

Abstract
We have investigated the folding dynamics of Thermus thermophilus cytochrome c552 by time-resolved fluorescence energy transfer between the heme and each of seven site-specific fluorescent probes. We have found both an equilibrium unfolding intermediate and a distinct refolding intermediate from kinetics studies. Depending on the protein region monitored, we observed either two-state or three-state denaturation transitions. The unfolding intermediate associated with three-state folding exhibited native contacts in β-sheet and C-terminal helix regions. We probed the formation of a refolding intermediate by time-resolved fluorescence energy transfer between residue 110 and the heme using a continuous flow mixer. The intermediate ensemble, a heterogeneous mixture of compact and extended polypeptides, forms in a millisecond, substantially slower than the ∼100-μs formation of a burst-phase intermediate in cytochrome c. The surprising finding is that, unlike for cytochrome c, there is an observable folding intermediate, but no microsecond burst phase in the folding kinetics of the structurally related thermostable protein.
Keywords: FRET, microfluidic mixing, protein refolding
A folding intermediate can be Jekyll or Hyde: it can be good (“on-pathway”), seeking out native structure by avoiding traps in the folding energy landscape, or bad, being misfolded in ways that lead to disease states (1–6). Among the more powerful methods that have been developed to probe these landscapes, time-resolved fluorescence energy transfer (trFET) deserves special mention, as analyses of such experiments have shed new light both on the structures and dynamics of intermediates along folding pathways (7–13).
We report here trFET studies of the folding energetics and dynamics of cytochrome c552 (c552), a small, thermally stable protein containing a single heme covalently bound via two cysteines and axially ligated by His15 and Met69 (14–16); c552 is the electron donor to cytochrome ba3 oxidase during the terminal step of respiration in the hyperthermophile Thermus thermophilus (17). Although c552 is a class I c-type cytochrome in which helices αI and αIV cross each other (Fig. 1A), it has distinct structural components, notably a β-sheet around the heme propionate and extra helices in the C-terminal region (Fig. 1A). Of interest is that Brunori and coworkers, who examined the c552 folding kinetics by native tryptophan (Trp91) fluorescence, found an on-pathway intermediate (18); in this work, Trp91 probed structures near the crossed helices of c552, but did not report on the heme β-sheet or the C-terminal helices.
Fig. 1.

(A) Crystal structure of WT c552 obtained at pH 5.44 (PDB ID code 3VNW). Dns-labeled sites are shown as spheres: group I (cyan) and group II (yellow). Helix αVI and β-sheets I and II are distinct structural features of c552 that exhibit group II folding behavior (green). For reference, Trp91 is also highlighted in cyan. (B) Dns-modified cysteine. (C) Red dashed lines indicate electrostatic interactions in the region of distinctive structures of c552.
Our trFET experiments have shed light on site-specific protein conformational changes that occur upon increasing denaturant concentrations in c552 solutions. Dansyl (Dns) probes at seven different residues in the protein reveal the existence of a loosely packed unfolding intermediate structure. Two sites associated with the distinct structural features of this class I cytochrome were also shown to be more resistant to unfolding than the rest of the protein (Fig. 1A). Moreover, the use of a continuous-flow mixer with a 150-μs dead time enabled us to study the formation kinetics of a refolding intermediate. Overall, the refolding steps in c552 are substantially slower than those of mitochondrial c-type cytochromes, a finding that presents a challenge to theoretical models that account for contact order (19) as well as hydrophobic and electrostatic forces (20).
Results and Discussion
Cytochrome c552 Structure and Variants.
We expressed T. thermophilus c552 in Escherichia coli and determined its crystal structure [Fig. 1A, Protein Data Bank (PDB) ID code 3VNW]. Amino acids at positions 1 and 2 were eliminated; Asp3 is the N-terminal residue. A few of the hydrogen bonds around the heme crevice differ from those reported previously (PDB ID codes 1C52 and 1DT1) (16, 21); however, the overall fold is virtually identical to those of structures 1C52 and 1DT1 (Fig. 1A). Our crystal was obtained at pH 5.44, which is substantially lower than reported for related structures: pH 7.6 for 1C52; pH 6.3 for 1DT1. We will refer to our c552 as wild type (WT).
