

Abstract
Venoms consist of a complex mixture of toxic components that are used by a variety of animal species for defense and predation. Envenomation of mammalian species leads to an acute inflammatory response and can lead to the development of IgE-dependent venom allergy. However, the mechanisms by which the innate immune system detects envenomation and initiates inflammatory and allergic responses to venoms remain largely unknown. Here we show that bee venom is detected by the NOD-like receptor family, pyrin domain-containing 3 inflammasome and can trigger activation of caspase-1 and the subsequent processing and unconventional secretion of the leaderless proinflammatory cytokine IL-1β in macrophages. Whereas activation of the inflammasome by bee venom induces a caspase-1–dependent inflammatory response, characterized by recruitment of neutrophils to the site or envenomation, the inflammasome is dispensable for the allergic response to bee venom. Finally, we find that caspase-1–deficient mice are more susceptible to the noxious effects of bee and snake venoms, suggesting that a caspase-1–dependent immune response can protect against the damaging effects of envenomation.
Keywords: melittin, phospholipase a2, mast cells, detoxification, noxious xenobiotics
A variety of evolutionarily divergent animals, including insect, arachnid, reptilian, and mammalian species, use venoms for defense and predation (1). These venoms, which consist of a complex mixture of toxic components, are usually delivered into their victims via bites or stings (2). The targeting of mammalian species by venomous animals has presumably introduced evolutionarily relevant selective pressures to either avoid or minimize the toxic effects of envenomation (3).
Animal venoms contain various components that are responsible for their toxicity and for triggering the inflammatory response to envenomation (1, 4). A large class of cytotoxic venom components consists of proteins with direct cytolytic activities that are dependent on the ability of these compounds to bind and disrupt cellular membranes either through direct pore formation or cleavage of membrane phospholipids (5–7). This class of toxins includes various cytolytic cationic peptides, such as melittin in bee venom, scorpion toxins in scorpion venom, lycotoxins in wolf spider venom (7, 8), direct lytic factor in Indian Cobra venom, and Crotamine in South American rattlesnake venom (9, 10). Furthermore, nearly all insect and snake venoms contain phospholipase A2s (PLA2s), which can lead to cell lysis by cleaving plasma membrane phospholipids (1).
Venoms induce a variety of immune responses, including both acute inflammatory responses, such as mast cell degranulation, and adaptive immune responses, such as T helper type 2 responses and IgE production (11, 12). However, the molecular mechanisms by which the innate immune system detects envenomation and initiates inflammatory and allergic responses remain largely unknown.
Stings by hymenoptera species, including the European honey bee (Apis mellifera), are a common environmental cause of inflammatory and allergic responses (11). Bee venoms typically cause an immediate local inflammatory reaction consisting of redness, pain, heat, and swelling, but can also lead to an allergic response in a subset of individuals—approximately 3% of the population is allergic to hymenoptera stings and 100 people die each year from hypmenoptera sting-induced anaphylaxis in the United States (13).
Large, multiprotein complexes responsible for the activation of caspase-1, termed inflammasomes, are activated in response to various infectious and noninfectious stimuli (14). The activation of inflammasomes culminates in the autocatalytic cleavage and activation of the proenzyme caspase-1 and the subsequent caspase-1–dependent cleavage and noncanonical (endoplasmic-reticulum– and Golgi-independent) secretion of the proinflammatory cytokines IL-1β and IL-18, which lack leader sequences. In addition, activation of caspase-1 leads to a proinflammatory cell death termed pyroptosis. The NLRP3 inflammasome consists of the “sensor” protein NLRP3, the adaptor apoptosis-associated speck-like protein (ASC) and caspase-1. Damage to cellular membranes is a common trigger of the NLRP3 inflammasome (15).
Because common components of venoms exhibit cytotoxic activities that depend on their ability to bind and disrupt cellular membranes, we hypothesized that venoms might also lead to activation of the inflammasome. We focused our studies on venom from the European honey bee because its composition and activities are well characterized and it is an environmentally common and medically relevant allergen.
We found that bee venom and the pore-forming bee venom peptide melittin could induce inflammasome activation both in vitro and in vivo. Furthermore, whereas caspase-1 was dispensable for the allergic response to bee venom, we found that caspase-1 was critical for neutrophil recruitment to the site of envenomation. Finally, we found that caspase-1–dependent inflammatory responses protected mice from the pathological effects of envenomation.
