

Abstract
Bacterial-derived lipopolysaccharides (LPS) play an essential role in the inflammatory process of inflammatory bowel disease. A defective intestinal tight junction (TJ) barrier is an important pathogenic factor of inflammatory bowel disease and other inflammatory conditions of the gut. Despite its importance in mediating intestinal inflammation, the physiological effects of LPS on the intestinal epithelial barrier remain unclear. The major aims of this study were to determine the effects of physiologically relevant concentrations of LPS (0 to 1 ng/mL) on intestinal barrier function using an in vitro (filter-grown Caco-2 monolayers) and an in vivo (mouse intestinal perfusion) intestinal epithelial model system. LPS, at physiologically relevant concentrations (0 to 1 ng/mL), in the basolateral compartment produced a time-dependent increase in Caco-2 TJ permeability without inducing cell death. Intraperitoneal injection of LPS (0.1 mg/kg), leading to clinically relevant plasma concentrations, also caused a time-dependent increase in intestinal permeability in vivo. The LPS-induced increase in intestinal TJ permeability was mediated by an increase in enterocyte membrane TLR-4 expression and a TLR-4–dependent increase in membrane colocalization of membrane-associated protein CD14. In conclusion, these studies show for the first time that LPS causes an increase in intestinal permeability via an intracellular mechanism involving TLR-4–dependent up-regulation of CD14 membrane expression.
An integral function of intestinal epithelial cells is to act as a physical barrier, separating the noxious luminal environment from the underlying lamina propria and the deeper intestinal layers.1,2 The apically located tight junctions (TJs) form a paracellular seal between the lateral membranes of adjacent intestinal epithelial cells, and act as a structural and functional barrier against paracellular flux of luminal substances. Defective intestinal epithelial TJ barrier has been shown to be an important pathogenic factor of inflammatory bowel disease (IBD) and necrotizing enterocolitis (NEC) by allowing paracellular permeation of luminal antigens that elicit and promote inflammatory response.1,2 Both clinical and animal studies have shown the importance of a defective intestinal TJ barrier in the development and prolongation of intestinal inflammation in IBD and NEC.1–5 These studies have shown that normalization of intestinal barrier in patients with active Crohn’s disease predicts prolonged clinical remission, whereas a persistent increase in intestinal permeability portends poor clinical outcome with rapid recurrence of the disease.6,7 Additionally, animal studies have also shown that a primary defect in intestinal junctional complexes was sufficient to induce or aggravate intestinal inflammation in murine models of IBD,8,9 whereas therapeutic tightening or enhancement of the intestinal TJ barrier prevented the development of intestinal inflammation.3,10
The terms endotoxin and lipopolysaccharide (LPS) are used interchangeably and refer to the major cell wall component of Gram-negative bacteria.11,12 LPS are complex amphiphilic molecules having a hydrophobic (consisting of lipid A) and a hydrophilic (consisting of carbohydrate core and polysaccharide O-antigen) component and are released from bacterial cell wall by shedding or through bacterial lysis.11–13 LPS concentrations are highest in the gut lumen, where many trillions of commensal bacteria reside. Normally, LPS in the gut lumen do not penetrate across the healthy intestinal epithelium14,15; however, in intestinal permeability disorders, the defective TJ barrier allows paracellular flux of LPS and other luminal antigens.11–13,16–19 The intestinal tissue and circulating LPS levels are markedly elevated in IBD and NEC, and play an important role in mediating inflammatory response.11–13,16–18 The involvement of LPS in the initiation and propagation of intestinal inflammation in IBD and NEC has been well demonstrated.20–23 These studies have shown LPS to be an important contributing factor of intestinal inflammation, and removal of circulating LPS accelerated the clinical improvement of IBD and NEC.20,22,23 Despite the importance of a defective intestinal barrier in the accentuation and prolongation of intestinal inflammation in IBD and NEC,3,6,9,20,22 the effects of circulating levels of LPS on the intestinal epithelial barrier remain unknown. Because LPS levels are markedly elevated in these diseases and play an important role in the inflammatory process, understanding the effects of LPS on intestinal barrier function has important potential clinical significance.
In normal healthy individuals, plasma concentrations of LPS range from undetectable levels up to 0.2 ng/mL.11,12,20,22 A variety of physiological factors such as prolonged physical exertion, high-fat diet, physiological stresses, or heat can lead to elevated plasma LPS levels as high as 1 to 2 ng/mL.24–27 Patients with intestinal permeability disorders such as Crohn’s disease, NEC, acute pancreatitis, alcoholic liver disease, and critical illnesses also have elevated plasma LPS levels ranging up to 2 to 10 ng/mL.11–13,20,22,28 Based on these reports, we consider LPS levels of 0 to 1.0 ng/mL to be physiologically relevant and 0 to 10 ng/mL to be clinically relevant. (For reference, the concentration of LPS in the gut lumen has been reported to be 1.8 μg/mL in the rat distal ileum.29,30) Inexplicably, in most of the published studies, extreme pharmacological concentrations of LPS ranging between 50 and 1000 μg/mL, which exceed the physiologically achievable concentrations by 104- to 107-fold, have been used to assess various biological responses.30–34 At these extreme concentrations, LPS causes rapid cell death in various cell types studied, including in intestinal and immune cells,30,33–35 and does not provide accurate depiction of biological activity of LPS. Herein, we show that LPS, at physiologically and clinically relevant concentrations (0 to 10 ng/mL), does not cause intestinal epithelial cell death, but causes a selective increase in intestinal TJ permeability in vitro and in vivo. These studies also show for the first time that pattern recognition receptors Toll-like receptor 4 (TLR-4) and CD14 play a central role in the modulation of the intestinal epithelial TJ barrier.
Materials and Methods
Reagents
Dulbecco’s modified Eagle’s medium, trypsin, fetal bovine serum, glutamine, penicillin, streptomycin, PBS, and horseradish peroxidase–conjugated secondary antibodies for Western blot analysis were purchased from Invitrogen/Life Technologies (Carlsbad, CA). Small-interfering RNA (siRNA) of TLR-4, CD14, and transfection reagents were from Dharmacon/Thermo Fisher Scientific (Lafayette, CO). LPS (O111:B4) was purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were of reagent grade and were purchased from Sigma-Aldrich, VWR (Aurora, CO), or Fisher Scientific (Pittsburgh, PA).
Cell Culture
Caco-2 cells (passage 20) were purchased from ATCC (Manassas, VA) and maintained at 37°C in a culture medium composed of Dulbecco’s modified Eagle’s medium with 4.5 mg/mL glucose, 50 U/mL penicillin, 50 U/mL streptomycin, 4 mmol/L glutamine, 25 mmol/L HEPES, and 10% fetal bovine serum as previously described.36,37 Caco-2 cells were used between passages 22 and 28 in this study. The human-derived, nontransformed colonic epithelial cell line NCM460 was maintained in M3:10 medium (INCELL, San Antonio, TX), but was shifted to Ham’s F-12 culture medium supplemented with 20% fetal bovine serum and antibiotics as previously described.38 NCM460 cells were used between passages 29 and 39 in this study. T84 intestinal epithelial cells (passage 53) were purchased from ATCC (Rockville, MD) and maintained at 37°C in Dulbecco’s modified Eagle’s medium/Ham’s F-12 medium supplemented with 16 mmol/L NaHCO3, antibiotics, and 5% fetal bovine serum, as previously described.39 Only T84 cells from passages 55 to 61 were used to maintain consistency. The cells were kept at 37°C in a 5% CO2 environment. For growth on filters, high-density cells (1 × 105 cells) were plated on Transwell filters with 0.4-μm pore size (Corning Incorporated, Corning, NY) and monitored regularly by visualization with an inverted microscope (Eclipse TS100/100-F; Nikon, Melville, NY) and by epithelial resistance measurements.
Determination of Epithelial Monolayer Resistance and Paracellular Permeability
Caco-2 transepithelial electrical resistance (TER) was measured by using an epithelial voltohmeter (World Precision Instruments, Sarasota, FL) as previously reported.36,37 Both apical and basolateral sides of the epithelium were bathed with buffer solution. Electrical resistance was measured until similar values were recorded on three consecutive measurements. Caco-2 paracellular permeability was determined by using an established paracellular marker inulin. Known concentrations of permeability marker (10 μmol/L) and its radioactive tracer were added to the apical solution.36 Low concentrations of permeability markers were used to ensure that negligible osmotic or concentration gradients were introduced.
