proteins and scaffolds. This in turn is determined by the relative concentrations of each protein and the balance of pro- and anti-apoptotic members is an important feature of the regulation. The major function of the Bcl-2 family is to regulate the activity of mitochondrial permeability (MOMP and MPTP) thereby determining programmed necrosis and apoptosis. Bax and Bak are the principal effector pro-apoptotic Bcl-2 family members and contain three BH domains (BH1–BH3). Bax is either loosely attached to the OMM or sequestered in the cytosol through interactions with protein chaperones (reviewed in [1–7]). Activation of Bax involves translocation into the mitochondrial membrane where it homo-oligomerizes or interacts with other proteins to induce MOMP and release apoptogenic proteins (Figure 5). The activation of Bax and Bak is regulated by the third class of Bcl-2 proteins that contain a single BH3 domain, known as BH3-only proteins. These proteins sense and transmit the death stimulus, are regulated pre- and post-transcriptionally by intermediate signaling molecules and transmit the signals of ischemia–reperfusion to the death channels.
Figure 5. Induction and activation of Bcl-2 proteins during ischemia and reperfusion.

Ischemia causes hypoxia, inhibition of mitochondrial electron transport and transcriptional induction of Bnip3, PUMA and Noxa. During ischemia, ATP levels and intracellular pH decrease, cytosolic calcium increases and the MAPKs, ERK and p38 are activated. The Bnip3 death pathway is activated by hypoxia-acidosis and can be blocked by neutralization of pH or inhibition of the mitochondrial pore. Calcium accumulates in mitochondria during ischemia. Reperfusion activates electron transport and stimulates ROS and further calcium uptake. Post-ischemic stunning and cardiac myocyte death are both linked to elevated ROS and calcium. Multiple kinases are stimulated further by reperfusion. BH3-only proteins activated during ischemia–reperfusion include Bnip3, tBid, BAD and PUMP and NOXA. Block bars indicate possible points for therapeutic intervention to inhibit programmed cell death.
ER: Endoplasmic reticulum; OM: Outer membrane; ROS: Reactive oygen species; SR: Sarcoplasmic reticulum.
BH3-only proteins
Five BH3-only proteins including Bid, Bad, Bnip3, Bim and PUMA have been shown to contribute to myocardial infarction caused by ischemia–reperfusion (reviewed in [2–7]). Each of these proteins is a potential therapeutic target for reducing cell loss associated with reperfusion injury, as well as the low level apoptosis that develops in the heart during failure [157].
BID
Bcl-2 Interacting Domain (BID) is one of the most abundant and widespread mammalian BH3-only proteins. It is strongly expressed during development and remains high in many adult tissues including the heart. In healthy cells, Bid is cytosolic or loosely membrane-associated and functionally inert (reviewed in [156,158]). It is cleaved at the N-terminus by caspase-8, granzyme B, or calpain creating an active form known as truncated Bid (tBid). tBid translocates to the mitochondria where it mobilizes cytochrome c and other apoptogenic proteins and facilitates their release through the OM, possibly by interacting with Bax. Caspase-8 is part of the extrinsic death pathway and its cleavage of tBid allows extrinsic pathway signals to cross-activate the intrinsic pathway thereby amplifying the extrinsic death response. tBid-mediated apoptosis has been demonstrated to contribute significantly to both neuronal and myocardial ischemic injury (reviewed in [158,159]). Studies have shown that calpain (but not caspase) inhibitors prevented cleavage of Bid during reperfusion of the heart, and significantly reduced infarct size [160]. Calpain inhibitors have also been shown to slow heart failure progression in rats [156]. Owing to its pleiotropic roles in both death pathways, Bid is an attractive therapeutic target. Inhibition of Bid cleavage by selective calpain and caspase-8 inhibitors presents a strategy for acute and possibly chronic protection against infarction and adverse remodeling. Calpain inhibitors are under development for the treatment of other chronic conditions including Parkinson’s disease and Duchenne muscular dystrophy [160,161].
BAD
Bcl-xL/Bcl-2 associated death protein (BAD) is another BH3-only protein that resides in the cytoplasm in healthy cells and translocates to the mitochondria in response to death signals. Like Bid, the BH3 domain of BAD mediates its death-promoting activities by binding and neutralizing prosurvival Bcl-2 proteins and activating Bax and Bak (reviewed in [162]). BAD is phosphorylated on Serines-112, -136 and -155 by growth factor and insulin-dependent kinases including Akt, PKA and RSK1. Phosphorylated BAD is sequestered away from the mitochondria through binding to the scaffold protein 14-3-3. Phosphorylation of BAD by Akt is an important point of cross-talk between metabolic pathways and programmed death. Akt is a central regulator of insulin signaling and protects the heart against multiple stresses including oxidative stress and ischemia [163–166]. In addition to BAD, the targets of Akt include FOXO transcription factors, caspases, glucose transport proteins and the mTOR complex [165]. Akt phosphorylation is part of the IPC signaling pathway and is probably an important component of the protection mediated by glucose, potassium and insulin infusions [165,166]. A possible role for PKC-δ in the downregulation of Akt and activation of BAD during ischemia–reperfusion was recently reported [167]. Using rodent and pig models, the authors demonstrated that infusion of a PKC-δ-selective peptide at the onset of reperfusion activated Akt, blocked BAD translocation and reduced infarct size, thereby identifying PKC-δ and BAD as possible clinical targets. Results of a clinical trial, the DELTA-MI trial, were recently reported and indicated safety of the procedure in patients but a nonsignificant reduction of infarction [168]. The disappointing results may be related to dosing [169].