We selected Dns as the fluorescence donor to monitor trFET. Solvent-exposed residues that form no intramolecular bonds were chosen to minimize structural perturbations upon labeling. Seven single-mutation variants of c552 were prepared; in each mutant a residue (Lys6, Glu40, Glu59, Lys76, Lys95, Lys110, or Lys126) was mutated to cysteine and further modified by covalent Dns attachment (Fig. 1 A and B).
Circular dichroism (CD) spectra of all Dns variants were virtually identical to that of WT (Fig. S1), indicating that the native secondary structure was preserved in the Dns-labeled proteins. Denaturation curves of the Dns variants, obtained from Dns fluorescence, UV-visible heme absorption, and CD measurements, showed midpoints ([Gdn]1/2) close to 4 M guanidine hydrochloride (Gdn), consistent with that of WT (Table 1 and Fig. S2). Standard denaturation parameters m and −ΔGw (22), determined from these curves, also showed no substantial deviation from those of WT (Table 1). We conclude from these results that Dns labeling does not perturb the stability or secondary structure of any of the variants.
Table 1.
Denaturation parameters for Dns c552 variants
| Fluorescence* |
Circular dichroism§ |
Absorption¶ |
|||||||
| Variant | [Gdn]1/2 (M) | M (kJ/mol/M) | −ΔGw (kJ/mol) | [Gdn]1/2 (M) | M (kJ/mol/M) | −ΔGw (kJ/mol) | [Gdn]1/2 (M) | M (kJ/mol/M) | −ΔGw (kJ/mol) |
| WT | 4.1 ± 0.02† | 11.9 ± 0.8† | 49 ± 4† | 4.1 ± 0.03 | 13.2 ± 1.6 | 54 ± 7 | 4.3 ± 0.02 | 11.9 ± 0.8 | 49 ± 4 |
| Dns6 | n.d.‡ | n.d.‡ | n.d.‡ | 4.0 ± 0.06 | 9.3 ± 1.8 | 37 ± 8 | 4.3 ± 0.02 | 7.1 ± 0.3 | 31 ± 1 |
| Dns40 | 4.3 ± 0.05 | 9.1 ± 1.2 | 39 ± 6 | 4.1 ± 0.04 | 7.6 ± 0.8 | 31 ± 4 | 4.2 ± 0.05 | 6.4 ± 0.6 | 27 ± 3 |
| Dns59 | 4.1 ± 0.05 | 7.9 ± 1.1 | 32 ± 5 | 3.9 ± 0.05 | 11.1 ± 1.9 | 43 ± 8 | 4.3 ± 0.06 | 5.8 ± 0.5 | 25 ± 3 |
| Dns76 | 4.4 ± 0.02 | 11.0 ± 0.7 | 48 ± 3 | 4.3 ± 0.05 | 11.0 ± 2.0 | 47 ± 9 | 4.4 ± 0.03 | 8.6 ± 0.7 | 38 ± 3 |
| Dns95 | 4.2 ± 0.02 | 12.2 ± 0.9 | 51 ± 4 | 4.1 ± 0.04 | 9.6 ± 1.1 | 39 ± 5 | 4.2 ± 0.03 | 9.7 ± 0.8 | 41 ± 4 |
| Dns110 | 4.2 ± 0.02 | 12.8 ± 1.5 | 51 ± 6 | 3.9 ± 0.03 | 13.2 ± 2.0 | 51 ± 8 | 4.1 ± 0.03 | 7.7 ± 0.6 | 32 ± 3 |
| Dns126 | 4.0 ± 0.01 | 12.0 ± 0.5 | 48 ± 2 | 3.9 ± 0.04 | 10.7 ± 1.5 | 42 ± 6 | 4.2 ± 0.03 | 7.7 ± 0.6 | 32 ± 3 |
Measured at 25 °C in pH 3.0 citrate buffer (10 mM for circular dichroism and 100 mM for fluorescence and heme absorption).
*Steady-state measurements of Dns fluorescence intensities. Excitation and emission wavelength are 355 and 513 nm, respectively.
†Data of WT were taken from Trp91 fluorescence. Excitation and emission wavelength are 290 and 360 nm, respectively.
‡Not determined (n.d.) because of negligible difference between native and denatured states.
§Ellipticity measurements at 222 nm.
¶Heme absorbance measurements at 391 nm.
Equilibrium Unfolding.