Assessment of Protein Expression by Western Blot Analysis
Protein expression from Caco-2 cells and mouse tissue was assessed by Western blot as previously described.36,37 Cells and mouse tissue were lysed with lysis buffer [50 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 500 μmol/L NaF, 2 mmol/L EDTA, 100 μmol/L vanadate, 100 μmol/L phenylmethylsulfonyl fluoride, 1 μg/mL leupeptin, 1 μg/mL pepstatin A, 40 mmol/L para-nitrophenyl phosphate, 1 μg/mL aprotinin, and 1% Triton X-100]. The lysates were centrifuged at 10,000 × g for 10 minutes in an Eppendorf Centrifuge (5417R; Hauppauge, NY) to obtain a clear lysate. The supernatant was collected and protein concentration was determined using the Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Hercules, CA). Laemmli gel loading buffer (Bio-Rad Laboratories) was added to the lysate containing 10 to 20 μg of protein and boiled at 100°C for 7 minutes, after which proteins were separated on an SDS-PAGE gel. Proteins from the gel were transferred to the membrane (Trans-Blot Transfer Medium, nitrocellulose membrane; Bio-Rad Laboratories) overnight. The membrane was incubated for 2 hours in blocking solution (5% dry milk in TBS-Tween 20 buffer). The membrane was then incubated with antibody in blocking solution. After a wash in TBS-1% Tween buffer, the membrane was incubated in secondary antibody and developed using Santa Cruz Western Blotting Luminol Reagents (Santa Cruz Biotechnology, Santa Cruz, CA) on Kodak BioMax MS film (Fisher Scientific). The films were exposed between 5 seconds and 10 minutes.
Assessment of Apoptosis
Caco-2 apoptosis was assessed using the CytoGLO Annexin V-FITC Apoptosis Detection kit from Imgenex as previously described.37 Following appropriate treatments, apoptosis was measured by flow cytometry using a FACScan flow cytometer (BD Biosciences, San Jose, CA). For each experimental sample, a total of 10,000 cells were counted for Annexin V-FITC and propidium iodide stain.
Proliferation Assay
Cell proliferation was measured using Quick Cell Proliferation Assay kit (ab65473; Abcam, Cambridge, MA) according to the manufacturer’s instructions. This assay is based on cleavage of the tetrazolium salt WST-1 to formazan by cellular mitochondrial dehydrogenases. Cell proliferation is accompanied by an increase in the activity of the mitochondrial dehydrogenases, which leads to an increase in conversion of tetrazolium salt, WST-1, to formazan. Briefly, Caco-2 cells (1 × 105 cells) were plated on Transwell filters. After LPS treatment, tetrazolium salt WST-1 solution was added to each filter, and the reaction was allowed to occur in standard culture condition for 4 hours. The optical density (formazan formation) was measured at 440 nm with a reference wavelength of 650 nm using a precision microplate reader (Molecular Devices, Sunnyvale, CA) as a measure of mitochondrial dehydrogenase activity.
Immunostaining
Colocalization of TLR-4 and CD14 was assessed by immunofluorescent antibody labeling.40 Caco-2 monolayers or frozen mouse intestinal sections were washed and fixed with 2% paraformaldehyde. After being permeabilized with 0.1% Triton X-100 in PBS, Caco-2 monolayers or intestinal tissues were incubated in blocking solution composed of bovine serum albumin and normal donkey serum in PBS for 1 hour, then labeled with primary antibodies in blocking solution overnight at 4°C, followed by fluorescein isothiocyanate or Cy3-conjugated secondary antibodies for 1 hour at room temperature. ProLong Gold Antifade reagent (Invitrogen) was used to mount the filters onto coverslips. Immunolocalizations of TLR-4 and CD14 were visualized using a confocal fluorescence microscope (LSM 510 Meta Confocal; University of New Mexico Fluorescence Microscopy Shared Resource) equipped with a Hamamatsu Digital Camera 9 (Hamamatsu Photonics, Hamamatsu, Japan). Images were processed with LSM software (Carl Zeiss, Oberkochen, Germany).
siRNA Transfection
Caco-2 monolayers were transiently transfected with siRNA using DharmaFect transfection reagent (Dharmacon/Thermo Fisher Scientific) as previously described.40 Briefly, cells (1 × 105/filter) were seeded into a 12-well Transwell plate and grown to confluency. Caco-2 monolayers were then washed with PBS twice; 0.5 mL Accell medium (Thermo Fisher Scientific) was added to the apical compartment of each filter, and 1.5 mL were added to the basolateral compartment of each filter. siRNA (5 ng) of interest and DharmaFect reagent (2 μL) were preincubated in Accell medium. After 5 minutes of incubation, the two solutions were mixed, and the mixture was added to the apical compartment of each filter. For LPS experiments, Caco-2 cells were transfected with siRNA for 24 hours before the LPS treatment. The Caco-2 TJ barrier assessments were performed at the end of day 5 of LPS treatment.
In Vivo Determination of Mouse Intestinal Permeability and LPS Plasma Level
The Laboratory Animal Care and Use Committee at the University of New Mexico approved all experimental protocols. Male C57BL/6 mice, TLR-4 knockout mice, and CD14 knockout mice (9 to 10 weeks) were purchased from the Jackson Laboratories (Bar Harbor, ME). The mice were kept two per cage in a temperature-controlled room at 25°C with a 12:12-hour light–dark cycle. Diet and drinking water were provided ad libitum.
LPS effect on intestinal permeability in an in vivo mouse model system was determined using a recycling intestinal perfusion method as previously described.36,40,41 Mice were injected with varying concentrations of LPS intraperitoneally (i.p.) every 24 hours for up to 5 days of the experimental period. A 6-cm segment of mouse small intestine was isolated and cannulated with a small-diameter plastic tube (in an anesthetized mouse maintained on 1% isoflurane in oxygen) and continuously perfused with 5 mL of Krebs-phosphate saline buffer for a 2-hour perfusion period.36,40 An external recirculating pump (Econo Pump; Bio-Rad Laboratories) was used to recirculate the perfusate at a constant flow rate (0.75 mL/min). The body temperature of the mouse was maintained at 37°C with a temperature-controlled warming blanket. The intestinal permeability was assessed by measuring the flux rate of the paracellular probe, Texas Red–labeled dextran (MW = 10,000 g/mol). Water absorption was determined by using a nonabsorbable marker, sodium ferrocyanide, or by measuring the difference between the initial and final volumes of the perfusate. The plasma LPS level was measured using Limulus Amebocyte Lysate Assay (Associates of Cape Cod, E. Falmouth, MA).
Statistical Analysis
Results are expressed as means ± SE, and analyzed using Student’s t-test for unpaired data (GraphPad Prism version 5.00 for Windows; GraphPad Software, La Jolla, CA). A P value of ≤0.05 was used to indicate statistical significance. All experiments were repeated at least three times to ensure reproducibility.
Results
LPS Effect on Caco-2 Intestinal Epithelial TJ Permeability
The effects of physiologically and clinically achievable concentrations of LPS (0 to 10 ng/mL) on intestinal epithelial TJ permeability was determined in vitro by measuring TER and mucosal-to-serosal flux of paracellular marker inulin in filter-grown Caco-2 intestinal monolayers, over a 5-day experimental period. LPS (0, 0.1, 0.3, 0.5, 1, 5, and 20 ng/mL) caused a concentration-dependent drop in Caco-2 TER (Figure 1A) and an increase in inulin flux (Figure 1B). LPS caused a significant drop in TER and an increase in inulin flux at a concentration of 0.3 ng/mL. Increasing the LPS concentration above 0.3 ng/mL caused only a minimal additional drop in Caco-2 TER (Figure 1A) or increase in inulin flux (Figure 1B). These results indicated that LPS at concentrations of 0.3 ng/mL or above cause an increase in Caco-2 TJ permeability. (In subsequent experiments, a LPS concentration of 0.3 ng/mL was used, unless stated otherwise.)
Figure 1.