Results
Bee Venom Induces Caspase-1 Activation and IL-1β Secretion by Macrophages.
We first tested whether A. mellifera venom could trigger IL-1β release from LPS-primed bone-marrow–derived macrophages (BMDMs). We found that bee venom triggered robust IL-1β secretion by LPS-primed macrophages, whereas LPS alone and the protease allergen papain failed to induce IL-1β secretion (Fig. 1 A and B). As expected from this result, bee venom efficiently induced caspase-1 activation in LPS-primed macrophages, as measured by the presence of the caspase-1 p10 cleavage product by Western blotting (WB) (Fig. 1C).
Fig. 1.

Bee venom and the bee venom component melittin induce inflammasome activation. (A) Bone-marrow–derived macrophages (BMDMs) were pretreated for 6 h with LPS (100 ng/mL) and then stimulated with bee venom (10 μg/mL) or papain (100 μg/mL) for 1 h. IL-1β secretion was measured by ELISA. (B) BMDMs were pretreated with LPS and then stimulated for 1 h with ATP (5 mM), bee venom (6.25 μg/mL), melittin (3.125 μg/mL), or PLA2 (250 μg/mL). IL-1β secretion was measured by ELISA. (C) BMDMs were stimulated as in B and caspase-1 activation was measured by Western blotting of total cell lysates. NS, nonspecific band. (D and E) WT and caspase-1–deficient BMDMs were treated as in B. Caspase-1 activation was measured by WB. IL-1β and IL-6 secretion were measured by ELISA.
The two major proteinaceous components of bee venom are the small cationic peptide melittin and the enzyme PLA2, which comprise ∼50 and 12%, respectively, of the protein in bee venom (16). Both melittin and PLA2 have direct cell lytic activities that depend on their ability to bind and disrupt membranes. We tested whether one or both of these components of bee venom were responsible for activation of the inflammasome. We found that melittin that was purified from whole bee venom (Fig. 1 B and C) and synthetically produced melittin (Fig. S1) could trigger caspase-1 activation and IL-1β secretion at doses that were equal to or lower than those required for whole bee venom. Purified bee venom PLA2 also triggered IL-1β release, but only did so at high doses relative to the doses of bee venom or melittin that could efficiently activate the inflammasome (Fig. 1 B and C). Furthermore, the activation of caspase-1 by bee venom PLA2, as measured by Western blotting, was near the limit of detection (Fig. 1 C and D). Because bee venom PLA2 is biochemically purified from total bee venom, high doses of PLA2 may trigger inflammasome activation due to residual contamination with melittin after purification. Therefore, melittin appears to be the main trigger of inflammasome activation in bee venom. Consistent with other triggers of inflammasome activation, such as uric acid crystals and ATP (15), caspase-1 activation in response to bee venom, required LPS priming (Fig. 1A). Furthermore, IL-1β secretion in response to bee venom, melittin, and PLA2 was caspase-1 dependent, whereas IL-6 secretion (due to LPS priming) was caspase-1 independent, as expected (Fig. 1E).
Bee Venom-Induced Caspase-1 Activation in Macrophages Is NLRP3- and ASC-Dependent.
We next tested which inflammasome components were required for caspase-1 activation and IL-1β release in response to bee venom. Consistent with our hypothesis that venom components cause a membrane damage that would lead to NLRP3 activation, bee venom-induced IL-1β release was dependent on NLRP3 and the inflammasome adaptor ASC, but independent of NLRC4 (Fig. 2). IL-6 secretion was unaffected by deficiencies in inflammasome components, as expected (Fig. 2). Inflammasome activation by bee venom could also be blocked by a high concentration of extracellular potassium (Fig. S2), which is known to prevent NLRP3-inflammasome activation. These data are consistent with a model in which bee venom melittin induces membrane damage that results in activation of the NLRP3 inflammasome.
Fig. 2.

Bee venom activates a NLRP3- and ASC-containing inflammasome. BMDMs from WT, Asc−/−, Nlrp3−/−, and Nlrc4−/− mice were prestimulated with LPS (100 ng/mL) for 6 h and then treated with ATP, bee venom, melittin, or PLA2 for 1 h as in Fig. 1. IL-1β and IL-6 secretions were measured by ELISA.