Bnip3
Bcl-xL or E1B 19K-binding protein (Bnip3) is expressed at low levels in most organs under normal (nonischemic) conditions, but is induced by hypoxia and ischemia [7,55,170–173]. Bnip3 is an atypical BH3-only protein that drives an atypical death pathway. The BH3 domain only has a low homology with other BH3 domains and is not essential for the death function. Deletion of the transmembrane domain eliminates the death function and converts the remaining N-terminal portion into a dominant negative that is protective. The components of the Bnip3-mediated death pathway are controversial with caspase-dependent and independent pathways reported [54,174]. Bnip3 has the dubious distinction of promoting cell death during both phases of ischemia–reperfusion injury. It is induced by hypoxia and activated by acidosis [54] or reperfusion [175]. Our laboratory described the dual roles of hypoxia and acidosis in activating Bnip3 and promoting a CsA-dependent death pathway with apoptosis-like DNA fragmentation, but no caspase activation [54]. Hypoxia and acidosis are integral features of ischemia and Bnip3 is strongly induced in ischemic hearts. Hamacher-Brady et al. recently reported that Bnip3 was activated by ischemia–reperfusion and mediated a classical intrinsic death pathway [175]. Bnip3 gene ablation and overexpression studies were also reported by Diwan et al. [56]. Bnip3 overexpression in the heart mediates an age-dependent progressive dilated cardiomyopathy. Infarct sizes after acute ischemia–reperfusion were similar in wild-type mice and mice with heart-specific Bnip3 gene ablation; hovever, the Bnip3−/− hearts had less apoptosis in the peri-infarct zone and preserved long-term cardiac functions post-MI. Other studies suggest that Bnip3 may activate parallel pathways of apoptosis, necrosis, autophagy and mitophagy that may protect or increase injury during ischemia–reperfusion [171,173]. Hamacher-Brady et al. infused a cell permeant TAT-Bnip3-ΔTM (dominant negative) peptide at the time of reperfusion and observed >50% reduction of infarct size with a parallel decrease of TUNEL-positive cells [175]. Whether hypoxia alone is sufficient to induce and activate Bnip3 during ischemia is not resolved. Diwan et al. reported that a second hit was not required in transgenic mice that overexpressed Bnip3 selectively in the heart because they observed significantly elevated apoptosis 2-month after birth without intervention, and the mice eventually developed dilated cardiomyopathy and heart failure [56]. In unpublished results, similar observations in the same transgenic mouse model created in our laboratory were made [Thompson JW et al., Unpublished Data]. However our interpretation is different; despite full activation of the Bnip3 transgene and >50-fold overexpression compared with wild-type hearts at 2-weeks after birth, there was no evidence of cell death or deterioration of function until 2–3 months. Even at this time TUNEL-positive cells were less than 0.5%, only marginally higher than wild-types. Therefore, it seems possible that transgenic Bnip3 is mostly quiescent, especially in the early stages (up to 2 months) and a second hit is required for activation. Different microenvironments, perhaps related to the physical location of myocytes within the heart and/or local microperfusion irregularities could provide a second hit such as hypoxia, acidosis or increased oxidative stress that activates Bnip3 in selected cells of these transgenic mice. A previous study suggested that skeletal muscle may be protected from elevated Bnip3 by inactive sequestration [176]. Another recent study reported that Bnip3 is induced during pressure-overload hypertrophy by JNK-1-mediated activation of the transcription factor FOXO3a [177]. Apoptosis was implicated and the hearts were protected by pharmacological or genetic suppression of Bnip3. Similar to ischemia, pressure-overload hypertrophy is a complex condition whereby stress-related signaling pathways are activated and heterogeneous microenvironments created that could activate quiescent Bnip3. Regardless of the mechanisms of action that are still disputed, the roles of Bnip3 in multiple death pathways and in multiple contexts of heart disease, including ischemia, reperfusion, hypertrophy and failure, single it out as a potentially important therapeutic target.
PUMA, NOXA & Bim
PUMA, NOXA and Bim are BH3-only proteins that are implicated in apoptotic pathways involving the cell cycle, DNA damage and other cellular stresses associated with ischemia–reperfusion [178]. PUMA and NOXA genes were identified as targets for the tumor suppressor transcription factor p53 and were demonstrated to play roles in p53-mediated apoptosis. PUMA can bind and sequester all of the prosurvival Bcl-2 proteins, whereas NOXA binds only a subset and probably requires partners to induce death; both work by activating Bax/Bak proteins [162,179]. PUMA and NOXA are transcriptionally activated by all stimuli that activate p53, including DNA damage and oxidative stress [179,180]. Both proteins also promote p53-independent death pathways; PUMA is activated by cytokine deprivation, staurosporine, phorbol esters and conditions that induce ER stress [181]. NOXA is activated by hypoxia through a 5′ promoter HIF-1-binding element [182]. PUMA is implicated in the apoptotic death of neurons during stroke and cardiac myocytes during ischemia–reperfusion [183,184]. NOXA is induced by ischemia in the rat brain and infusions of NOXA antisense oligonucleotides protect against infarction [182]. Changes in the levels of NOXA expression in the heart have not been reported, however in unpublished studies, work from this laboratory supported the possibility that the protein is present in the mouse heart, and is induced by ischemia [Webster KA, Unpublished Data]. Pro-apoptotic Bim is activated by ischemia–reperfusion and it was recently demonstrated that its expression in the infarct border zone could be lowered by targeting micro-RNA-24 providing another possible cardioprotective strategy [185].
Conclusion
Myocardial infarction involves death by necrosis and apoptosis through pathways that are substantially regulated by permeability changes of mitochondrial membranes. The CsA-sensitive MPTP and Bax/Bak-regulated MOMP have emerged as central regulators of preventable necrotic and apoptotic cell death and targets for reducing infarction. Classical unregulated necrosis and hyperactive autophagy may also contribute to cell death during infarction, and mitochondrial morphological changes including remodeling of the cristae, fission and fusion are involved in modulating each pathway. Calcium and ROS are still thought to play central roles in initiating all cell death pathways (Figure 6). Technically it has become difficult to separate the two individual pathways because infarction following ischemia–reperfusion is a complex and dynamic process and apoptosis, necrosis and autophagy may be activated simultaneously, possibly in the same cells. Despite large gaps in our knowledge of the cell death pathways, sufficient data are available to allow patient testing of new cardioprotective approaches targeting MOMP and MPTP, delivered at or prior to reperfusion (reviewed in [3]). While minimizing the time to reperfusion is still the primary goal, recent trials have tested the MPTP blocker CsA [100], and mimics of ischemic preconditioning or postconditioning (reviewed in [3]). Such trials may be expanded to include interventions at the level of BH3-only proteins, of which Bnip3 may be an exciting candidate. Many uncertainties exist regarding the mechanism, and early results, for example from the CsA [100] and DELTA-MI trials [170,171], confirm the trends of multiple previous AMI interventions (e.g., postconditioning, antioxidants anti-inflammatory agents) that major cardioprotection observed in preclinical trials does not translate well into patient treatments. Efficacy may be substantially influenced by the severity of the existing ischemia, baseline medications and comorbid conditions. There are reports that apoptosis continues for extended periods after a myocardial infarction, with loss of myocardium continuing for several months. Chronically enhanced NO availability, orally active calpain inhibitors, other CsA derivatives and/or Bax channel blockers may help prevent the chronic loss of myocytes post myocardial infarction.
Figure 6. Summary of events leading up to myocardial infarction.

Ischemia causes dysfunctional calcium handling such that calcium accumulates in the sarcoplasm and is taken up by mitochondria, followed by phosphate and water causing swelling. Reperfusion causes additional calcium and other ionic disturbances, and further uptake and swelling of mitochondria and ROS release that favors opening of the CypD/adenine nucleotide translocase-dependent MPTP. A series of parallel events including protein damage, ER stress, kinase activation, remodeling of the cristae and changes in mitochondrial fission and fusion may cause the release of sequestered BH3-only proteins, activation of pro-apoptotic Bak and Bax with increased MOMP, release of cytochrome c and other pro-apoptotic factors. Autophagy may be activated in parallel and excess autophagy may promote cell death by necrosis and/or apoptosis or an undefined pathway. Bax feedback may enhance remodeling of cristae and fission/fusion events and amplify death pathways. Changes of Bcl-2 proteins may regulate MOMP as well as MPTP, and primary cell damage can induce inflammation that activates extrinsic apoptosis. All of these evens may occur at different stages or simultaneously in the same dying cells and all contribute to infarction.