Effect of LPS on Caco-2 TJ permeability. A: Effect of increasing concentrations of LPS (0, 0.1, 0.3, 0.5, 1, 5, and 20 ng/mL) on Caco-2 TER. The mean TER for control (Cont) Caco-2 monolayers was 481 ± 5 Ω • cm2. B: Effect of increasing concentrations of LPS (0, 0.1, 0.3, 0.5, 1, 5, and 20 ng/mL) on Caco-2 transepithelial flux of paracellular marker inulin. Filter-grown Caco-2 monolayers were treated with LPS for a 5-day experimental period. C: Time-course effect of LPS (0.3 ng/mL) on Caco-2 TER. The mean TER for day 0 was 450 ± 14 Ω • cm2. D: Time-course effect of LPS (0.3 ng/mL) on Caco-2 inulin flux. The effect of LPS (0.3 ng/mL) on Caco-2 TER and inulin flux were measured over a 7-day experimental period. E: Graph of TER versus inulin flux (r = 0.93). F: Time course of Caco-2 TER recovery after LPS (0.3 ng/mL) removal. LPS was removed from the basolateral compartment after 5 days of treatment (arrow). The mean TER for control Caco-2 monolayers was 486 ± 28 Ω • cm2. *P < 0.0001 versus control.
Next, the time-course effect of LPS (0.3 ng/mL) on Caco-2 TJ permeability was determined. LPS did not have significant effect on Caco-2 TER up to day 3, but caused a drop in TER between days 4 and 5. The drop in TER reached a trough level by day 5 and remained unchanged up to day 7 (Figure 1C). Conversely, LPS caused an increase in mucosal-to-serosal inulin flux by day 4, and the increase in inulin flux persisted up to day 7 (Figure 1D). A similar time-course effect was also observed for higher concentrations of LPS (1 and 20 ng/mL) (data not shown). The plot of the LPS-induced drop in TER and increase in paracellular flux demonstrates a linear relationship (relative correlation coefficient, r = 0.93) between the drop in Caco-2 TER and the increase in inulin flux (Figure 1E), indicating that the LPS-induced drop in Caco-2 TER correlates with an increase in paracellular permeability. The time course of Caco-2 TER recovery following LPS removal was also examined. The removal of LPS after 5 days of treatment resulted in full restoration of the Caco-2 TER within 2 days of removal (Figure 1F).
To confirm that the LPS effect extends to other intestinal epithelial cell lines, we also examined the effect of LPS (0.3 ng/mL) on NCM460 and T84 monolayer TER. NCM460 is a human-derived, nontransformed colonic epithelial cell line, and T84 is a colon cancer–derived cell line having high TER. LPS caused a similar time-dependent drop in NCM460 and T84 TER (Figure 2) as in the Caco-2 monolayers. There was an approximately 30% drop in TER by day 4 or 5 of LPS treatment. Because LPS preparation is known to contain traces of impurities, including cytokines and other lipoproteins, the effect of heat-inactivated LPS (exposure to 121°C for 30 minutes) on Caco-2, NCM460, and T84 TER was also examined. Heat-inactivated LPS did not affect the TER of intestinal monolayers (data not shown), suggesting that the impurities in LPS did not affect the intestinal monolayer TER and that active LPS was required for the drop in TER.
Figure 2.
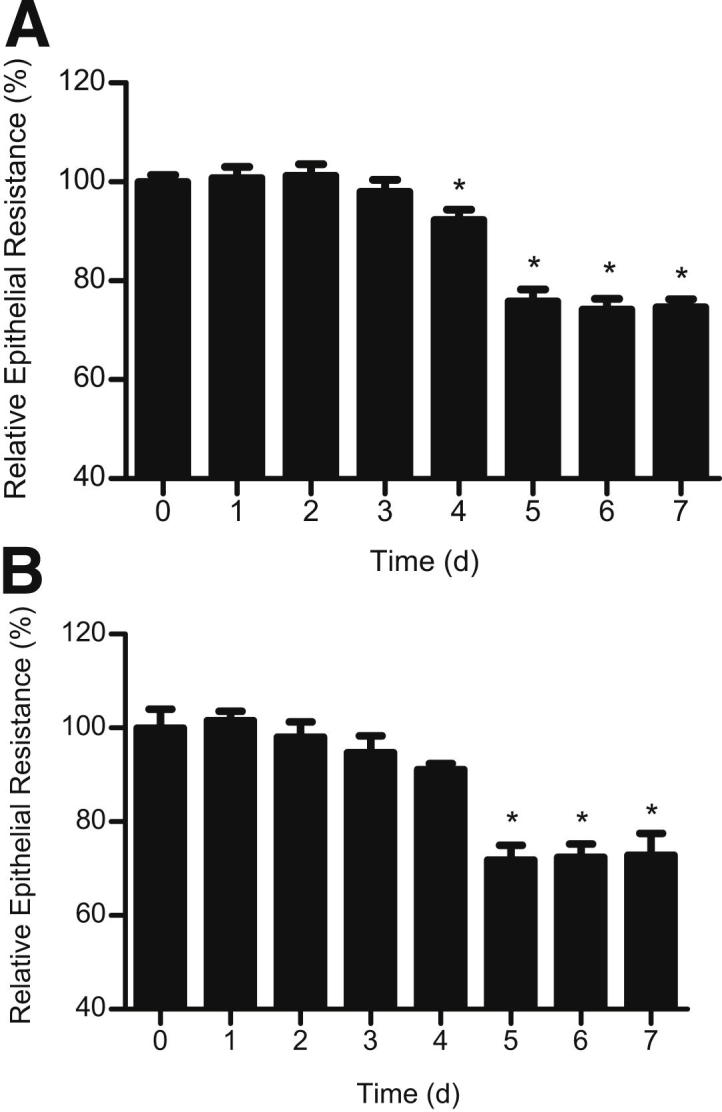
Time-course effect of LPS (0.3 ng/mL) on NCM460 TER (A) and T84 TER (B). The mean TER for NCM460 monolayers at day 0 was 418 ± 14 Ω • cm2, and the mean TER for T84 monolayers at day 0 was 1570 ± 108 Ω • cm2. *P < 0.0001 versus control.
To assess the membrane specificity of LPS effect, LPS (0.3 ng/mL) was added to either the apical, basolateral, or combined apical and basolateral compartments. The addition of LPS to the apical solution did not have a significant effect on Caco-2 TER (Figure 3A) or inulin flux (Figure 3B), whereas the addition of LPS to the basolateral solution caused a drop in Caco-2 TER and an increase in inulin flux. The addition of LPS to both apical and basolateral compartments caused a similar change in TJ permeability as the basolateral compartment alone (Figure 3, A and B). To investigate the possibility that the LPS effect on the apical membrane may be concentration related, higher concentrations of LPS were also added to the apical solution. The higher concentrations (20 ng/mL, 1 μg/mL, and 50 μg/mL) of LPS in the apical membrane surface also did not affect Caco-2 TER or inulin flux (Figure 3, C and D), suggesting that the presence of high concentrations of LPS on the mucosal surface do not affect intestinal TJ barrier function.
Figure 3.

Membrane specificity of the LPS effect on Caco-2 permeability. A: Effect of LPS (0.3 ng/mL) on Caco-2 TER. The mean TER for control (Cont) Caco-2 monolayers was 475 ± 12 Ω • cm2. B: Effect of LPS (0.3 ng/mL) on Caco-2 inulin flux. LPS (0.3 ng/mL) was added to either apical (AP), basolateral (BL), or combined apical and basolateral compartments (Both) for the 5-day experimental period. C: Effect of increasing concentrations of LPS (0, 0.1, 0.3, 1, 20, 1000, and 50,000 ng/mL) on Caco-2 TER. The mean TER for control Caco-2 monolayers was 454 ± 19 Ω • cm2. D: Effect of increasing concentrations of LPS (0, 0.1, 0.3, 1, 20, 1000, and 50,000 ng/mL) on Caco-2 inulin flux. LPS was added to the apical compartment at each 24 hours treatment period for 5 days. *P < 0.0001 versus control.