Bee Venom Triggers Inflammasome- and IL-1–Dependent Neutrophil Recruitment in Vivo.
Because we found that bee venom activated the NLRP3 inflammasome in vitro, we next tested whether bee venom would also trigger inflammasome activation in vivo. Inflammasome activation and the IL-1 receptor (IL-1R) are critical for the recruitment of neutrophils to sites of inflammasome activation after exposure to activators of the NLRP3 inflammasome, such as uric acid crystals (17). We therefore examined recruitment of neutrophils to the peritoneum after i.p. injection of bee venom in inflammasome-deficient mice, as well as IL-1R–deficient mice. We found that caspase-1, NLRP3, ASC, and the IL-1 receptor were all required for recruitment of neutrophils to the peritoneum after i.p. injection of bee venom (Fig. 3). Importantly, NLRP3, caspase-1, and the IL-1R are dispensable for neutrophil recruitment to the peritoneum in response to zymosan and thioglycollate, demonstrating that not all neutrophil recruitment is inflammasome dependent (17, 18). These data suggest that envenomation can be detected by the NLRP3 inflammasome both in vitro and in vivo.
Fig. 3.

Bee-venom–induced neutrophil recruitment is caspase-1, NLRP3, and IL-1R dependent. Casp1−/−, Nlrp3−/−, Il1r1−/−, and control C57BL/6 mice were injected intraperitoneally with 50 μg bee venom and peritoneal cells were collected by lavage 18 h later, counted, and analyzed by flow cytometry. Neutrophils were defined as Ly6G+ and 7/4+.
Inflammasome Components Are Dispensable for the Allergic Response to Bee Venom.
Venoms are well known immunologically as potent triggers of the allergic response. Envenomation is known to trigger mast cell degranulation and local or systemic anaphylaxis. Furthermore, bee venom exposure can lead to the production of venom-specific IgE in humans, and approximately 2% of humans are allergic to bee stings (11). Bee venom immunizations also effectively trigger IgE responses in mice (19). Because caspase-1 activation is known to influence the initiation and outcome of the adaptive immune response (20), we tested whether the allergic response to bee venom was dependent on the inflammasome. Whereas bee venom was indeed a potent inducer of IgE production, caspase-1–deficient and ASC-deficient mice showed no apparent defects in total IgE production after immunization with bee venom (Fig. 4A). Bee venom and purified melittin can also be used as adjuvant to induce antigen-specific IgE production to an admixed antigen, such as ovalbumin (OVA) (21). However, we found that caspase-1 was also dispensable for the induction of anti-OVA IgE after immunization with OVA admixed with melittin (Fig. 4B).
Fig. 4.

Caspase-1 and ASC are dispensable for the allergic response to bee venom. (A) WT (n = 5), Casp1−/− (n = 5), and Asc−/− (n = 5) mice were immunized s.c. with Human Serum Albumin (HSA) (100 μg per mouse) or bee venom (100 μg per ) in PBS on days 0 and 21. Serum IgE production was measured by ELISA on days 0, 14, and 28. (B) WT (n = 4) and Casp1−/− (n = 4) mice were immunized with endotoxin-free OVA (100 μg per mouse) and melittin (100 μg per mouse) on days 0 and 21, and OVA-specific IgE in the serum was measured on day 28.
The Caspase-1 Inflammasome Protects Mice from the Noxious Effects of Envenomation.
Envenomation with bee venom can induce various pathologies in mice, including localized skin necrosis and acute hypothermia (22). We therefore tested the effect of caspase-1 deficiency on these pathological consequences of envenomation. Surprisingly, caspase-1–deficient mice showed increased susceptibility to hypothermia induced by i.p. injection with bee venom (Fig. 5A) and increased formation of necrotic lesions after s.c. injection with bee venom or melittin (Fig. 5 B and C) compared with wild-type (WT) mice, suggesting that caspase-1 somehow protects mice from the pathological effects of envenomation.
Fig. 5.