We used trFET to acquire site-specific D–A (Dns–heme) distance distributions [P(rDA)] upon denaturation with Gdn as a method to detect unfolding intermediates. A Tikhonov regularization (TR) method was used to extract distance distributions (Fig. 2) from the fluorescence decays shown in Fig. S3. Measurements were performed at pH 3 to prevent misligation of the heme by histidine residues or the N-terminal amino group. In most cases, Dns fluorescence decays could not be fit to single-exponential functions as rDA components exist with various probabilities, consistent with the presence of multiple conformations.
Fig. 2.

Gdn-induced changes in the Dns–heme distance distributions [P(rDA)] for Dns c552 variants at pH 3.0, extracted from TR fitting of fluorescence decay curves (Fig. S3): Gdn 0 M (black), 3 M (red), 3.5 M (yellow), 4 M (green), 4.5 M (cyan), 5 M (blue), and 7 M (magenta). The area of each bar reflects the probability amplitude over the corresponding distance range. Group I and group II variants display three- and two-state folding transitions, respectively.
At 0 M Gdn, rDA for all Dns variants obtained from TR fitting are consistent with the WT crystal structure (Fig. 2 and Table S1). The most probable D–A distances extracted from the fluorescence decays (rmode) are slightly longer (1–4 Å) than the Cγ–Fe distances in the crystal structure, likely owing to the length of linker between the Dns fluorophore and the Cys sulfur atom.
Denaturation with Gdn slowed the fluorescence decay in all Dns variants (Fig. S3), consistent with an increase in the average distance between D and A. Extended populations were observed in all but one of the Dns variants upon the addition of Gdn (Fig. 2). Because Dns6 is located close to the heme and thus exhibited only a modest broadening of the D-A distribution when denatured (Fig. S4), we will not consider it further.
We defined three categories of structural conformations from the distance distributions: native/compact (C: 18 ≤ rDA ≤ 22 Å), intermediate (I: 25 ≤ rDA ≤ 35 Å), extended (E: 35 Å ≤ rDA), and we plotted their relative populations as functions of [Gdn] (Fig. 3). Population of E conformations in all variants occurs at Gdn concentrations greater than 4 M (Fig. 3), which is consistent with the denaturation curves determined using absorption, CD, and steady-state fluorescence spectroscopic methods (Fig. S2). Interestingly, the Dns variants can be divided into two groups that differ in how C, I, and E populations change with Gdn concentration; Dns40, 76, 95, and 110 are in group I, and Dns59 and 126 are in group II (Figs. 2 and 3).
Fig. 3.
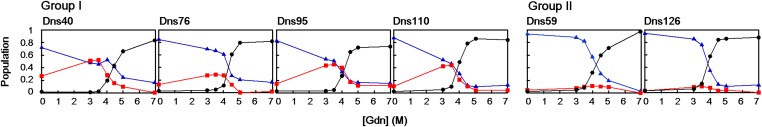
Population changes of structural conformations as a function of the Gdn concentration: native/compact (N), blue triangle; intermediate (I), red square; and extended (E), black circle. Group I and group II variants display three- and two-state folding transitions, respectively.
C populations in group I transform into I components when denatured by 3–4 M Gdn, which then disappear at higher concentrations (≥ 5 M, Fig. 3). The findings for group I variants are consistent with a three-state transition among the C, I, and E populations. The appearance and disappearance of I structures indicate that they arise from an unfolding intermediate state and are not simply constituents of the denatured protein ensemble. The I state has a folding stability similar to that of the native structure as I appears at relatively low [Gdn] in the presence of C (Fig. 2). Absorption spectroscopy indicates that the heme environment in the intermediate is structurally similar to the native state; the heme Soret peak maxima are comparable at 0 and 3 M Gdn, indicating that the heme is low-spin with intact, native Fe–S(Met) ligation (Fig. S5). It is possible that I corresponds to a local minimum on the native protein folding energy landscape. In the absence of denaturant, its population is too small to be detected, but the introduction of a low concentration of denaturant shifts the equilibrium to populate this extended conformation rather than the compact native structure.
With fluorescent probes at Dns59 and Dns126, we found no evidence for the existence of I components during Gdn denaturation; these two derivatives form group II (Fig. 3). Extended structures appear in both groups around 4 M. If we exclude the possibility that mutation and labeling at residues 59 and 126 have altered the unfolding pathway, then we can conclude that the unfolding intermediate formed at low Gdn concentrations involves little structural rearrangement from the native structure in the vicinity of Dns59 and Dns129 with slight expansion in the rest of the protein.