CypD: Cyclophilin D; ER: Endoplasmic reticulum; MOMP: Mitochondrial outer membrane permeability; MPTP: Mitochondrial permeability transition pore; ROS: Reactive oxygen species.
Future perspective
Ischemic cardiovascular disease continues to be a major global cause of morbidity and mortality. Salvage of tissue by interventions delivered at or before reperfusion that target programmed death is still the major objective of research in this area. We are likely to see additional trials of compounds based on preclinical results of pre- and postconditioning, as well as candidate molecules for blocking programmed death pathways. These may include volatile anesthetics, other NOS regulators (sildenafil), calpain inhibitors and interventions at the level of BH3-only proteins. Experience from clinical trials cautions that we can anticipate the recurrence of disappointing outcomes in many cases [3]. Gene therapy and stem cell therapy will be added to pharmacology. Bnip3 is an exciting target for gene therapy owing to its reported activity in multiple death pathways and multiple forms of heart disease including ischemia, postinfarct remodeling, pressure overload hypertrophy and heart failure. A recent report that cardiac stem cell infusions markedly improved the function of post-infarcted myocardium, perhaps by promoting repair, may be the most promising approach for the treatment of heart failure to date [186]. This combined with new pharmacology and other biotechnological approaches; peptides, siRNA, micro-RNA and gene therapy will be pursued aggressively, and will probably provide positive clinical results within the next 10 years.
Executive summary.
Overview of death pathways activated during acute myocardial infarction
-
Classical necrosis
Caused by energy failure, irreversible.
-
Programmed necrosis
Associated with reperfusion, possibly preventable.
-
Extrinsic apoptosis
Inflammatory response, possibly preventable.
-
Intrinsic apoptosis
Major programmed death pathway, possibly preventable.
-
Autophagy
Initially protective but over-activation may initiate programmed cell death.
Normal calcium and reactive oxygen species handling, and dysregulation by ischemia–reperfusion
-
Normal calcium handling by the plasma membrane, sarcoplasmic reticulum and myofilament
Calcium fluxes are tightly regulated during normal excitation–contraction coupling by L-type Ca2+ channels, ryanodine receptors, SR-calcium ATPase channels and the sarcolemmal calcium ATPase. Regions of high calcium flux between the sarcolemma and the SR are known as Ca2+ microdomains.
-
Calcium handling during ischemia–reperfusion
Defective calcium handling causes reversible and irreversible myocardial injury and is a primary therapeutic target.
-
Mitochondrial transients in the junctional cleft
Mitochondria in the junctional cleft cycle calcium in synchrony with contraction cycle and this contributes importantly to myocardial bioenergetics.
-
Oxidative stress of reperfusion
Reperfusion causes more than an order of magnitude increase of electron leakage by mitochondria and reactive oxygen species (ROS) production. ROS generators include ubiquinone–ubiquinol mobile electron carriers known as coenzymes Q10.
-
Hypercontracture and cell death during acute myocardial infarction (AMI)
The links between defective calcium handling, elevated ROS and contracture are compelling, as are the links with defective mitochondrial function, membrane permeabilization and programmed cell death.
Death channels
-
Mitochondrial permeability transition pore (MPTP) structure and mechanism
No structural component has been identified as essential for activity of the MPTP. Modulatory roles have been described for the voltage-dependent anion channel, adenine nucleotide translocase and phosphate ion channel. Accessory proteins include cyclophilin D, hexokinase-II and the peripheral-type benzodiazepine receptor.
-
MPTP opening by calcium and ROS
MPTP opening by elevated calcium and ROS correlates with the extent of infarction. The MPTP may fluctuate rapidly between open and closed states in intact cells.
-
Mitochondrial outer membrane permeabilization (MOMP)
Homo- or hetero-dimerization of Bax, Bak, and possibly Bid through BH3 domains may be a prerequisite for MOMP. There are established roles for mitochondrial fission and fusion. The molecular mechanism of MOMP opening is still unclear.
Regulation of infarction by mitochondrial permeability channels
-
Relative contributions of necrosis and apoptosis during AMI
During AMI, cardiac myocytes are subjected to a graded level of oxidative stress determined by their distance from capillaries. This may cause independent initiation of necrotic and apoptotic pathways in different regions during reperfusion.
-
Regulation by Bcl-2 proteins
Pro-apoptotic Bax and Bak are normally in equilibrium with Bcl-2 and Bcl-XL that prevent MOMP. Cellular insults disturb the equilibrium allowing Bax and Bid translocation, Bak conformational change and activation of the intrinsic death pathway.
-
Overall regulation by BH3-only proteins
Five BH3-only proteins Bid, Bad, Bnip3, Bim and PUMA may contribute to myocardial infarction during AMI. Bnip3 promotes cell death during both phases of ischemia–reperfusion, as well as in association with pathological hypertrophy and heart failure.
Conclusion and future perspective
MPTP and MOMP have emerged as central regulators of ‘preventable’ necrotic and apoptotic cell death and are major therapeutic targets for reducing infarction.