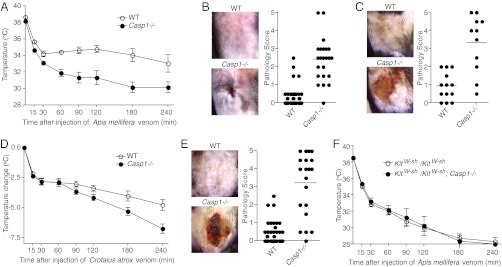
Caspase-1–deficient mice are hypersensitive to the toxic effects of bee and snake venom. (A) WT C57BL/6 (n = 4) and Casp1−/− (n = 4) mice were injected intraperitoneally with 50 μg bee (Apis mellifera) venom, and their body temperature was followed for 4 h. (B) WT C57BL/6 and Casp1−/− mice were injected s.c. with 250–500 μg bee venom and skin lesions were scored 7 d postinjection. (C) WT C57BL/6 and Casp1−/− mice were injected s.c. with 500 μg purified melittin from bee venom and skin lesions were scored 7 d postinjection. (D) WT C57BL/6 (n = 15) and Casp1−/− (n = 15) mice were injected intraperitoneally with 65 μg Western Diamondback (Crotalus atrox) venom, and their body temperature was followed for 4 h. (E) WT C57BL/6 and Casp1−/− mice were injected s.c. with 65 μg Western Diamondback venom and lesions were photographed and scored on day 5 after injection. (F) Mast-cell–deficient (KitW-sh/KitW-sh) and KitW-sh/KitW-sh; Casp1−/− mice were injected intraperitoneally with 50 μg bee venom, and their body temperature was followed for 4 h.
In addition to bee venom, many other animal venoms contain cell lytic peptides or enzymes that might be predicted to activate the inflammasome. To test whether caspase-1 is also critical for protection from the toxic effects of venoms from other animals, we examined the response to Western Diamondback (Crotalus atrox) rattlesnake venom (WD venom) in caspase-1–deficient and control WT mice. Similar to what we observed after exposure to bee venom, caspase-1–deficient mice showed more severe lesion formation and hypothermia after exposure to WD venom (Fig. 5 D and E). Thus, caspase-1 inflammasomes enhance resistance to the toxic effects of animal venoms from evolutionarily divergent organisms.
There are a few possible mechanisms by which caspase-1 might protect from the noxious effects of envenomation. These mechanisms can be separated into two major classes—tissue protection/repair and venom detoxification. In addition to its role in secretion of IL-1β and IL-18, caspase-1 also can protect from cell death induced by the bacterial pore-forming toxin aerolysin by initiating membrane repair through the activation of sterol regulatory element binding proteins (23). Thus, we examined the possibility that caspase-1 might prevent venom-induced tissue damage by inducing membrane repair. In contrast to what was observed for aerolysin, we were unable to observe a role for caspase-1 in protection of Chinese hamster ovary cells from melittin- or bee-venom–induced death (Fig. S3). Neutrophils are known to play a critical role in the resolution of inflammation and, thus, the initiation of tissue repair (24). Because the inflammasome is required for recruitment of neutrophils after envenomation, we also tested the role of neutrophils in protection from envenomation. However, antibody-mediated depletion of neutrophils had no effect on the formation of necrotic lesions after s.c. injection with snake venom (Fig. S4).
The second mechanism by which the inflammasome might protect against envenomation is by participating in detoxification of venom components. Mast cell proteases released during degranulation can detoxify venom components via direct cleavage and inactivation of venom toxins (22, 25). Furthermore, heparin, which is one of the major components of mast cell granules, can detoxify bee venom by binding and neutralizing cationic venom toxins (26, 27). Mast-cell–deficient mice thus show increased susceptibility to the noxious effects of envenomation (22). To address the role of mast cells in caspase-1–dependent protection from envenomation, we crossed caspase-1–deficient mice with mast-cell–deficient mice (C57BL/6 KitW-sh/KitW-sh). In the absence of mast cells, caspase-1 deficiency had no effect on hypothermia induced by i.p. injection with bee venom (Fig. 5F). This suggests that mast cells are essential players in caspase-1–dependent protection from envenomation.
Discussion
Animal venoms are ubiquitous in nature and are potent inducers of tissue damage and inflammatory responses. However, the mechanisms whereby venoms trigger inflammatory reactions are poorly understood. Here we examined the mechanism by which the innate immune system detects and responds to animal venoms. Because many types of venom contain pore-forming components, and the inflammasome can be activated in response to pore formation, we hypothesized that venoms might be detected by the inflammasome. Indeed, we found that bee venom and melittin from bee venom induced NLRP3 inflammasome activation in vitro and in vivo. Furthermore, caspase-1 appeared to be involved in protection from the noxious effects of envenomation as mice lacking caspase-1 were hypersensitive to venom-induced pathologies (Fig. 6).