The two group II residues are located in the distinct protein structural region that differentiates c552 from other class I c-type cytochromes (position 59 on β-sheet βI and 126 on the extra helix αVI). The extra helix αVI and the β-sheet βI of c552 form a network of interactions (via residues 59, 125, and 131) linking them to the heme propionate (Fig. 1C). The WT crystal structure reveals that the amide nitrogen and carbonyl oxygen of Glu59 can hydrogen bond with the Lys131 carbonyl oxygen and the Arg125 side chain, respectively. Additionally, the Arg125 side chain forms salt bridges with the heme propionate and the carbonyl oxygen of Lys131. It is likely that these interactions stabilize this region of the polypeptide chain near the heme, making it more resistant to denaturation than the parts of the protein containing group I residues. By way of contrast, the other distinct helix in c552 (αV) does not exhibit enhanced stability, insofar as the Dns110 label, located in αV, displays a three-state folding transition (Figs. 2 and 3).
In the original report of the T. thermophilus c552 X-ray crystal structure, it was proposed that extra structural elements in the protein were responsible for its stability at high temperatures (16). The inherent stability of the c552 fold is reflected by the finding that the apoprotein retains the same secondary structure as the holoprotein (23). In marked contrast, the apoprotein of mitochondrial c is unstructured; in this case, the heme, which acts as a hydrophobic nucleation site, is needed to stabilize the native fold (24). Our c552 unfolding data confirm that the extra C-terminal helices and β-sheet enhance the stability of the native structure.
Refolding Kinetics.
To monitor the kinetics of c552 refolding, we selected the Dns110 derivative (located on helix αV) as representative of the three-state transition folding variants from group I. We triggered refolding using a continuous-flow mixer (11) to dilute denatured protein ([Gdn]= 6 M) with folding buffer ([Gdn]= 0 M) on a submillisecond timescale ([Gdn]final= 1 M) and monitored the reaction progress using trFET. The Dns110 fluorescence decay rate increases as the protein folds, indicating more efficient energy transfer to the heme (Fig. 4A). Very little compaction of the protein occurs on timescales shorter than 200 μs. The distance distributions [P(rDA), Fig. 4C] reveal I and C components developing in the first millisecond of folding. Populations of C gradually increase with time, but some E and I components still remain at 3.8 ms, the longest measurable reaction time.
Fig. 4.

Folding kinetics of Dns110–c552 triggered by Gdn jump from 6 to 1 M in a continuous-flow mixer (pH 3.0, ambient temperature). (A) Fluorescence decay curve (7 of the 23 measured decays are displayed): 0 ms (unfolded in 6 M Gdn, red), 0.2 ms (magenta), 0.6 ms (yellow), 1.0 ms (green), 2.0 ms (cyan), 3.4 ms (blue), and >30 min (folded state in 1 M Gdn, black) after the initiation of folding reaction. (B) Integrated Dns fluorescence intensity (M0) as a function of folding time. The solid line is a monoexponential fit with a rate constant of 1,230 s−1. (C) Distributions of P(rDA) for refolding extracted from TR fitting of the fluorescence decay curve (A). For clarity, only 7 of the 23 observed decays are displayed: unfolded in 6 M Gdn (red), 0.2 (magenta), 0.6 (yellow), 1.0 (green), 2.0 (cyan), 3.4 ms (blue), and folded in 1 M Gdn (black).
The kinetics of P(rDA) evolution were evaluated by moment (M) analysis (Materials and Methods). The time course of the normalized integrated fluorescence intensity (M0) can be fit to a monoexponential function (kobs = 1,230 s−1, Fig. 4B). For a two-state process, the time course of the first (M1) and second (M2) moments will be exponential, with a rate constant corresponding to kobs for the reaction (12); we find kobs of 1,060 and 1,140 s−1 from the time courses of M1 and M2, respectively (Fig. 5 A and B). The time course of the variance (V) in a two-state transition is biphasic, with a growth rate constant equal to 2kobs and a decay equal to kobs (12); we find that V agrees with this model (kobs = 1,100 s−1, Fig. 5C). These results indicate that Dns110 exhibits a two-state transition with a rate constant of 1,100 s−1 from the unfolded conformations.
Fig. 5.