During the next decade, pharmacological preconditioning agents will be combined with gene therapy, microRNA manipulation and stem cells.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Financial & competing interests disclosure
Supported by National Institutes of Health grant HL44578 and by a WG Ross Chair in vascular biology. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Adams JM, Cory S. The Bcl-2-regulated apoptosis switch: mechanism and therapeutic potential. Curr Opin Immunol. 2007;19(5):488–496. doi: 10.1016/j.coi.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Konstantinidis K, Whelan RS, Kitsis RN. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol. 2012;32(7):1552–1562. doi: 10.1161/ATVBAHA.111.224915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oerlemans MI, Koudstaal S, Chamuleau SA, de Kleijn DP, Doevendans PA, Sluijter JP. Targeting cell death in the reperfused heart: pharmacological approaches for cardioprotection. Int J Cardiol. 2012 doi: 10.1016/j.ijcard.2012.03.055. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 4.Machado NG, Alves MG, Carvalho RA, Oliveira PJ. Mitochondrial involvement in cardiac apoptosis during ischemia and reperfusion: can we close the box? Cardiovasc Toxicol. 2009;9(4):211–227. doi: 10.1007/s12012-009-9055-1. [DOI] [PubMed] [Google Scholar]
- 5.Dorn GW., 2nd Apoptotic and non-apoptotic programmed cardiomyocyte death in ventricular remodelling. Cardiovasc Res. 2009;81(3):465–473. doi: 10.1093/cvr/cvn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamacher-Brady A, Brady NR, Gottlieb RA. The interplay between pro-death and pro-survival signaling pathways in myocardial ischemia/reperfusion injury: apoptosis meets autophagy. Cardiovasc Drugs Ther. 2006;20(6):445–462. doi: 10.1007/s10557-006-0583-7. [DOI] [PubMed] [Google Scholar]
- 7.Webster KA, Graham RM, Thompson JW, et al. Redox stress and the contributions of BH3-only proteins to infarction. Antioxid Redox Signal. 2006;8(9–10):1667–1676. doi: 10.1089/ars.2006.8.1667. [DOI] [PubMed] [Google Scholar]
- 8.Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol. 2008;9(5):378–390. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- 9.Jung JE, Kim GS, Chen H, et al. Reperfusion and neurovascular dysfunction in stroke: from basic mechanisms to potential strategies for neuroprotection. Mol Neurobiol. 2010;41(2–3):172–179. doi: 10.1007/s12035-010-8102-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nieminen AL. Apoptosis and necrosis in health and disease: role of mitochondria. Int Rev Cytol. 2003;224:29–55. doi: 10.1016/s0074-7696(05)24002-0. [DOI] [PubMed] [Google Scholar]
- 11.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baines CP, Kaiser RA, Purcell NH, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 13.Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434(7033):652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 14.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 15.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9(5):550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gustafsson AB, Gottlieb RA. Bcl-2 family members and apoptosis, taken to heart. Am J Physiol Cell Physiol. 2006;292:C45–C51. doi: 10.1152/ajpcell.00229.2006. [DOI] [PubMed] [Google Scholar]
- 17.Juhaszova M, Zorov DB, Kim SH, et al. Glycogen synthase kinase-3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113(11):1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem. 1976;1251(16):5069–5077. [PubMed] [Google Scholar]
- 19.Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria Nature of the Ca2+ trigger site. Arch Biochem Biophys. 1979;195(2):460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- 20.Crompton M, Costi A. Kinetic evidence for a heart mitochondrial pore activated by Ca2+ inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca2+ overload. Eur J Biochem. 1988;178:489–501. doi: 10.1111/j.1432-1033.1988.tb14475.x. [DOI] [PubMed] [Google Scholar]
- 21.Nazareth W, Yafei N, Crompton M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin-A. J Mol Cell Cardiol. 1991;23:1351–1354. doi: 10.1016/0022-2828(91)90181-k. [DOI] [PubMed] [Google Scholar]
- 22.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 23.Tsujimoto Y, Nakagawa T, Shimizu S. Mitochondrial membrane permeability transition and cell death. Biochim Biophys Acta. 2006;1757:1297–1300. doi: 10.1016/j.bbabio.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 24.Whelan RS, Konstantinidis K, Wei AC, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci USA. 2012;109:6566–6571. doi: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, Tsujimoto Y. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol. 2001;152:237–250. doi: 10.1083/jcb.152.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crompton M. On the involvement of mitochondrial intermembrane junctional complexes in apoptosis. Curr Med Chem. 2003;10:1473–1484. doi: 10.2174/0929867033457197. [DOI] [PubMed] [Google Scholar]
- 28.Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator – thinking outside the box. Biochim Biophys Acta. 2006;1762:1181–1190. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 29.Majewski N, Nogueira V, Bhaskar P, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 30.Shoshan-Barmatz V, Israelson A, Brdiczka D, Sheu SS. The voltage-dependent anion channel (VDAC): function in intracellular signaling, cell life and cell death. Curr Pharm Design. 2006;12:2249–2270. doi: 10.2174/138161206777585111. [DOI] [PubMed] [Google Scholar]
- 31.Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005;12:751–760. doi: 10.1038/sj.cdd.4401599. [DOI] [PubMed] [Google Scholar]
- 32.Cheng EHY, Sheiko T, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;5632:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 33.Kim R, Emi M, Tanabe K, Murakami S, Uchida Y, Arihiro K. Regulation and interplay of apoptotic and non-apoptotic cell death. J Pathol. 2006;208:319–326. doi: 10.1002/path.1885. [DOI] [PubMed] [Google Scholar]
- 34.Rostovtseva TK, Antonsson B, Suzuki M, Youle RJ, Colombini M, Bezrukov SM. Bid, but not Bax, regulates VDAC channels. J Biol Chem. 2004;279:13575–13583. doi: 10.1074/jbc.M310593200. [DOI] [PubMed] [Google Scholar]
- 35.Szabó I, Zoratti M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J Biol Chem. 1991;266(6):3376–3379. [PubMed] [Google Scholar]
- 36.Kaufmann T, Strasser A, Jost PJ. Fas death receptor signalling: roles of Bid and XIAP. Cell Death Differ. 2012;19(1):42–50. doi: 10.1038/cdd.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeremias I, Kupatt C, Martin-Villaba A, et al. Involvement of CD95/Apo1/Fas in cell death after myocardial ischemia. Circulation. 2000;102:915–920. doi: 10.1161/01.cir.102.8.915. [DOI] [PubMed] [Google Scholar]
- 38.Lee P, Sata M, Lefer DJ, et al. Fas pathway is a critical mediator of cardiac myocyte death and MI during ischemia-reperfusion in vivo. Am J Physiol Heart Circ Physiol. 2003;284:H456–H463. doi: 10.1152/ajpheart.00777.2002. [DOI] [PubMed] [Google Scholar]