LPS Effect on Caco-2 Apoptosis and Cell Death
In previous studies, the effects of high pharmacological concentrations (50 μg/mL to 1 mg/mL) of LPS on intestinal epithelial barrier integrity were evaluated.30–32 These studies indicated that at these extreme concentrations (which exceed the physiological levels by 104- to 107-fold), LPS causes a rapid apoptosis and death of intestinal epithelial cells and apoptosis-dependent loss of epithelial integrity.30–32 However, the effects of physiologically relevant concentrations of LPS (0 to 1 ng/mL) on intestinal epithelial cell apoptosis or death remain unclear. The LPS effect on Caco-2 apoptosis and necrosis was determined over a 5-day experimental period by Annexin V-FITC labeling of phosphatidylserine on plasma membrane of apoptotic cells and by propidium iodide labeling of DNA in necrotic cells using flow cytometry37 and by caspase-3 expression in filter-grown Caco-2 monolayers.30 Increasing the concentration of LPS up to 100 ng/mL did not induce Caco-2 cell apoptosis (Figure 4A) or necrosis (data not shown). As a positive control, Caco-2 monolayers were also treated with high LPS concentration (1 mg/mL) that had been previously shown to induce Caco-2 cell apoptosis (Figure 4A). In separate experiments, the time-course effect of LPS (0.3 ng/mL) on caspase-3 expression in Caco-2 monolayers was also examined. LPS did not cause an increase in caspase-3 expression (Figure 4B). Additionally, the LPS effect on morphology of Caco-2 monolayers was examined using a Nikon inverted microscope (×400 magnification). LPS (0 to 100 ng/mL) did not cause morphological changes to the epithelial monolayer surface or detachment of epithelial cells (data not shown). To examine the effect on cell turnover, we also measured the effect of LPS (0.3 ng/mL) on cell proliferation over a 5-day experimental period by assessing mitochondrial dehydrogenase activity. LPS did not have a significant effect on Caco-2 cell proliferation (Figure 4C). Together, these results indicate that LPS at concentrations up to 100 ng/mL does not induce Caco-2 cell death and that the LPS effect on Caco-2 TJ permeability is not due to cell death leading to a breach in the epithelial integrity.
Figure 4.

Effect of LPS on Caco-2 cell apoptosis and proliferation. A: Effect of increasing concentrations of LPS on Caco-2 cell apoptosis was assessed by flow cytometry. LPS concentration of 1 mg/mL was used as a positive control of apoptosis.23 There were a total of 10,000 events for each sample. B: Effect of LPS on caspase-3 expression assessed by Western blot analysis. C: Effect of LPS on Caco-2 cell proliferation as assessed by mitochondrial dehydrogenase conversion of tetrazolium salt WST-1 to formazan. *P < 0.0001 versus control.
LPS-Induced Increase in Caco-2 TJ Permeability Requires an Increase in TLR-4 Expression
In the following studies, the possible involvement of TLR-2, TLR-4, or TLR-5 in the LPS-induced increase in Caco-2 TJ permeability was determined. The basal expressions of TLR-2 and TLR-4 were low in unstimulated Caco-2 monolayers, whereas TLR-5 levels were higher (Figure 5A). LPS (0.3 ng/mL) treatment resulted in a time-dependent increase in TLR-4 expression (Figure 5A), but did not significantly affect TLR-2 or TLR-5 levels. LPS did not cause an increase in TLR-4 expression until day 4 of the treatment. The time course of the LPS-induced increase in Caco-2 TLR-4 expression paralleled the drop in Caco-2 TER and the increase in paracellular permeability (Figure 1, C and D), suggesting a possible cause-and-effect relationship. To determine the possible causal role of TLR-4 in the LPS-induced increase in Caco-2 TJ permeability, the effect of siRNA-induced silencing of TLR-4 expression was examined. The TLR-4 siRNA transfection resulted in an inhibition of the LPS-induced increase in Caco-2 TLR-4 expression (Figure 5B), drop in Caco-2 TER (Figure 5C), and increase in inulin flux (Figure 5D), confirming the specific requirement of TLR-4 in LPS-induced increase in Caco-2 TJ permeability. In a separate set of experiments, the requirement of TLR-2 or TLR-5 in the LPS-induced increase in Caco-2 TJ permeability was also examined. The siRNA-induced knockdown of TLR-2 or TLR-5 did not affect the LPS-induced drop in Caco-2 TER or the increase in inulin flux (Figure 6). To further validate the involvement of TLR-4 signal transduction pathway, the effect of a pharmacological inhibitor of the TLR-4 pathway, TAK-242,42 on the LPS-induced increase in Caco-2 TJ permeability was examined. TAK-242 (3 μmol/L) prevented the LPS-induced drop in Caco-2 TER (Figure 5E) and the increase in inulin flux (Figure 5F). Together, these findings suggest that the LPS effect on Caco-2 TJ permeability is mediated by an increase in TLR-4 expression and activation of the TLR-4 signal transduction pathway.
Figure 5.

Effect of LPS on Toll-like receptors. A: Time-course effect of LPS on TLR-2, TLR-4, and TLR-5 expression in Caco-2 monolayers. B: Effect of TLR-4 siRNA transfection on TLR-4 expression in Caco-2 monolayers. Effect of inhibition of TLR-4 by siRNA silencing on Caco-2 TER (C) and inulin flux (D). The Caco-2 cells were transfected with TLR-4 siRNA, 24 hours before the LPS treatment. The mean TER for control (Cont) Caco-2 monolayers was 441 ± 4 Ω • cm2. Effect of TLR-4 inhibitor, TAK-242 (3 μmol/L), on Caco-2 TER (E) and inulin flux (F). TAK-242 was added to the apical compartment 1 hour before the LPS treatment. The mean TER for control Caco-2 monolayers was 463 ± 15 Ω • cm2. Scr, scrambled. *P < 0.001 versus control; **P < 0.001 versus LPS treatment.
Figure 6.

Effect of siRNA silencing of TLR-2 or TLR-5 on LPS-induced alteration in Caco-2 TER and inulin flux. Caco-2 cells were transfected with TLR-2 siRNA or TLR-5 siRNA for 24 hours before the LPS treatment. siRNA-induced silencing of TLR-2 did not affect Caco-2 TER (A) or inulin flux (B). The mean TER for control Caco-2 monolayers was 468 ± 17 Ω • cm2. siRNA-induced silencing of TLR-5 also did not affect Caco-2 TER (C) or inulin flux (D). The mean TER for control Caco-2 monolayers was 492 ± 24 Ω • cm2. Scr, scrambled. *P < 0.001 versus control.
LPS Causes an Increase in Mouse Intestinal Permeability in Vivo
In the following studies, an in vivo model of the LPS-induced increase in mouse intestinal permeability was established, using a modification of a previously described method of recycling mouse intestinal perfusion.36,40,41 To determine the appropriate LPS concentration to use in vivo, the effects of increasing i.p. doses of LPS (0, 0.05, 0.1, 1.0, or 10 mg/kg body weight) on mouse plasma LPS level were determined. The basal plasma LPS level in control mice (C57BL/6, the Jackson Laboratory, Bar Harbor, ME) was 0.017 ng/mL; increasing the i.p. LPS concentration resulted in a dose-dependent increase in mouse plasma LPS levels (Table 1). The LPS concentration of 0.1 mg/kg body weight produced a 1-hour peak level of 2.230 ng/mL and a 24-hour steady-state plasma level of 0.405 ng/mL (Table 1), which were within physiologically and clinically relevant plasma concentrations.11–13,20,22 By contrast, the lethal LPS dose (10 mg/kg) used in previous animal studies32 resulted in very high peak (13.49 μg/mL) and trough (252.4 ng/mL) plasma LPS levels, which produced a rapid intestinal mucosal damage (Table 1). On the basis of the above data, the LPS dose of 0.1 mg/kg was selected for the mouse intestinal permeability studies. Over a 5-day treatment period (LPS injection every 24 hours), the steady-state plasma LPS level remained between 0.4 and 0.5 ng/mL (Figure 7A). Similar to the in vitro studies, intraperitoneal LPS (0.1 mg/kg) injection did not have a significant effect on mouse intestinal permeability up to day 3, as assessed by mucosal-to-serosal flux of Texas Red–dextran 10 kDa, but caused an increase in intestinal permeability at day 5 (Figure 7B). LPS (0.1 mg/kg) treatment did not cause intestinal mucosal damage (Figure 7C) or changes in mouse behavior or body weight (data not shown). These data indicate that LPS, at clinically relevant plasma concentrations, causes an increase in mouse intestinal permeability without causing acute intestinal mucosal damage.