Folding kinetics of Dns110-c552. (A) Mean distance between Dns and heme (M1) as a function of folding time. The solid line is a monoexponential fit with rate constant 1,060 s−1. This fit extrapolates to 31.5 Å, in contrast with M1 for the folded protein of 23 Å. (B) Time course of the second moment (M2). The solid line is a monoexponential fit with rate constant 1,140 s−1. This fit extrapolates to 1,200 Å2, in contrast with M2 for the folded protein of 560 Å2. (C) Time course of the variance (V). The solid line is a double-exponential fit with rate constants of 2,070 and 1,100 s−1. This fit extrapolates to 200 Å2, in contrast with V for the folded protein of 31 Å2.
The millisecond folding process does not appear to produce the native state, as roughly one-fourth (24%) of the normalized integrated fluorescence intensity persists at the end of this kinetics phase (Fig. 4B). Because the extrapolated endpoints to the fits of the moments (M1 = 31.5 Å, M2 = 1,200 Å2, and V = 200 Å2) are not in accord with values for the native protein (M1 = 23 Å, M2 = 560 Å2, and V = 31 Å2, Fig. 5), the millisecond process must correspond to formation of an intermediate folding ensemble. Notably, this ensemble has a greater population of extended components (3.4 ms, Fig. 4C) than that observed under equilibrium unfolding conditions. The early appearance of structures with 30–35 Å Dns110–heme distances suggests partial collapse involving the heme and helix αV, but the scarcity of 20-Å structures indicates little native-state formation.
Concluding Remarks.
We have found evidence for distinct intermediates on the folding energy landscape of c552. A kinetic refolding intermediate forms rapidly and partially comprises extended structures, whereas an equilibrium unfolding intermediate is structurally and energetically similar to the native state. The equilibrium unfolding intermediate, which contains native heme ligation, is not observed during refolding and thus likely occurs after the rate-determining step.
Brunori found evidence for an on-pathway folding intermediate in c552 using native Trp91 fluorescence as a probe (18). The difference between the previously reported rate constant for intermediate formation (kUI = 400 s−1, [Gdn] = 1 M) and ours can be rationalized by the differing pH conditions and temperatures used (pH 2.1, 10 °C versus pH 3.0, 18 °C). The final step in c552 folding, the transition from intermediate to native structure, was reported to have a 1-s time constant ([Gdn] = 1 M) (18). The native protein contains five proline residues, all in the more stable trans configuration in the crystal structure. Brunori and coworkers observed a third refolding phase (k ∼ 10−2 s−1) which, on the basis of interrupted unfolding experiments, they attributed to proline isomerization steps (18). Hence, the early folding intermediate that we and Brunori have observed is not likely due to the presence of cis-Xaa-Pro traps.
Although the folding intermediate was proposed to be a single compact structure in which 60% of the exposed surface in the unfolded protein had been buried (18), our trFET data suggest instead that it is a heterogeneous ensemble of compact and extended structures, and moment analysis of the Dns110 folding kinetics indicates that this ensemble has developed about 75% of native structure (Fig. 4B). For comparison, the burst-phase ensemble of yeast c contains a comparable amount of native character, 60–75%, depending on residue (7).
The remarkable difference between the two proteins is that in yeast c this structure develops in less than 150 μs (25), whereas comparable structure development in c552 is almost 10 times slower. The sluggish refolding of c552 carries over into the final phase as well, as a full second is required for formation of the native state; in yeast c, the folded state develops in tens to hundreds of milliseconds in the absence of misligation (7).
Analogous features in yeast c behave similarly under equilibrium unfolding conditions. In our work on yeast c, I components were observed at moderate-to-high Gdn concentrations (2.7–5.9 M Gdn) for Dns39 and 50 near the heme propionate site and for Dns66 near the Met80 axial ligand site (8), indicating that compact structure is preserved in the heme region when unfolded. Our work on c552 reveals that heme region variants (Dns59 and 126) do not develop I or E populations at mild Gdn concentrations where intermediate structures are found in other regions of the protein (3–4 M Gdn, Figs. 2 and 3). We conclude that β-sheet βI (Dns59) and helix αVI (Dns126) are substantially more resistant to denaturant-induced unfolding.
Materials and Methods
Protein Preparation and Crystallography.