Fig. 6.

Bee venom induces caspase-1–dependent and –independent immune responses. Caspase-1 activation by bee venom leads to neutrophil recruitment to the site of envenomation. Caspase-1 is also necessary for protection from the noxious effects of envenomation, including venom-induced hypothermia and cutaneous lesion formation. This protection is likely mediated by mast cells, which can directly detoxify venom components after degranulation via degradation and neutralization of venom toxins. In contrast to neutrophil recruitment and protection from envenomation, the allergic (IgE) response to bee venom is caspase-1 independent, and instead likely requires alternative signals from the innate immune system.
We found that the pore-forming cationic peptide melittin was the major inducer of NLRP3 inflammasome activation in bee venom and that inflammasome activation could be blocked by high concentrations of extracellular potassium. Our data are thus consistent with a model where venoms are detected by the innate immune system through sensing of membrane disruption, which triggers assembly and activation of the inflammasome; thus, components of the innate immune system may have evolved to detect a noxious feature of venoms that is common to many animal venoms, thereby enabling the detection of envenomation through a single sensing pathway. Notably, whereas low concentrations of bee venom and melittin efficiently induced inflammasome activation in macrophages, higher concentrations simply led to cell lysis and therefore did not trigger caspase-1 activation. Thus, inflammasome activation in vivo is likely restricted to cells adjacent to the sting site.
Bee venom induces an acute inflammatory response that is characterized by the classical signs of inflammation including pain, redness, heat and swelling around the site of the sting. We found that bee venom induced neutrophil recruitment to the site of envenomation and that this recruitment was dependent upon caspase-1 and the IL-1 receptor. However, we were unable to find a role for neutrophils in caspase-1–dependent protection from venom-induced hypothermia and tissue damage. Thus, it appears that neutrophils are not critical for the caspase-1–dependent protection from envenomation we describe here. Regardless, neutrophil recruitment after envenomation may play an important role in the acute inflammatory response to bee venom by maintaining the sterility of the site of envenomation, removing debris that results from cell death after envenomation, and participating in resolution of the inflammatory response and, therefore, tissue repair (24).
Venoms from hymenoptera species, including the European honey bee, are a significant environmental cause of allergic responses. Approximately 3% of the population is allergic to hymenoptera stings and about 100 people die each year from hypmenoptera sting-induced anaphylaxis in the United States (13). Bee venom immunizations also efficiently induce IgE responses in mice (19). However, the mechanism by which venoms are sensed by the innate immune system and induce an IgE response remains undiscovered. Because we found that bee venom induces caspase-1 activation, we tested whether caspase-1 is involved in the allergic response to bee venom. We found that mice lacking caspase-1 showed normal IgE responses to bee venom, suggesting that caspase-1–independent innate immune pathways are involved in the allergic response to bee venom.
As venoms are well known as triggers of the allergic response, immune responses to venoms are often considered to be pathological rather than beneficial (3, 28). However, data obtained more than four decades ago by Higginbotham suggested that some immune responses to venoms might act to control the toxic effects of envenomation through detoxification of venom components rather than cause immunopathology (26, 27). Higginbotham observed that mast cells in the skin rapidly degranulated after s.c. venom injection and hypothesized that components of those mast cell granules might be involved in protection against venom toxins. In support of this hypothesis, he showed that the anionic glycosaminoglycan heparin, which is one of the major components of mast cell granules, could neutralize the toxic activity of bee venom by complexing with cationic venom toxins. More recently, mast-cell–deficient mice were found to be hypersensitive to the toxic effects of scorpion, bee, and snake venoms, thus providing in vivo support for Higginbotham’s hypothesis (22, 25, 29). Furthermore, these studies demonstrated that mast cell proteases released during degranulation could detoxify specific venom components through proteolytic degradation. These data suggest that the immune response can enhance resistance to the noxious effects of envenomation by directly detoxifying venom components. Thus, at least some immune responses to venoms are likely to be intentional and beneficial rather than accidental and pathological (3, 28).