Table 1.
Effect of Increasing Concentration of i.p. LPS (0, 0.05, 0.1, 1.0, 10.0 mg/kg Body Weight) on Mouse Plasma LPS Level in Vivo
| LPS (mg/kg) | 0 hours (ng/mL) | 1 hour (ng/mL) | 24 hours (ng/mL) |
|---|---|---|---|
| 0.05 | 0.017 | 0.366 | 0.020 |
| 0.1 | 0.017 | 2.230 | 0.405 |
| 1 | 0.018 | 1327.1 | 20.732 |
| 10 | 0.018 | 13,491.0 | 252.427 |
Plasma LPS level was measured prior to i.p. LPS injection and 1 hour and 24 hours post-LPS injection (n = 3 to 4).
Figure 7.

Effect of i.p. LPS (0.1 mg/kg body weight) on mouse intestinal permeability and plasma TLR-4 level. A: Time-course effect of i.p. LPS on plasma LPS level in mice. B: Time-course effect of i.p. LPS on intestinal permeability. C: Effect of LPS administration on intestinal tissue: wild-type (WT); LPS 1 day treatment; LPS 5 days treatment. H&E stain, original magnification, ×200. D: Time-course effect of LPS on TLR-4 expression in mice. E: Effect of LPS on intestinal permeability in TLR-4−/− mice. *P < 0.001 versus WT control; **P < 0.001 versus WT LPS treatment.
Next, the involvement of TLR-4 in the LPS-induced increase in mouse intestinal permeability was examined. Intraperitoneal LPS (0.1 mg/kg) caused a time-dependent increase in intestinal tissue TLR-4 expression (Figure 7D). In the control mice, there was only minimal expression of TLR-4 in the intestinal tissue. LPS treatment did not affect TLR-4 expression up to day 3; however, by day 5, there was a marked increase in TLR-4 expression (Figure 7D). The time course of TLR-4 expression paralleled the increase in mouse intestinal permeability (Figure 7B). To determine the requirement of TLR-4 in the LPS-induced increase in mouse intestinal permeability, the LPS effect was examined in TLR-4 deficient mice (C57BL/6 Tlr4lps-d, the Jackson Laboratory). Intraperitoneal LPS did not cause an increase in intestinal permeability in TLR-4−/− mice (Figure 7E), indicating that TLR-4 was also required for the LPS-induced increase in mouse intestinal permeability.
LPS-Induced Increase in Mouse Intestinal TJ Permeability Is Mediated by a TLR-4–Dependent Up-Regulation and Membrane Recruitment of CD14
TLR-4 forms an aggregate receptor complex with membrane-associated proteins CD14 and myeloid differentiation protein 2 (MD2) and other adaptor proteins.43,44 CD14 appears to be the receptor protein involved in the binding of LPS/LPS binding protein complex.43,44 In the following studies, the LPS effect on expression and localization of TLR-4 and CD14 in Caco-2 monolayers was examined by immunostaining and imaging by confocal microscopy. In the control Caco-2 monolayers, there was only minimal TLR-4 and CD14 expression (Figure 8). LPS (0.3 ng/mL) treatment caused a time-dependent increase in TLR-4 expression, both in the cytoplasm and at the level of the plasma membrane; and by day 5, a marked increase in plasma membrane localization of TLR-4 was seen (Figure 8). Corresponding to the increase in TLR-4 expression and membrane localization, there was also an increase in cellular expression and membrane colocalization of CD14 by day 5 (Figure 8). The time course of the LPS-induced increase in CD14 expression was also confirmed by immunoblot analysis (Figure 9A). Next, the involvement of CD14 in the LPS-induced increase in Caco-2 TJ permeability was examined. The siRNA-induced knockdown of CD14 prevented the LPS-induced drop in Caco-2 TER (Figure 9B) and increase in paracellular permeability (Figure 9C), indicating that CD14 expression was also required for the LPS-induced increase in Caco-2 TJ permeability. In the following studies, the possible dependence of CD14 expression and membrane localization on TLR-4 expression was examined. The siRNA-induced knockdown of TLR-4 inhibited the LPS-induced increase in Caco-2 CD14 expression (Figure 9D) and membrane localization (Figure 8), suggesting that the increase in TLR-4 was required for the increase in CD14 expression.
Figure 8.

Effect of LPS on TLR-4 and CD14 localization in filter-grown Caco-2 monolayers as assessed by immunostaining. Original magnification, ×600.
Figure 9.

Effect of LPS on CD14 expression in filter-grown Caco-2 monolayers. A: Time-course effect of LPS on CD14 expression. B: Effect of siRNA CD14 transfection on Caco-2 TER. The mean TER for control Caco-2 monolayers was 482 ± 15 Ω • cm2. C: Effect of siRNA CD14 transfection on Caco-2 inulin flux. D: Effect of siRNA TLR-4 transfection on expression of CD14 in Caco-2 cells. Scr, scrambled. *P < 0.001 versus control; **P < 0.001 versus LPS treatment.
Next, the role of TLR-4/CD14 receptor complex in the LPS-induced increase in intestinal permeability was also determined in vivo. In the control mice, there was only a minimal expression of TLR-4 or CD14 in mouse enterocytes (Figure 10A). LPS administration (0.1 mg/kg i.p.) resulted in a marked increase in mouse enterocyte TLR-4 and CD14 expression, both in the cytoplasm and at the level of the plasma membrane (Figure 10A). LPS treatment did not cause an increase in enterocyte CD14 expression in TLR-4−/− mice. Additionally, LPS did not cause an increase in intestinal permeability in CD14−/− mice (C57BL/6 CD14−/−; the Jackson Laboratory) (Figure 10B). Together, these data suggest that the LPS-induced increase in enterocyte CD14 expression and membrane localization is dependent on TLR-4 expression and that CD14 expression is necessary for the LPS-induced increase in mouse intestinal permeability.
Figure 10.

Effect of LPS (0.1 mg/kg body weight) on TLR-4 and CD14 expression on mouse intestinal tissue in vivo. A: Effect of LPS on TLR-4 and CD14 localization on mouse intestinal tissue as assessed by immunostaining. Original magnification, ×600. B: Effect of LPS on intestinal permeability in CD14−/− mice. *P < 0.001 versus wild-type (WT) control; **P < 0.001 versus WT LPS treatment.
Discussion
A defective intestinal epithelial TJ barrier plays an important pathogenic role in intestinal inflammation by allowing increased intestinal permeation and systemic circulation of gut-derived bacterial antigens.1,2 Previous clinical studies have demonstrated a correlation between an increase in intestinal permeability and plasma LPS levels and an increase in disease activity in IBD.20–23 Although the cause-and-effect relationship between a defective intestinal TJ barrier and intestinal penetration of LPS and systemic endotoxemia is well established,11,13,16–20,22 the effects of clinically relevant concentrations of LPS on the intestinal barrier remain unknown. Because LPS levels are significantly elevated during physiological stresses and in intestinal permeability disorders,20,22,24–28 and because circulating LPS are an important determinant of inflammatory response and multiorgan failure,11–13,16–19,22 understanding the effects of clinically relevant concentrations of LPS on intestinal barrier function has important potential clinical significance. Herein, we show for the first time that physiologically and clinically relevant concentrations (0 to 10 ng/mL) of LPS cause an increase in intestinal epithelial TJ permeability in vitro and in vivo, when present in the interstitial fluid or basolateral membrane compartment. Consistent with the presence of high concentrations of bacteria and LPS in the gut lumen, LPS in the apical membrane surface did not affect the intestinal barrier function. These data suggest that the high concentrations of LPS present in the normal gut lumen do not affect intestinal epithelial barrier function,14,15,25 but that relatively low levels of LPS in the interstitial fluid, which are readily achievable during physiological stresses or in intestinal permeability disorders, lead to an increase in intestinal permeability.