Plasmid construction (Table S2), expression, and purification of c552 variants were performed according to previously published procedures with minor alterations. The crystal structure of the WT protein was determined (Table S3), and the coordinates were deposited in the Protein Data Bank (PDB ID code 3VNW). Variants were labeled with Dns following published methods. The stability of the seven Dns labeled variants was confirmed by denaturation curves, generated using fluorescence, UV-visible, and CD spectroscopy. Detailed descriptions of conditions are in Supporting Information.
Fluorescence Decay Kinetics.
For trFET measurements, samples were excited at 355 nm by the third harmonic of pulses (50 ps, 0.5 mJ) from a regeneratively amplified (Continuum), mode-locked picosecond neodymium-doped yttrium–aluminum–garnet laser (Vanguard 2000-HM532; Spectra-Physics). Luminescence was collected by a picosecond streak camera (C5680; Hamamatsu Photonics) in the photon-counting and analog integration modes for equilibrium unfolding and refolding kinetics experiments, respectively (12). A long-pass cutoff filter (>430 nm) was selected for Dns fluorescence. The decay data were collected on short- (1 or 5 ns) and long (20 or 50 ns) timescales.
Experiments were carried out at pH 3.0 in 100 mM sodium citrate buffer with varying Gdn concentration, which was measured by refractive index measurements (26). Protein concentrations were confirmed by absorption spectroscopy (ɛ552red = 21.1 mM−1) (14); sample concentrations were 2 μM for equilibrium unfolding experiments.
For refolding kinetics experiments, a T-shaped continuous-flow mixer was used as described in cytochromes c′ and cb562 analyses (11, 12). Folding reactions were initiated by mixing a solution of denatured c552 in 6 M Gdn in buffer (100 mM sodium citrate, pH 3.0) with buffer in a volume ratio of 1:5. Background measurements of buffer with the corresponding concentration of Gdn were subtracted from the data. Final protein concentrations were 12–15 μM. All measurements were conducted at ambient temperature (∼18 °C).
Data Fitting and Analysis.
The resulting short- and long-timescale data were spliced together, and the combined traces were compressed logarithmically before fitting (70 points per decade). This compression does not alter the interpretation of data (12). Fluorescence energy transfer analysis involves the numerical inversion of a Laplace transform [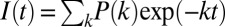 ] (27, 28). We have fit the kinetics data using MATLAB (Mathworks) with a TR and a maximum-entropy (ME) fitting algorithm with regularization methods that impose additional constraints on the properties of P(k) ≥ 0. TR fitting minimizes the sum of the squares of the deviations between calculated and observed fluorescence intensities (χ2), as well as the gradient of the P(k) distribution [
] (27, 28). We have fit the kinetics data using MATLAB (Mathworks) with a TR and a maximum-entropy (ME) fitting algorithm with regularization methods that impose additional constraints on the properties of P(k) ≥ 0. TR fitting minimizes the sum of the squares of the deviations between calculated and observed fluorescence intensities (χ2), as well as the gradient of the P(k) distribution [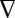 P(k)]. ME fitting minimizes the sum of the squares of the deviations between calculated and observed fluorescence intensities (χ2), while maximizing the entropy of the rate-constant distribution {
P(k)]. ME fitting minimizes the sum of the squares of the deviations between calculated and observed fluorescence intensities (χ2), while maximizing the entropy of the rate-constant distribution {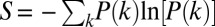 }. TR and ME fitting yields stable and reproducible numerical inversions of the kinetics data. The balance between minimization of χ2 and
}. TR and ME fitting yields stable and reproducible numerical inversions of the kinetics data. The balance between minimization of χ2 and  P(k) minimization or S maximization was determined by L-curve analysis (29).
P(k) minimization or S maximization was determined by L-curve analysis (29).
The probability distributions of rate constant P(k) were converted to probability distributions of distance rDA using the Förster equation [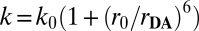 ] (30). The decay rate of the unquenched Dns fluorophore k0 is 1.0 × 108 s−1, determined using Dns-modified cysteines. The Förster critical length r0 for the Dns–heme pair is 39 Å under both native and denatured conditions (8). At distances longer than 1.5r0, energy transfer quenching of D is not competitive with excited-state decay; D–A distances cannot be obtained reliably (31, 32) and different structures in the protein ensemble with r ≥ 59 Å cannot be resolved.