We found that caspase-1–deficient mice, like mast-cell–deficient mice, were hypersensitive to the noxious effects of envenomation as measured by venom-induced hypothermia and tissue damage. We explored several mechanisms by which caspase-1 might enhance resistance to the noxious effects of venoms. We found that mast-cell–deficient mice and mast-cell–deficient mice lacking caspase-1 were equally susceptible to the noxious effects of envenomation, suggesting that mast cells are required for the caspase-1–mediated resistance to envenomation we describe here. Mast cells themselves express components of the inflammasome, including caspase-1, ASC, and NLRP3 (30, 31), raising the interesting possibility that caspase-1 in mast cells may mediate protection from envenomation.
In an attempt to dissect the relative contributions of caspase-1 in hematopoietic and nonhematopoietic cells in protection from envenomation, we performed bone marrow chimera studies using WT and caspase-1–deficient mice. We found that deficiency in caspase-1 in nonhematopoietic cells, but not hematopoietic cells, led to increased susceptibility to lesion formation after s.c. injection with Western Diamondback venom (Fig. S5). However, these data were somewhat difficult to interpret because many of the control chimeras lacking caspase-1 in both hematopoietic and nonhematopoietic compartments died during reconstitution; furthermore, the remaining caspase-1–deficient control chimeras were not appreciably more susceptible to lesion formation. Nonetheless, these data imply that caspase-1 in nonhematopoietic cells may be more critical than caspase-1 in hematopoietic cells for protection from envenomation. Notably, a recent study reported that melittin can induce activation of the absent in melanoma 2 (AIM-2) inflammasome in human keratinocytes (32). Together with our study, this suggests that bee venom induces caspase-1 activation in both hematopoietic and nonhematopoietic cells, and that inflammasome activation in nonhematopoietic cells may play a critical role in the immune response to envenomation. These data also imply that caspase-1 in mast cells does not play a critical role in resistance to envenomation; instead, we hypothesize that caspase-1 activation in nonhematopoietic cells (e.g., keratinocytes) may influence mast-cell–mediated venom detoxification. However, additional work will be required to determine the precise mechanism by which caspase-1 mediates protection from the noxious effects of envenomation.
It was recently discovered that the caspase-1–deficient mice used in these studies also lack caspase-11, and that at least some phenotypes that have previously been attributed to caspase-1 are actually dependent on caspase-11 (33). Thus, it is formally possible that the phenotypes observed in these studies are in fact due to caspase-11 deficiency rather than caspase-1 deficiency. Further experiments will be required to determine whether the increased susceptibility to the noxious effects of venom seen in caspase-1/11–deficient mice is actually attributable to caspase-1.
Inflammasomes are activated in response to various infectious microbes, including bacterial, viral, and fungal pathogens, and in response to various exogenous (e.g., alum crystals, silica) and endogenous (e.g., uric acid, amyloid-beta) noninfectious agents (15). Inflammasome-dependent responses to infectious microbes are often critical for microbial defense and, therefore, represent an evolutionarily adaptive role of the inflammasome (34). In contrast, inflammasome activation in response to both exogenous and endogenous nonmicrobial agents is responsible for multiple inflammatory pathologies, such as silicosis and gout (15, 17). Whether the inflammasome plays a beneficial role in responses to noninfectious triggers of inflammasome activation remains unknown. Our data showing that caspase-1–deficient mice are hypersensitive to the noxious effects of envenomation suggest that the inflammasome can also play a beneficial role in responses to noninfectious stimuli.
Taken together, our data demonstrate that animal venoms, likely because of their membrane damaging activity, can be detected by the innate immune system through activation of a NLRP3-containing inflammasome. Surprisingly, the inflammasome-dependent immune response to venoms reduces the noxious effects of envenomation rather than causing immunopathology. To our knowledge, this represents a unique example of a beneficial, rather than pathological, role for the inflammasome in response to a noninfectious stimulus. In addition, it provides further evidence of a role for the immune system in general, and unique evidence of a role for the inflammasome in particular, in detoxification and/or clearance of noxious xenobiotics such as animal venoms.
Materials and Methods
Mice.