In the healthy intestine, luminal LPS do not permeate the intact intestinal epithelial barrier to any significant extent.14,15 However, in intestinal permeability disorders, the defective TJ barrier allows paracellular permeation of LPS, leading to elevated levels of LPS in the intestinal tissue and in systemic circulation.11,16,17,20,22 LPS is a prototypical pathogen-associated molecular pattern (PAMP) and binds to a pattern recognition receptor (PRR).43,44 In the enterocytes, TLRs play an integral role in the recognition of PAMPs. Among the TLRs, TLR-4 is known to be the PRR for LPS.43,45 Our data indicate that the TLR-4 levels are low in quiescent Caco-2 cells and healthy mouse enterocytes. LPS produced a time-dependent increase in TLR-4 expression in Caco-2 cells, and by day 4 to 5, a marked increase in TLR-4 expression was observed. The time course of TLR-4 expression paralleled the increase in Caco-2 TJ permeability; targeted knockdown of TLR-4 expression or pharmacological inhibition of the TLR-4 signal transduction pathway prevented the LPS-induced increase in Caco-2 TJ permeability, demonstrating the requirement of TLR-4 and the activation of the signal transduction pathway in mediating the LPS-induced increase in Caco-2 TJ permeability. The possibility of TLR-2 or TLR-5 involvement was also considered, but LPS did not affect the expression of TLR-2 or TLR-5, and siRNA-induced silencing of TLR-2 or TLR-5 did not prevent the LPS-induced increase in intestinal TJ permeability, confirming that the LPS effect is regulated specifically by the TLR-4 signal transduction pathway. Consistent with our data, previous studies have also shown low or undetectable levels of TLR-4 in healthy human intestine and markedly elevated TLR-4 expression in patients with IBD or NEC.43,45 An important potential implication of these findings is that the increase in TLR-4 expression could be an important pathogenic mechanism contributing to the persistent increase in intestinal permeability and prolongation of intestinal inflammation in IBD and NEC.22
Sheth et al46 previously reported that a LPS concentration of 500 ng/mL causes an acute drop in NRC-1 cholangiocyte monolayer TER, with associated changes in junctional localization of TJ proteins [occludin and Zona occludens protein 1 (ZO-1)]. The LPS effect on NRC-1 TJ permeability also required a rapid increase (within minutes) in tyrosine phosphorylation of TJ proteins and myosin light chain kinase activation, suggesting that the LPS-induced increase in NRC-1 TJ permeability was mediated by an acute signaling process leading to an activation of tyrosine kinase and myosin light chain kinase.46 By contrast, in our studies, the lower concentration of LPS (0 to 20 ng/mL) did not cause an acute change in TJ permeability or TJ protein localization in intestinal monolayers, but did cause a delayed increase in intestinal TJ permeability (4 to 5 days), which was dependent on TLR-4 expression. In our preliminary studies, we also examined the possibility that NF-κB activation may play a role in LPS modulation of Caco-2 TJ permeability. These studies suggest that the LPS-induced increase in TLR-4 expression leads to an activation of NF-κB. Our initial studies also suggests that siRNA-induced knockdown of NF-κB and/or p65 prevents the LPS-induced increase in Caco-2 TJ permeability. These studies suggest the possibility that NF-κB may be an important down-stream mediator of LPS. An important focus of our future studies will be to validate the role of NF-κB and to delineate the molecular mechanisms by which LPS modulates the intestinal TJ barrier.
Previous studies have indicated that the membrane-associated protein CD14 forms a receptor complex with TLR-4 and MD2, and plays an important role in the recognition of LPS/LPS binding protein (LBP) complex and in facilitating the interaction with TLR-4.43,44 LBP is a lipid transfer protein that catalyzes the transfer of LPS monomer to a binding site on CD14 via a ternary complex of LBP-LPS-CD14, to form a monomeric LPS-CD14 complex. CD14 then transfers LPS to TLR-4/MD2.47–52 The LPS effect on biological activity may be CD14 dependent or independent.44,53 Our data suggest that CD14 also plays an integral role in the modulation of intestinal TJ barrier. Immunostaining studies have shown that the LPS-induced increase in plasma membrane TLR-4 expression and aggregation is associated with an increase in CD14 membrane expression and colocalization, consistent with the formation of the TLR-4 receptor cluster.43,44 The requirement of CD14 membrane localization in LPS modulation of the Caco-2 TJ barrier was confirmed by siRNA silencing studies, which showed that CD14 knockdown inhibited the LPS-induced increase in Caco-2 TJ permeability. These findings indicate that the plasma membrane expression and colocalization of CD14 and TLR-4 are prerequisites for the LPS modulation of the Caco-2 TJ barrier. Another novel finding related to the CD14/TLR-4 interaction in the modulation of Caco-2 TJ barrier is the dependence of enterocyte CD14 expression and membrane localization on TLR-4 expression. Our results show that CD14 expression and membrane localization are dependent on TLR-4 expression; and that targeted knockdown of TLR-4 prevents the LPS-induced increase in CD14 expression and membrane localization in Caco-2 cells and mouse enterocytes. Thus, our data suggest for the first time that TLR-4 is an important regulator of CD14 expression in enterocytes.
Previous studies have shown that in Caco-2 monolayers, high pharmacological concentrations of LPS (50 μg/mL to 1 mg/mL) cause rapid intestinal epithelial cell death (within 1 to 2 hours of LPS treatment), leading to a loss of epithelial integrity.30,31 Similarly, intraperitoneal injection of a high lethal dose (10 mg/kg) of LPS in mice and rats also resulted in rapid (within 2 to 3 hours) intestinal mucosal damage, with epithelial cell apoptosis and rapid loss of intestinal barrier function.32 However, these high LPS concentrations are not clinically achievable.33,34 At these high pharmacological concentrations, LPS-treated mice were very sick, and most died shortly after the treatment.32,54,55 Our in vitro studies indicate that LPS, at physiologically or clinically relevant concentrations (0 to 10 ng/mL), does not cause cell death, but leads to a selective increase in Caco-2 paracellular permeability. To assess the in vivo effects of LPS, we also developed an in vivo mouse model of LPS modulation of intestinal barrier function, which closely recapitulates the plasma LPS levels reported in the clinical studies.11–13,24–27 To establish the appropriate LPS dose to use in the in vivo studies, mice were injected i.p. with increasing concentrations of LPS. These studies indicate that a LPS dose of 0.1 mg/kg produces LPS plasma levels (peak level of 2.23 ng/mL and trough level of 0.41 ng/mL) that are achievable in clinical studies (Table 1). The LPS dose of 10 mg/kg, which had been used in previous animal studies,32 resulted in a very high plasma LPS level (peak level of 13.49 μg/mL and trough level of 252.4 ng/mL) that was not achievable clinically and led to rapid intestinal mucosal damage and apoptosis. LPS at the dose used (0.1 mg/kg) in this study did not cause any apparent illness or intestinal mucosal damage in the LPS-treated mice (data not shown), and produced a similar time-course effect on mouse intestinal permeability as the in vitro studies with Caco-2 monolayers. The increase in mouse intestinal permeability was associated with an increase in TLR-4 expression in mouse enterocytes; and the increase in intestinal permeability was inhibited in TLR-4−/− mice, indicating the requirement of TLR-4 in the LPS-induced increase in mouse intestinal permeability. Our in vivo studies also suggest that the increase in mouse intestinal permeability is due to a TLR-4–dependent increase in enterocyte CD14 expression and membrane colocalization. Together, these in vivo studies show that the LPS-induced increase in mouse intestinal permeability is mediated by an increase in enterocyte membrane expression of TLR-4, and TLR-4–dependent expression and colocalization of CD14.
In conclusion, LPS at physiologically relevant concentrations cause an increase in intestinal TJ permeability in vitro and in vivo, without inducing epithelial cell death or intestinal mucosal damage. The LPS effect on intestinal TJ permeability in vitro and in vivo is mediated by an increase in enterocyte expression and membrane localization of TLR-4, and TLR-4–dependent expression and colocalization of CD14. An important potential clinical implication of these studies is that LPS, at physiologically relevant concentrations, causes an increase in intestinal permeability and could play an important role in further deterioration and prolongation of intestinal TJ barrier defect in intestinal permeability disorders and inflammatory diseases of the gut. These data also show for the first time that TLR-4 signal transduction pathway plays an important role in intestinal barrier regulation and may be targeted therapeutically.