] (30). The decay rate of the unquenched Dns fluorophore k0 is 1.0 × 108 s−1, determined using Dns-modified cysteines. The Förster critical length r0 for the Dns–heme pair is 39 Å under both native and denatured conditions (8). At distances longer than 1.5r0, energy transfer quenching of D is not competitive with excited-state decay; D–A distances cannot be obtained reliably (31, 32) and different structures in the protein ensemble with r ≥ 59 Å cannot be resolved.
The moments (Mn) of the P(r) distributions were calculated according to the equation [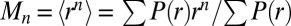 ]. The variances (V), which reflect the breadth of the P(r) distributions, were calculated according to this equation [
]. The variances (V), which reflect the breadth of the P(r) distributions, were calculated according to this equation [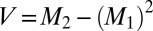 ].
].
Supplementary Material
Acknowledgments
S.Y. thanks Dr. Julie A. Hoy (Caltech) for X-ray data collection; Dr. Hiroshi Nakajima (Nagoya University) for advice on expression and purification of c552; and Drs. Yuichi Tokita and Yoshio Goto (Sony Corporation) for the management, promotion, and kind support of this study. S.Y. was supported by a Research Fellowship of Sony Corporation. Research at Caltech was supported by National Institutes of Health Grants DK019038 (to H.B.G. and J.R.W.) and GM068461 (to J.R.W.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3VNW).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1221832110/-/DCSupplemental.
References
- 1.Gomes CM, Wittung-Stafshede P. Protein Folding and Metal Ions: Mechanisms, Biology and Disease. New York: CRC; 2011. [Google Scholar]
- 2.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95(23):13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lacor PN, et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci. 2004;24(45):10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins. 1995;21(3):167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 5.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: The energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 6.Oliveberg M, Wolynes PG. The experimental survey of protein-folding energy landscapes. Q Rev Biophys. 2005;38(3):245–288. doi: 10.1017/S0033583506004185. [DOI] [PubMed] [Google Scholar]
- 7.Pletneva EV, Gray HB, Winkler JR. Snapshots of cytochrome c folding. Proc Natl Acad Sci USA. 2005;102(51):18397–18402. doi: 10.1073/pnas.0509076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pletneva EV, Gray HB, Winkler JR. Many faces of the unfolded state: Conformational heterogeneity in denatured yeast cytochrome C. J Mol Biol. 2005;345(4):855–867. doi: 10.1016/j.jmb.2004.10.085. [DOI] [PubMed] [Google Scholar]
- 9.Faraone-Mennella J, Gray HB, Winkler JR. Early events in the folding of four-helix-bundle heme proteins. Proc Natl Acad Sci USA. 2005;102(18):6315–6319. doi: 10.1073/pnas.0502301102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faraone-Mennella J, Tezcan FA, Gray HB, Winkler JR. Stability and folding kinetics of structurally characterized cytochrome c-b562. Biochemistry. 2006;45(35):10504–10511. doi: 10.1021/bi060242x. [DOI] [PubMed] [Google Scholar]
- 11.Kimura T, Lee JC, Gray HB, Winkler JR. Site-specific collapse dynamics guide the formation of the cytochrome c’ four-helix bundle. Proc Natl Acad Sci USA. 2007;104(1):117–122. doi: 10.1073/pnas.0609413103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimura T, Lee JC, Gray HB, Winkler JR. Folding energy landscape of cytochrome cb562. Proc Natl Acad Sci USA. 2009;106(19):7834–7839. doi: 10.1073/pnas.0902562106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivarsson Y, et al. An on-pathway intermediate in the folding of a PDZ domain. J Biol Chem. 2007;282(12):8568–8572. doi: 10.1074/jbc.M611026200. [DOI] [PubMed] [Google Scholar]
- 14.Hon-Nami K, Oshima T. Purification and some properties of cytochrome c-552 from an extreme thermophile, Thermus thermophilus HB8. J Biochem. 1977;82(3):769–776. doi: 10.1093/oxfordjournals.jbchem.a131753. [DOI] [PubMed] [Google Scholar]