- 39.Zhao WS, Xu L, Wang LF, et al. A 60-s postconditioning protocol by percutaneous coronary intervention inhibits myocardial apoptosis in patients with acute myocardial infarction. Apoptosis. 2009;14(10):1204–1211. doi: 10.1007/s10495-009-0387-x. [DOI] [PubMed] [Google Scholar]
- 40.Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102(1):43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 41.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8(3):613–621. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 43.Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003;17(12):1487–1496. doi: 10.1101/gad.1097903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oakes SA, Scorrano L, Opferman JT, et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102(1):105–110. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23(5):1207–1216. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oakes SA, Opferman JT, Pozzan T, Korsmeyer SJ, Scorrano L. Regulation of endoplasmic reticulum Ca2+ dynamics by proapoptotic BCL-2 family members. Biochem Pharmacol. 2003;66(8):1335–1340. doi: 10.1016/s0006-2952(03)00482-9. [DOI] [PubMed] [Google Scholar]
- 47.Scorrano L, Oakes SA, Opferman JT, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300(5616):135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 48.Thompson JW, Graham RM, Webster KA. DNase activation by hypoxia-acidosis parallels but is independent of programmed cell death. Life Sci. 2012;91(7–8):223–229. doi: 10.1016/j.lfs.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007;7(12):964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 50.Dutta D, Calvani R, Bernabei R, Leeuwenburgh C, Marzetti E. Contribution of impaired mitochondrial autophagy to cardiac aging: mechanisms and therapeutic opportunities. Circ Res. 2012;110(8):1125–1138. doi: 10.1161/CIRCRESAHA.111.246108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER stress to autophagy: potential implications for cancer therapy. Int J Cell Biol. 2010;2010:930509. doi: 10.1155/2010/930509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circ Res. 2009;104(2):150–158. doi: 10.1161/CIRCRESAHA.108.187427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sadoshima J. The role of autophagy during ischemia/reperfusion. Autophagy. 2008;4(4):402–403. doi: 10.4161/auto.5924. [DOI] [PubMed] [Google Scholar]
- 54.Kubasiak LA, Hernandez OM, Bishopric NH, Webster KA. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci USA. 2002;99(20):12825–12830. doi: 10.1073/pnas.202474099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Webster KA, Graham RM, Bishopric NH. BNip3 and signal-specific programmed death in the heart. J Mol Cell Cardiol. 2005;38(1):35–45. doi: 10.1016/j.yjmcc.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 56.Diwan A, Krenz M, Syed FM, et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007;117(10):2825–2833. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Groenendyk J, Sreenivasaiah PK, Do HK, Agellon LB, Michalak M. Biology of endoplasmic reticulum stress in the heart. Circ Res. 2010;107(10):1185–1197. doi: 10.1161/CIRCRESAHA.110.227033. [DOI] [PubMed] [Google Scholar]
- 58.Giricz Z, Mentzer RM, Jr, Gottlieb RA. Autophagy, myocardial protection and the metabolic syndrome. J Cardiovasc Pharmacol. 2012;60(2):125–132. doi: 10.1097/FJC.0b013e318256ce10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Q, Zhou LY, Gao GF, Jiao JQ, Li PF. Mitochondrial network in the heart. Protein Cell. 2012;3(6):410–418. doi: 10.1007/s13238-012-2921-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inserte J, Barrabés JA, Hernando V, Garcia-Dorado D. Orphan targets for reperfusion injury. Cardiovasc Res. 2009;83(2):169–178. doi: 10.1093/cvr/cvp109. [DOI] [PubMed] [Google Scholar]
- 61.Pott C, Eckardt L, Goldhaber JI. Triple threat: the Na+/Ca2+ exchanger in the pathophysiology of cardiac arrhythmia, ischemia and heart failure. Curr Drug Targets. 2011;12(5):737–747. doi: 10.2174/138945011795378559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bers DM. Cardiac excitation contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 63.Chen-Izu Y, McCulle SL, Ward CW, et al. Three-dimensional distribution of ryanodine receptor clusters in cardiac myocytes. Biophys J. 2006;91:1–13. doi: 10.1529/biophysj.105.077180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Collins TJ, Lipp P, Berridge MJ, Bootman MD. Mitochondrial Ca(2+) uptake depends on the spatial and temporal profile of cytosolic Ca(2+) signals. J Biol Chem. 2001;276:26411–26420. doi: 10.1074/jbc.M101101200. [DOI] [PubMed] [Google Scholar]
- 65.Jones PP, Bazzazi H, Kargacin GJ, Colyer J. Inhibition of cAMP dependent protein kinase under conditions occurring in the cardiac dyad during a Ca2+ transient. Biophys J. 2006;91:433–443. doi: 10.1529/biophysj.106.083931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peskoff A, Post JA, Langer GA. Sarcolemmal calcium binding sites in heart: II Mathematical model for diffusion of calcium released from the sarcoplasmic reticulum into the dyadic region. J Membr Biol. 1992;129:59–69. doi: 10.1007/BF00232055. [DOI] [PubMed] [Google Scholar]
- 67.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–3371. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weber CR, Piacentino V, 3rd, Ginsburg KS, Houser SR, Bers DM. Na(+)-Ca(2+) exchange current and submembrane [Ca(2+)] during the cardiac action potential. Circ Res. 2002;90:182–189. doi: 10.1161/hh0202.103940. [DOI] [PubMed] [Google Scholar]
- 69.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 70.Lee JA, Allen DG. Mechanisms of acute ischemic contractile failure of the heart Role of intracellular calcium. J Clin Invest. 1991;88(2):361–367. doi: 10.1172/JCI115311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arnaudeau S, Kelley WL, Walsh JV, Jr, Demaurex N. Mitochondria recycle Ca(2þ) to the endoplasmic reticulum and prevent the depletion of neighboring endoplasmic reticulum regions. J Biol Chem. 2001;276:29430–29439. doi: 10.1074/jbc.M103274200. [DOI] [PubMed] [Google Scholar]
- 72.Cox DA, Matlib MA. A role for the mitochondrial Na(+)-Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. J Biol Chem. 1993;268:938–947. [PubMed] [Google Scholar]
- 73.Territo PR, French SA, Balaban RS. Simulation of cardiac work transitions, in vitro: effects of simultaneous Ca2+ and ATPase additions on isolated porcine heart mitochondria. Cell Calcium. 2001;30:19–27. doi: 10.1054/ceca.2001.0211. [DOI] [PubMed] [Google Scholar]
- 74.Maack C, O’Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. 2007;102(5):369–392. doi: 10.1007/s00395-007-0666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 76.Maak C, O’Rourke B. Excitation-contraction coupling and mitochondrial bioenergetics. Basic Res Cardiol. 2007;102:369–392. doi: 10.1007/s00395-007-0666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation- contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–182. doi: 10.1161/01.RES.0000232546.92777.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Skulachev VP. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci. 2001;26(1):23–29. doi: 10.1016/s0968-0004(00)01735-7. [DOI] [PubMed] [Google Scholar]