Acknowledgment
Images in this article were generated in the UNM & Cancer Center Fluorescence Microscopy Shared Resource.
Footnotes
Supported by a Veterans Affairs (VA) Merit Review grant from the VA Research Service and by National Institute of Diabetes and Digestive and Kidney Diseases grants RO 1-DK-64165–01 and RO 1-DK-081429 (T.Y.M.). The UNM & Cancer Center Fluorescence Microscopy Shared Resource is supported by NCIP30 CA118100 (C. Willman), NCRR1S10RR025540 (A. Wandinger-Ness), NIDDK3R301 DK050141 (A. Wandinger-Ness), and NIGMSP50 GM085273 (J. Oliver).
References
- 1.Turner J.R. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 2.Ma T.Y., Anderson J.M. Tight junctions and the intestinal barrier. Physiology of the Gastrointestinal Tract, vol. 1–2, ed 4, ch 61. In: Johnson L.R., Barrett K.E., Gishan F.K., Merchant J.L., Said H.M., Wood J.D., editors. Elsevier Academic Press; Burlington MA: 2006. pp. 1559–1594. [Google Scholar]
- 3.Arrieta M.C., Madsen K., Doyle J., Meddings J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–48. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berman L., Moss R.L. Necrotizing enterocolitis: an update. Semin Fetal Neonatal Med. 2011;16:145–150. doi: 10.1016/j.siny.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Clark J.A., Doelle S.M., Halpern M.D., Saunders T.A., Holubec H., Dvorak K., Boitano S.A., Dvorak B. Intestinal barrier failure during experimental necrotizing enterocolitis: protective effect of EGF treatment. Am J Physiol Gastrointest Liver Physiol. 2006;291:G938–G949. doi: 10.1152/ajpgi.00090.2006. [DOI] [PubMed] [Google Scholar]
- 6.Wyatt J., Vogelsang H., Hubl W., Waldhoer T., Lochs H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet. 1993;341:1437–1439. doi: 10.1016/0140-6736(93)90882-h. [DOI] [PubMed] [Google Scholar]
- 7.Wild G.E., Waschke K.A., Bitton A., Thomson A.B. The mechanisms of prednisone inhibition of inflammation in Crohn’s disease involve changes in intestinal permeability, mucosal TNFalpha production and nuclear factor kappa B expression. Aliment Pharmacol Ther. 2003;18:309–317. doi: 10.1046/j.1365-2036.2003.01611.x. [DOI] [PubMed] [Google Scholar]
- 8.Hermiston M.L., Gordon J.I. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science. 1995;270:1203–1207. doi: 10.1126/science.270.5239.1203. [DOI] [PubMed] [Google Scholar]
- 9.Vetrano S., Rescigno M., Cera M.R., Correale C., Rumio C., Doni A., Fantini M., Sturm A., Borroni E., Repici A., Locati M., Malesci A., Dejana E., Danese S. Unique role of junctional adhesion molecule-A in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology. 2008;135:173–184. doi: 10.1053/j.gastro.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Mennigen R., Nolte K., Rijcken E., Utech M., Loeffler B., Senninger N., Bruewer M. Probiotic mixture VSL#3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1140–G1149. doi: 10.1152/ajpgi.90534.2008. [DOI] [PubMed] [Google Scholar]
- 11.Hurley J.C. Endotoxemia: methods of detection and clinical correlates. Clin Microbiol Rev. 1995;8:268–292. doi: 10.1128/cmr.8.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andreasen A.S., Krabbe K.S., Krogh-Madsen R., Taudorf S., Pedersen B.K., Moller K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem. 2008;15:1697–1705. doi: 10.2174/092986708784872393. [DOI] [PubMed] [Google Scholar]
- 13.Marshall J.C., Walker P.M., Foster D.M., Harris D., Ribeiro M., Paice J., Romaschin A.D., Derzko A.N. Measurement of endotoxin activity in critically ill patients using whole blood neutrophil dependent chemiluminescence. Crit Care. 2002;6:342–348. doi: 10.1186/cc1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benoit R., Rowe S., Watkins S.C., Boyle P., Garrett M., Alber S., Wiener J., Rowe M.I., Ford H.R. Pure endotoxin does not pass across the intestinal epithelium in vitro. Shock. 1998;10:43–48. doi: 10.1097/00024382-199807000-00008. [DOI] [PubMed] [Google Scholar]
- 15.Ge Y., Ezzell R.M., Warren H.S. Localization of endotoxin in the rat intestinal epithelium. J Infect Dis. 2000;182:873–881. doi: 10.1086/315784. [DOI] [PubMed] [Google Scholar]
- 16.Ammori B.J., Leeder P.C., King R.F., Barclay G.R., Martin I.G., Larvin M., McMahon M.J. Early increase in intestinal permeability in patients with severe acute pancreatitis: correlation with endotoxemia, organ failure, and mortality. J Gastrointest Surg. 1999;3:252–262. doi: 10.1016/s1091-255x(99)80067-5. [DOI] [PubMed] [Google Scholar]
- 17.Keshavarzian A., Holmes E.W., Patel M., Iber F., Fields J.Z., Pethkar S. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. Am J Gastroenterol. 1999;94:200–207. doi: 10.1111/j.1572-0241.1999.00797.x. [DOI] [PubMed] [Google Scholar]
- 18.Lambert G.P. Stress-induced gastrointestinal barrier dysfunction and its inflammatory effects. J Anim Sci. 2009;87:E101–E108. doi: 10.2527/jas.2008-1339. [DOI] [PubMed] [Google Scholar]
- 19.Lambert G.P. Intestinal barrier dysfunction, endotoxemia, and gastrointestinal symptoms: the ‘canary in the coal mine’ during exercise-heat stress? Med Sport Sci. 2008;53:61–73. doi: 10.1159/000151550. [DOI] [PubMed] [Google Scholar]
- 20.Wellmann W., Fink P.C., Benner F., Schmidt F.W. Endotoxaemia in active Crohn’s disease. Treatment with whole gut irrigation and 5-aminosalicylic acid. Gut. 1986;27:814–820. doi: 10.1136/gut.27.7.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gardiner K.R., Halliday M.I., Barclay G.R., Milne L., Brown D., Stephens S., Maxwell R.J., Rowlands B.J. Significance of systemic endotoxaemia in inflammatory bowel disease. Gut. 1995;36:897–901. doi: 10.1136/gut.36.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma R., Tepas J.J., 3rd, Hudak M.L., Mollitt D.L., Wludyka P.S., Teng R.J., Premachandra B.R. Neonatal gut barrier and multiple organ failure: role of endotoxin and proinflammatory cytokines in sepsis and necrotizing enterocolitis. J Pediatr Surg. 2007;42:454–461. doi: 10.1016/j.jpedsurg.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 23.Pastor Rojo O., Lopez San Roman A., Albeniz Arbizu E., de la Hera Martinez A., Ripoll Sevillano E., Albillos Martinez A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:269–277. doi: 10.1002/ibd.20019. [DOI] [PubMed] [Google Scholar]
- 24.Brock-Utne J.G., Gaffin S.L., Wells M.T., Gathiram P., Sohar E., James M.F., Morrell D.F., Norman R.J. Endotoxaemia in exhausted runners after a long-distance race. S Afr Med J. 1988;73:533–536. [PubMed] [Google Scholar]
- 25.Amar J., Burcelin R., Ruidavets J.B., Cani P.D., Fauvel J., Alessi M.C., Chamontin B., Ferrieres J. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr. 2008;87:1219–1223. doi: 10.1093/ajcn/87.5.1219. [DOI] [PubMed] [Google Scholar]
- 26.Lira F.S., Rosa J.C., Pimentel G.D., Souza H.A., Caperuto E.C., Carnevali L.C., Jr., Seelaender M., Damaso A.R., Oyama L.M., de Mello M.T., Santos R.V. Endotoxin levels correlate positively with a sedentary lifestyle and negatively with highly trained subjects. Lipids Health Dis. 2010;9:82. doi: 10.1186/1476-511X-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bosenberg A.T., Brock-Utne J.G., Gaffin S.L., Wells M.T., Blake G.T. Strenuous exercise causes systemic endotoxemia. J Appl Physiol. 1988;65:106–108. doi: 10.1152/jappl.1988.65.1.106. [DOI] [PubMed] [Google Scholar]