- 15.Hon-nami K, Oshima T. Denaturation of thermophilic ferricytochrome c-552 by acid, guanidine hydrochloride, and heat. Biochemistry. 1979;18(25):5693–5697. doi: 10.1021/bi00592a027. [DOI] [PubMed] [Google Scholar]
- 16.Than ME, et al. Thermus thermophilus cytochrome-c552: A new highly thermostable cytochrome-c structure obtained by MAD phasing. J Mol Biol. 1997;271(4):629–644. doi: 10.1006/jmbi.1997.1181. [DOI] [PubMed] [Google Scholar]
- 17.Muresanu L, et al. The electron transfer complex between cytochrome c552 and the CuA domain of the Thermus thermophilus ba3 oxidase. A combined NMR and computational approach. J Biol Chem. 2006;281(20):14503–14513. doi: 10.1074/jbc.M601108200. [DOI] [PubMed] [Google Scholar]
- 18.Travaglini-Allocatelli C, et al. Exploring the cytochrome c folding mechanism: Cytochrome c552 from thermus thermophilus folds through an on-pathway intermediate. J Biol Chem. 2003;278(42):41136–41140. doi: 10.1074/jbc.M303990200. [DOI] [PubMed] [Google Scholar]
- 19.Clementi C, Nymeyer H, Onuchic JN. Topological and energetic factors: What determines the structural details of the transition state ensemble and “en-route” intermediates for protein folding? An investigation for small globular proteins. J Mol Biol. 2000;298(5):937–953. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- 20.Weinkam P, Pletneva EV, Gray HB, Winkler JR, Wolynes PG. Electrostatic effects on funneled landscapes and structural diversity in denatured protein ensembles. Proc Natl Acad Sci USA. 2009;106(6):1796–1801. doi: 10.1073/pnas.0813120106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fee JA, et al. Integrity of thermus thermophilus cytochrome c552 synthesized by Escherichia coli cells expressing the host-specific cytochrome c maturation genes, ccmABCDEFGH: Biochemical, spectral, and structural characterization of the recombinant protein. Protein Sci. 2000;9(11):2074–2084. doi: 10.1110/ps.9.11.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pace NC, Shirley BA, Thomson TF. Protein Structure: A Practical Approach. Oxford: IRL; 1990. pp. 311–330. [Google Scholar]
- 23.Ibrahim SM, et al. Cytochrome c552 from Thermus thermophilus engineered for facile substitution of prosthetic group. Biochemistry. 2011;50(45):9826–9835. doi: 10.1021/bi201048e. [DOI] [PubMed] [Google Scholar]
- 24.Wain R, et al. The cytochrome c fold can be attained from a compact apo state by occupancy of a nascent heme binding site. J Biol Chem. 2001;276(49):45813–45817. doi: 10.1074/jbc.M107572200. [DOI] [PubMed] [Google Scholar]
- 25.Shastry MCR, Sauder JM, Roder H. Kinetic and structural analysis of submillisecond folding events in cytochrome c. Acc Chem Res. 1998;31(11):717–725. [Google Scholar]
- 26.Nozaki Y. The preparation of guanidine hydrochloride. Methods Enzymol. 1972;26:43–50. doi: 10.1016/s0076-6879(72)26005-0. [DOI] [PubMed] [Google Scholar]
- 27.Beals JM, Haas E, Krausz S, Scheraga HA. Conformational studies of a peptide corresponding to a region of the C-terminus of ribonuclease A: Implications as a potential chain-folding initiation site. Biochemistry. 1991;30(31):7680–7692. doi: 10.1021/bi00245a004. [DOI] [PubMed] [Google Scholar]
- 28.Beechem JM, Haas E. Simultaneous determination of intramolecular distance distributions and conformational dynamics by global analysis of energy transfer measurements. Biophys J. 1989;55(6):1225–1236. doi: 10.1016/S0006-3495(89)82918-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawson CL, Hanson RJ. Solving Least Squares Problems. Englewood Cliffs, NJ: Prentice-Hall; 1974. [Google Scholar]
- 30.Wu P, Brand L. Resonance energy transfer: Methods and applications. Anal Biochem. 1994;218(1):1–13. doi: 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- 31.Jordanides XJ, Scholes GD, Shapley WA, Reimers JR, Fleming GR. Electronic couplings and energy transfer dynamics in the oxidized primary electron donor of the bacterial reaction center. J Phys Chem B. 2004;108(5):1753–1765. [Google Scholar]
- 32.Scholes GD, Jordanides XJ, Fleming GR. Adapting the Forster theory of energy transfer for modeling dynamics in aggregated molecular assemblies. J Phys Chem B. 2001;105(8):1640–1651. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.