- 79.Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16(1):59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 80.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med. 2009;47(4):333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 82.Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 83.Dougherty CJ, Kubasiak LA, Frazier DP, et al. Mitochondrial signals initiate the activation of c-Jun N-terminal kinase (JNK) by hypoxia-reoxygenation. FASEB J. 2004;18(10):1060–1070. doi: 10.1096/fj.04-1505com. [DOI] [PubMed] [Google Scholar]
- 84.Frazier DP, Wilson A, Dougherty CJ, et al. PKC-α and TAK-1 are intermediates in the activation of c-Jun NH2-terminal kinase by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2007;292(4):H1675–H1684. doi: 10.1152/ajpheart.01132.2006. [DOI] [PubMed] [Google Scholar]
- 85.Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta. 2007;1767(8):1007–1031. doi: 10.1016/j.bbabio.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aon MA, Cortassa S, Marban E, O’Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem. 2003;278:44735–44744. doi: 10.1074/jbc.M302673200. [DOI] [PubMed] [Google Scholar]
- 88.Inserte J, Barrabés JA, Hernando V, Garcia-Dorado D. Orphan targets for reperfusion injury. Cardiovasc Res. 2009;83(2):169–178. doi: 10.1093/cvr/cvp109. [DOI] [PubMed] [Google Scholar]
- 89.Garcia-Dorado D, Theroux P, Duran JM, et al. Selective inhibition of the contractile apparatus A new approach to modification of infarct size, infarct composition, and infarct geometry during coronary artery occlusion and reperfusion. Circulation. 1992;85:1160–1174. doi: 10.1161/01.cir.85.3.1160. [DOI] [PubMed] [Google Scholar]
- 90.Sebbag L, Verbinski SG, Reimer KA, Jennings RB. Protection of ischemic myocardium in dogs using intracoronary 2,3-butanedione monoxime (BDM) J Mol Cell Cardiol. 2003;35:165–176. doi: 10.1016/s0022-2828(02)00303-6. [DOI] [PubMed] [Google Scholar]
- 91.Schlack W, Uebing A, Schafer M, et al. Regional contractile blockade at the onset of reperfusion reduces infarct size in the dog heart. Pflugers Arch. 1994;428:134–141. doi: 10.1007/BF00374850. [DOI] [PubMed] [Google Scholar]
- 92.Siegmund B, Klietz T, Schwartz P, Piper HM. Temporary contractile blockade prevents hypercontracture in anoxic-reoxygenated cardiomyocytes. Am J Physiol. 1991;260:H426–H435. doi: 10.1152/ajpheart.1991.260.2.H426. [DOI] [PubMed] [Google Scholar]
- 93.Garcia-Dorado D, Inserte J, Ruiz-Meana M, et al. Gap junction uncoupler heptanol prevents cell-to-cell progression of hypercontracture and limits necrosis during myocardial reperfusion. Circulation. 1997;96:3579–3586. doi: 10.1161/01.cir.96.10.3579. [DOI] [PubMed] [Google Scholar]
- 94.Rodriguez-Sinovas A, Garcia-Dorado D, Ruiz-Meana M, Soler-Soler J. Enhanced effect of gap junction uncouplers on macroscopic electrical properties of reperfused myocardium during myocardial reperfusion. Circulation. 2004;96:3579–3586. doi: 10.1113/jphysiol.2004.065144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kokoszka JE, Waymire KG, Levy SE, et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rodriguez Enriquez S, He LH, Lemasters JJ. Role of mitochondrial permeability transition pores in mitochondrial autophagy. Int J Biochem Cell Biol. 2004;36:2463–2472. doi: 10.1016/j.biocel.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 97.Crompton M. On the involvement of mitochondrial intermembrane junctional complexes in apoptosis. Curr Med Chem. 2003;10:1473–1484. doi: 10.2174/0929867033457197. [DOI] [PubMed] [Google Scholar]
- 98.Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator – thinking outside the box. Biochim Biophys Acta. 2006;1762:1181–1190. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 99.Majewski N, Nogueira V, Bhaskar P, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 100.Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359(5):473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 101.Blachly-Dyson E, Forte M. VDAC channels. IUBMB Life. 2001;52:113–118. doi: 10.1080/15216540152845902. [DOI] [PubMed] [Google Scholar]
- 102.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 103.Vander Heiden MG, Chandel NS, Schumacker PT, Thompson CB. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol Cell. 1999;3:159–167. doi: 10.1016/s1097-2765(00)80307-x. [DOI] [PubMed] [Google Scholar]
- 104.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25(34):4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3β disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–10554. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- 106.Miura T, Miki T. GSK-3β, a therapeutic target for cardiomyocyte protection. Circ J. 2009;73(7):1184–1192. doi: 10.1253/circj.cj-09-0284. [DOI] [PubMed] [Google Scholar]
- 107.Chen Z, Chua CC, Ho YS, Hamdy RC, Chua BH. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol. 2001;280:H2313–H2320. doi: 10.1152/ajpheart.2001.280.5.H2313. [DOI] [PubMed] [Google Scholar]
- 108.Shimizu S, Konishi A, Kodama T, Tsujimoto Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci USA. 2000;97:3100–3105. doi: 10.1073/pnas.97.7.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gustafsson AB, Gottlieb RA. Bcl-2 family members and apoptosis, taken to heart. Am J Physiol Cell Physiol. 2006;292:C45–C51. doi: 10.1152/ajpcell.00229.2006. [DOI] [PubMed] [Google Scholar]
- 110.Juhaszova M, Zorov DB, Kim SH, et al. Glycogen synthase kinase-3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113(11):1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ruiz-Meana M, Garcia-Dorado D, Miro-Casas E, Abellan A, Soler-Soler J. Mitochondrial Ca2+ uptake during simulated ischemia does not affect permeability transition pore opening upon simulated reperfusion. Cardiovasc Res. 2006;71:715–724. doi: 10.1016/j.cardiores.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 112.Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89(7):1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 113.Petronilli V, Miotto G, Canton M, et al. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J. 1999;76(2):725–734. doi: 10.1016/S0006-3495(99)77239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18(4):157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lindsten T, Ross AJ, King A, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6(6):1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kinnally KW, Antonsson B. A tale of two mitochondrial channels, MAC and PTP, in apoptosis. Apoptosis. 2007;12(5):857–868. doi: 10.1007/s10495-007-0722-z. [DOI] [PubMed] [Google Scholar]
- 118.Peixoto PM, Ryu SY, Bombrun A, Antonsson B, Kinnally KW. MAC inhibitors suppress mitochondrial apoptosis. Biochem J. 2009;423(3):381–387. doi: 10.1042/BJ20090664. [DOI] [PubMed] [Google Scholar]