- 28.Liu H., Li W., Wang X., Li J., Yu W. Early gut mucosal dysfunction in patients with acute pancreatitis. Pancreas. 2008;36:192–196. doi: 10.1097/MPA.0b013e31815a399f. [DOI] [PubMed] [Google Scholar]
- 29.Yagi S., Takaki A., Hori T., Sugimachi K. Enteric lipopolysaccharide raises plasma IL-6 levels in the hepatoportal vein during non-inflammatory stress in the rat. Fukuoka Igaku Zasshi. 2002;93:38–51. [PubMed] [Google Scholar]
- 30.Yu L.C., Flynn A.N., Turner J.R., Buret A.G. SGLT-1-mediated glucose uptake protects intestinal epithelial cells against LPS-induced apoptosis and barrier defects: a novel cellular rescue mechanism? FASEB J. 2005;19:1822–1835. doi: 10.1096/fj.05-4226com. [DOI] [PubMed] [Google Scholar]
- 31.Forsythe R.M., Xu D.Z., Lu Q., Deitch E.A. Lipopolysaccharide-induced enterocyte-derived nitric oxide induces intestinal monolayer permeability in an autocrine fashion. Shock. 2002;17:180–184. doi: 10.1097/00024382-200203000-00004. [DOI] [PubMed] [Google Scholar]
- 32.Liu C., Li A., Weng Y.B., Duan M.L., Wang B.E., Zhang S.W. Changes in intestinal mucosal immune barrier in rats with endotoxemia. World J Gastroenterol. 2009;15:5843–5850. doi: 10.3748/wjg.15.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirotani Y., Ikeda K., Kato R., Myotoku M., Umeda T., Ijiri Y., Tanaka K. Protective effects of lactoferrin against intestinal mucosal damage induced by lipopolysaccharide in human intestinal Caco-2 cells. Yakugaku Zasshi. 2008;128:1363–1368. doi: 10.1248/yakushi.128.1363. [DOI] [PubMed] [Google Scholar]
- 34.Kim I.D., Ha B.J. Paeoniflorin protects RAW 264.7 macrophages from LPS-induced cytotoxicity and genotoxicity. Toxicol In Vitro. 2009;23:1014–1019. doi: 10.1016/j.tiv.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 35.Vogel S.N., Marshall S.T., Rosenstreich D.L. Analysis of the effects of lipopolysaccharide on macrophages: differential phagocytic responses of C3H/HeN and C3H/HeJ macrophages in vitro. Infect Immun. 1979;25:328–336. doi: 10.1128/iai.25.1.328-336.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye D., Guo S., Al-Sadi R., Ma T.Y. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology. 2011;141:1323–1333. doi: 10.1053/j.gastro.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma T.Y., Iwamoto G.K., Hoa N.T., Akotia V., Pedram A., Boivin M.A., Said H.M. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 38.Said H.M., Ortiz A., Moyer M.P., Yanagawa N. Riboflavin uptake by human-derived colonic epithelial NCM460 cells. Am J Physiol Cell Physiol. 2000;278:C270–C276. doi: 10.1152/ajpcell.2000.278.2.C270. [DOI] [PubMed] [Google Scholar]
- 39.Boivin M.A., Roy P.K., Bradley A., Kennedy J.C., Rihani T., Ma T.Y. Mechanism of interferon-gamma-induced increase in T84 intestinal epithelial tight junction. J Interferon Cytokine Res. 2009;29:45–54. doi: 10.1089/jir.2008.0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Al-Sadi R., Khatib K., Guo S., Ye D., Youssef M., Ma T. Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2011;300:G1054–G1064. doi: 10.1152/ajpgi.00055.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clayburgh D.R., Barrett T.A., Tang Y., Meddings J.B., Van Eldik L.J., Watterson D.M., Clarke L.L., Mrsny R.J., Turner J.R. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–2715. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsunaga N., Tsuchimori N., Matsumoto T., Ii M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol. 2011;79:34–41. doi: 10.1124/mol.110.068064. [DOI] [PubMed] [Google Scholar]
- 43.Abreu M.T. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;10:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 44.Triantafilou M., Triantafilou K. The dynamics of LPS recognition: complex orchestration of multiple receptors. J Endotoxin Res. 2005;11:5–11. doi: 10.1179/096805105225006641. [DOI] [PubMed] [Google Scholar]
- 45.Abreu M.T., Fukata M., Arditi M. TLR signaling in the gut in health and disease. J Immunol. 2005;174:4453–4460. doi: 10.4049/jimmunol.174.8.4453. [DOI] [PubMed] [Google Scholar]
- 46.Sheth P., Delos Santos N., Seth A., LaRusso N.F., Rao R.K. Lipopolysaccharide disrupts tight junctions in cholangiocyte monolayers by a c-Src-. TLR4-, and LBP-dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2007;293:G308–G318. doi: 10.1152/ajpgi.00582.2006. [DOI] [PubMed] [Google Scholar]
- 47.Brito B.E., Zamora D.O., Bonnah R.A., Pan Y., Planck S.R., Rosenbaum J.T. Toll-like receptor 4 and CD14 expression in human ciliary body and TLR-4 in human iris endothelial cells. Exp Eye Res. 2004;79:203–208. doi: 10.1016/j.exer.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 48.Schroder N.W., Opitz B., Lamping N., Michelsen K.S., Zahringer U., Gobel U.B., Schumann R.R. Involvement of lipopolysaccharide binding protein. CD14, and Toll-like receptors in the initiation of innate immune responses by Treponema glycolipids. J Immunol. 2000;165:2683–2693. doi: 10.4049/jimmunol.165.5.2683. [DOI] [PubMed] [Google Scholar]
- 49.Plotz S.G., Lentschat A., Behrendt H., Plotz W., Hamann L., Ring J., Rietschel E.T., Flad H.D., Ulmer A.J. The interaction of human peripheral blood eosinophils with bacterial lipopolysaccharide is CD14 dependent. Blood. 2001;97:235–241. doi: 10.1182/blood.v97.1.235. [DOI] [PubMed] [Google Scholar]
- 50.Asai Y., Hashimoto M., Ogawa T. Treponemal glycoconjugate inhibits Toll-like receptor ligand-induced cell activation by blocking LPS-binding protein and CD14 functions. Eur J Immunol. 2003;33:3196–3204. doi: 10.1002/eji.200324219. [DOI] [PubMed] [Google Scholar]
- 51.Schroder N.W., Heine H., Alexander C., Manukyan M., Eckert J., Hamann L., Gobel U.B., Schumann R.R. Lipopolysaccharide binding protein binds to triacylated and diacylated lipopeptides and mediates innate immune responses. J Immunol. 2004;173:2683–2691. doi: 10.4049/jimmunol.173.4.2683. [DOI] [PubMed] [Google Scholar]
- 52.Lizundia R., Sauter K.S., Taylor G., Werling D. Host species-specific usage of the TLR4-LPS receptor complex. Innate Immun. 2008;14:223–231. doi: 10.1177/1753425908095957. [DOI] [PubMed] [Google Scholar]
- 53.Hritz I., Mandrekar P., Velayudham A., Catalano D., Dolganiuc A., Kodys K., Kurt-Jones E., Szabo G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. 2008;48:1224–1231. doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Radnai B., Tucsek Z., Bognar Z., Antus C., Mark L., Berente Z., Gallyas F., Jr., Sumegi B., Veres B. Ferulaldehyde, a water-soluble degradation product of polyphenols, inhibits the lipopolysaccharide-induced inflammatory response in mice. J Nutr. 2009;139:291–297. doi: 10.3945/jn.108.097386. [DOI] [PubMed] [Google Scholar]
- 55.Aoshiba K., Onizawa S., Tsuji T., Nagai A. Therapeutic effects of erythropoietin in murine models of endotoxin shock. Crit Care Med. 2009;37:889–898. doi: 10.1097/CCM.0b013e31819b8371. [DOI] [PubMed] [Google Scholar]