- 119.Westphal D, Dewson G, Czabotar PE, Kluck RM. Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta. 2011;1813(4):521–531. doi: 10.1016/j.bbamcr.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 120.Hom J, Sheu SS. Morphological dynamics of mitochondria – a special emphasis on cardiac muscle cells. J Mol Cell Cardiol. 2009;46(6):811–820. doi: 10.1016/j.yjmcc.2009.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jourdain A, Martinou JC. Mitochondrial outer-membrane permeabilization and remodelling in apoptosis. Int J Biochem Cell Biol. 2009;41(10):1884–1889. doi: 10.1016/j.biocel.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 122.Yamaguchi R, Lartigue L. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell. 2008;31(4):557–569. doi: 10.1016/j.molcel.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Whelan RS, Konstantinidis K, Wei AC, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci USA. 2012;109(17):6566–6571. doi: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Guo L, Pietkiewicz D, Pavlov EV, et al. Effects of cytochrome c on the mitochondrial apoptosis-induced channel MAC. Am J Physiol. 2004;286:C1109–C1117. doi: 10.1152/ajpcell.00183.2003. [DOI] [PubMed] [Google Scholar]
- 125.Tissier R, Berdeaux A, Ghaleh B, et al. Making the heart resistant to infarction: how can we further decrease infarct size? Front Biosci. 2008;13:284–301. doi: 10.2741/2679. [DOI] [PubMed] [Google Scholar]
- 126.Skyschally A, Schulz R, Heusch G. Pathophysiology of myocardial infarction: protection by ischemic pre- and postconditioning. Herz. 2008;33(2):88–100. doi: 10.1007/s00059-008-3101-9. [DOI] [PubMed] [Google Scholar]
- 127.Zidar N, Jera J, Maja J, Dusan S. Caspases in myocardial infarction. Adv Clin Chem. 2007;44:1–33. doi: 10.1016/s0065-2423(07)44001-x. [DOI] [PubMed] [Google Scholar]
- 128.Churchill EN, Mochly-Rosen D. The roles of PKCδ and ε isoenzymes in the regulation of myocardial ischaemia/reperfusion injury. Biochem Soc Trans. 2007;35(Pt 5):1040–1042. doi: 10.1042/BST0351040. [DOI] [PubMed] [Google Scholar]
- 129.Palaniyandi SS, Sun L, Ferreira JC, Mochly-Rosen D. Protein kinase C in heart failure: a therapeutic target? Cardiovasc Res. 2009;82(2):229–239. doi: 10.1093/cvr/cvp001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gustafsson AB, Tsai JG, Logue SE, Crow MT, Gottlieb RA. Apoptosis repressor with caspase recruitment domain protects against cell death by interfering with Bax activation. J Biol Chem. 2004;279(20):21233–21238. doi: 10.1074/jbc.M400695200. [DOI] [PubMed] [Google Scholar]
- 131.Nam YJ, Mani K, Ashton AW, et al. Inhibition of both the extrinsic and intrinsic death pathways through nonhomotypic death-fold interactions. Mol Cell. 2004;15(6):901–912. doi: 10.1016/j.molcel.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 132.Liu HR, Gao E, Hu A, et al. Role of Omi/HtrA2 in apoptotic cell death after myocardial ischemia and reperfusion. Circulation. 2005;111(1):90–96. doi: 10.1161/01.CIR.0000151613.90994.17. [DOI] [PubMed] [Google Scholar]
- 133.Bhuiyan MS, Fukunaga K. Activation of HtrA2, a mitochondrial serine protease mediates apoptosis: current knowledge on HtrA2 mediated myocardial ischemia/ reperfusion injury. Cardiovasc Ther. 2008;26(3):224–232. doi: 10.1111/j.1755-5922.2008.00052.x. [DOI] [PubMed] [Google Scholar]
- 134.Chua CC, Gao J, Ho YS, et al. Overexpression of IAP-2 attenuates apoptosis and protects against myocardial ischemia/reperfusion injury in transgenic mice. Biochim Biophys Acta. 2007;1773(4):577–583. doi: 10.1016/j.bbamcr.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Roubille F, Combes S, Leal-Sanchez J, et al. Myocardial expression of a dominant-negative form of Daxx decreases infarct size and attenuates apoptosis in an in vivo mouse model of ischemia/reperfusion injury. Circulation. 2007;116(23):2709–2717. doi: 10.1161/CIRCULATIONAHA.107.694844. [DOI] [PubMed] [Google Scholar]
- 136.Hochhauser E, Cheporko Y, Yasovich N, et al. Bax deficiency reduces infarct size and improves long-term function after myocardial infarction. Cell Biochem Biophys. 2007;47(1):11–20. doi: 10.1385/cbb:47:1:11. [DOI] [PubMed] [Google Scholar]
- 137.Imahashi K, Schneider MD, Steenbergen C, Murphy E. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ Res. 2004;95(7):734–741. doi: 10.1161/01.RES.0000143898.67182.4c. [DOI] [PubMed] [Google Scholar]
- 138.Wei J, Wang W, Chopra I, et al. c-Jun N-terminal kinase (JNK-1) confers protection against brief but not extended ischemia during acute myocardial infarction. J Biol Chem. 2011;286(16):13995–14006. doi: 10.1074/jbc.M110.211334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Freude B, Masters TN, Robiczek F, et al. Apoptosis is initiated by myocardial ischaemia and executed during reperfusion. J Mol Cell Cardiol. 2000;32:197–208. doi: 10.1006/jmcc.1999.1066. [DOI] [PubMed] [Google Scholar]
- 140.Van Cruchten S, Van der Broeck W. Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat Histol Embryol. 2002;31:214–223. doi: 10.1046/j.1439-0264.2002.00398.x. [DOI] [PubMed] [Google Scholar]
- 141.Scarabelli TM, Stephanou A, Rayment N. Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischaemia/ reperfusion injury. Circulation. 2001;104:253–256. doi: 10.1161/01.cir.104.3.253. [DOI] [PubMed] [Google Scholar]
- 142.Dumont EA, Hofstra L, van Heerde WL, et al. Cardiomyocyte death induced by myocardial ischemia and reperfusion measurement with recombinant human annexin-V in a mouse model. Circulation. 2000;102:1564–1568. doi: 10.1161/01.cir.102.13.1564. [DOI] [PubMed] [Google Scholar]
- 143.Dumont EA, Reutelingsperger CP, Smits JF, et al. Real-time imaging of apoptotic cell-membrane changes at the single-cell level in the beating murine heart. Nat Med. 2001;7:1352–1355. doi: 10.1038/nm1201-1352. [DOI] [PubMed] [Google Scholar]
- 144.Hofstra L, Liem IH, Dumont EA, et al. Visualisation of cell death in vivo in patients with acute myocardial infarction. Lancet. 2000;356:209–212. doi: 10.1016/s0140-6736(00)02482-x. [DOI] [PubMed] [Google Scholar]
- 145.Nutt LK, Gogvadze V, Uthaisang W, Mirnikjoo B, McConkey DJ, Orrenius S. Indirect effects of Bax and Bak initiate the mitochondrial alterations that lead to cytochrome c release during arsenic trioxide-induced apoptosis. Cancer Biol Ther. 2005;4:459–467. doi: 10.4161/cbt.4.4.1652. [DOI] [PubMed] [Google Scholar]
- 146.Zheng Y, Shi Y, Tian C, et al. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene. 2004;23:1239–1247. doi: 10.1038/sj.onc.1207205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Przygodzki T, Sokal A, Bryszewska M. Calcium ionophore A23187 action on cardiac myocytes is accompanied by enhanced production of reactive oxygen species. Biochim Biophys Acta. 2005;1740:481–488. doi: 10.1016/j.bbadis.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 148.Lax A, Soler F, Fernandez-Belda F. Cytoplasmic Ca2+ signals and cellular death by apoptosis in myocardiac H9c2 cells. Biochim Biophys Acta. 2006;1736:937–947. doi: 10.1016/j.bbamcr.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 149.Wei MC, Zon WX, Cheng EHY, et al. Proapoptotic Bax and Bak; a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Capano M, Crompton M. Bax translocates to mitochondria of heart cells during simulated ischaemia: involvement of AMP-activated and p38 mitogen-activated protein kinases. Biochem J. 2006;395:57–64. doi: 10.1042/BJ20051654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lundberg KC, Szweda LI. Initiation of mitochondrial-mediated apoptosis during cardiac reperfusion. Arch Biochem Biophys. 2004;432:50–57. doi: 10.1016/j.abb.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 152.Huang J, Nakamura K, Ito Y, et al. Bcl-xL gene transfer inhibits Bax translocation and prolongs cardiac cold preservation time in rats. Circulation. 2005;112:76–83. doi: 10.1161/CIRCULATIONAHA.105.535740. [DOI] [PubMed] [Google Scholar]
- 153.Nutt LK, Pataer A, Pahler J, et al. Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J Biol Chem. 2002;277:9219–9225. doi: 10.1074/jbc.M106817200. [DOI] [PubMed] [Google Scholar]
- 154.Nutt LK, Chandra J, Pataer A, et al. Bax-mediated Ca2+ mobilization promotes cytochrome c release during apoptosis. J Biol Chem. 2002;277:20301–20308. doi: 10.1074/jbc.M201604200. [DOI] [PubMed] [Google Scholar]
- 155.Hetz C, Vitte PA, Bombrun A, et al. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J Biol Chem. 2005;280:42960–42970. doi: 10.1074/jbc.M505843200. [DOI] [PubMed] [Google Scholar]
- 156.Takahashi M, Tanonaka K, Yoshida H, et al. Possible involvement of calpain activation in pathogenesis of chronic heart failure after acute myocardial infarction. J Cardiovasc Pharacol. 2006;47:413–421. doi: 10.1097/01.fjc.0000210074.56614.3b. [DOI] [PubMed] [Google Scholar]
- 157.Wencker D, Chandra M, Nguyen K, et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111:1497–1404. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Saez ME, Ramirez-Lorca R, Moron FJ, Ruiz A. The therapeutic potential of the calpain family: new aspects. Drug Discov Today. 2006;11:917–923. doi: 10.1016/j.drudis.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 159.Engel T, Plesnila N, Prehn JH, Henshall DC. In vivo contributions of BH3-only proteins to neuronal death following seizures, ischemia, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31(5):1196–1210. doi: 10.1038/jcbfm.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chen M, Won DJ, Krajewski S, Gottlieb RA. Calpain and mitochondria in ischemia reperfusion injury. J Biol Chem. 2002;277:29282–29286. doi: 10.1074/jbc.M204951200. [DOI] [PubMed] [Google Scholar]
- 161.Saez ME, Ramirez-Lorca R, Moron FJ, Ruiz A. The therapeutic potential of the calpain family: new aspects. Drug Discov Today. 2006;11:917–923. doi: 10.1016/j.drudis.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 162.Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17:617–625. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Matsui T, Rosenzweig A. Convergent signal transduction pathways controlling cardiomyocyte survival and function: the role of PI 3-kinase and Akt. J Mol Cell Cardiol. 2005;38:63–71. doi: 10.1016/j.yjmcc.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 164.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–8998. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 165.Webster KA. Aktion in the nucleus. Circ Res. 2004;94:856–859. doi: 10.1161/01.RES.0000126699.49835.5D. [DOI] [PubMed] [Google Scholar]
- 166.Yamashita K, Kajstura J, Discher DJ, Wasserlauf BJ, Anversa P, Webster KA. Reperfusion-activated Akt kinase prevents apoptosis in transgenic mouse hearts overexpressing insulin-like growth factor-1. Circ Res. 2001;88:609–614. doi: 10.1161/01.res.88.6.609. [DOI] [PubMed] [Google Scholar]
- 167.Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D. Protein kinase Cδ activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J Biol Chem. 2004;279:47985–47991. doi: 10.1074/jbc.M405071200. [DOI] [PubMed] [Google Scholar]
- 168.The Direct Inhibition of ƒ-Protein Kinase C Enzyme to Limit Total Infarct Size in Acute Myocardial Infarction (DELTA MI) Investigators. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation. 2008;117:886–896. doi: 10.1161/CIRCULATIONAHA.107.759167. [DOI] [PubMed] [Google Scholar]
- 169.Metzler B, Xu Q, Mayr M. Circulation. 2008;118 doi: 10.1161/CIRCULATIONAHA.108.772772. (letter) [DOI] [PubMed] [Google Scholar]
- 170.Graham RM, Frazier D, Thompson JW, et al. A unique pathway of cardiac myocyte death caused by hypoxia-acidosis. J Exp Biol. 2004;207:3189–3200. doi: 10.1242/jeb.01109. [DOI] [PubMed] [Google Scholar]
- 171.Hamacher-Brady A, Brady NR, Gottlieb RA, Gustafsson AB. Autophagy as a protective response to Bnip3-mediated apoptotic signaling in the heart. Autophagy. 2006;2(4):307–309. doi: 10.4161/auto.2947. [DOI] [PubMed] [Google Scholar]
- 172.Shaw J, Kirshenbaum LA. Molecular regulation of autophagy and apoptosis during ischemic and non-ischemic cardiomyopathy. Autophagy. 2008;4(4):427–434. doi: 10.4161/auto.5901. [DOI] [PubMed] [Google Scholar]
- 173.Burton TR, Gibson SB. The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ. 2009;16(4):515–523. doi: 10.1038/cdd.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ray R, Chen G, Vande Velde C, et al. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem. 2000;275(2):1439–1448. doi: 10.1074/jbc.275.2.1439. [DOI] [PubMed] [Google Scholar]
- 175.Hamacher-Brady A, Brady NR, Logue SE, et al. Response to myocardial ischemia/ reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14(1):146–157. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 176.Ray R, Chen G, Vande Velde C, et al. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem. 2000;275(2):1439–1448. doi: 10.1074/jbc.275.2.1439. [DOI] [PubMed] [Google Scholar]
- 177.Chaanine AH, Jeong D, Liang L, et al. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012;3:265. doi: 10.1038/cddis.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Shibue T, Suzuki S, Okamoto H, et al. Differential contribution of Puma and Noxa in dual regulation of p53-mediated apoptotic pathways. EMBO J. 2006;25:4952–4962. doi: 10.1038/sj.emboj.7601359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yu J, Zhang L. No PUMA, no death: implications for p53-dependent apoptosis. Cancer Cell. 2003;4:248–249. doi: 10.1016/s1535-6108(03)00249-6. [DOI] [PubMed] [Google Scholar]
- 180.Erlacher M, Michalak EM, Kelly PN, et al. BH3-only proteins Puma and Bim are rate-limiting for γ-radiation- and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 2005;106:4131–4138. doi: 10.1182/blood-2005-04-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kim JY, Ahn HJ, Ryu JH, Suk K, Park JH. BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1α. J Exp Med. 2004;199:113–124. doi: 10.1084/jem.20030613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol. 2003;162:587–597. doi: 10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Toth A, Jeffers JR, Nickson P, et al. Targeted deletion of puma attenuates cardiomyocyte death and improves cardiac function during ischemia/reperfusion. Am J Physiol Heart Circ Physiol. 2006;291(1):H52–H60. doi: 10.1152/ajpheart.01046.2005. [DOI] [PubMed] [Google Scholar]
- 185.Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. MiR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med. 2011;14(208)(3):549–560. doi: 10.1084/jem.20101547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bolli R, Chugh AR, D’Amario D, et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO), initial results of a randomised Phase 1 trial. Lancet. 2011;378(9806):1847–1857. doi: 10.1016/S0140-6736(11)61590-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]