All mice were bred and maintained at the Yale University School of Medicine and were used in accordance with Yale Animal Research and Care Guidelines. Asc−/− mice were made by John Bertin and Anthony Coyle (Millennium Pharmaceuticals) and were a kind gift of Millennium Pharmaceuticals through Richard Flavell and Fayyaz Sutterwala (Yale University). Nlrp3−/− and Casp1−/− mice were a kind gift of Fayyaz Sutterwala and Richard Flavell. Il1r1−/−, and C57BL/6 KitW-sh/KitW-sh mice were from Jackson Laboratories. All knockout mice were backcrossed to the C57BL/6 background at least seven times. Wild-type C57BL/6 mice were purchased from the National Cancer Institute as controls. In vivo experiments measuring susceptibility to the noxious effects of envenomation were performed using male mice between 12 and 18 wk of age because older male mice showed more dramatic responses to envenomation.
Reagents.
Western Diamondback venom, purified melittin from bee venom, and phospholipase A2 from bee venom were purchased from Sigma-Aldrich. Synthetic melittin was purchased from Genscript. Bee venoms sourced from both Sigma-Aldrich and Venom Supplies were used for these studies. We noticed considerable variation between lots of bee and snake venom with regards to their effects in vivo—e.g., some lots of bee venom caused no lesions at all but very efficiently induced hypothermia, whereas other lots induced lesion formation but were less effective at inducing a drop in temperature. It is widely reported that the composition of collected venoms can vary widely depending on the age, location, and stress level of the snakes or bees at the time of venom collection. Therefore, multiple lots of venom were screened for their ability to induce lesions or a drop in temperature in both wild-type and caspase-1–deficient mice and venom lots that displayed the desired activities (e.g., lesion formation) were chosen for subsequent studies.
Bone-Marrow–Derived Macrophage Cultures.
To grow bone-marrow–derived macrophages, bone marrow cells were incubated in complete RPMI supplemented with 30% (vol/vol) L929 supernatant containing Macrophage colony-stimulating factor for 7–8 d. Cells were plated in non–tissue culture-treated dishes at ∼6 × 10^5 cells/mL and were fed with fresh media on day 4.
Caspase-1 Activation and IL-1β Secretion.
Bone marrow macrophages (3.5 × 10^5 per well in 500 μL in a 24-well plate) were prestimulated with 100 ng/mL LPS (Sigma-Aldrich) for 6 h then stimulated with ATP (5 mM; Sigma-Aldrich), or titrating doses of bee venom (25 μg/mL–3.125 μg/mL), bvPLA2 (500 μg/mL–62.5 μg/mL), or melittin (25 μg/mL–3.125 μg/mL) for 1 h. Caspase-1 activation was measured via Western blotting using 4–12% NuPAGE gradient gels (Invitrogen), PVDF membranes (Millipore) and rabbit antimouse caspase-1 (sc-514; Santa Cruz) as described (35). IL-1β and IL-6 in the supernatants were measured via sandwich ELISA using antibodies from BD Biosciences. The doses of bee venom, melittin, and bvPLA2 that induced the highest levels of IL-1β release are shown in all figures.
Neutrophil Recruitment.
Mice were injected intraperitoneally with 50 μg bee venom and cells were collected by peritoneal lavage using 5 mL of cold PBS 18 h later. Peritoneal lavage cells were counted and analyzed by flow cytometry and neutrophils were defined as Ly6G+ and 7/4+ cells (18).
Immunizations for IgE.
Mice were immunized with 100 μg human serum albumin (Sigma) or bee venom in 100 μL of PBS in the rear footpads on days 0 and 21, and total serum IgE was measured by ELISA (using coating and detection antibodies from Southern Biotech) on days 0, 14, and 28. Alternatively, mice were immunized with 100 μg endotoxin-low endograde ovalbumin (Biosource) with or without 100 μg melittin in PBS on days 0 and 21, and OVA-specific IgE in the serum on days 0 and 28 was measured using an anti-OVA IgE ELISA kit (Chondrex).
In Vivo Effects of Envenomation.
For lesion formation, mice of the indicated genotypes were shaved and injected s.c. in the flank with 250–500 μg of bee venom, 500 μg of melittin, or 50–70 μg of Western Diamondback venom in 100 μL of PBS. Mice were examined daily for lesion formation for 5–7 d. Mice were euthanized and photographs of the lesions were taken on day 5 or 7. Lesions were scored based on gross visible pathology using the following scale: 0, no visible lesion; 1, small but visible skin destruction with no visible myonecrosis; 2, visible necrotic lesion (both skin and muscle) smaller than 0.5 cm; 3, necrotic lesion between 0.5 and 1 cm; 4, necrotic lesion between 1 and 1.5 cm; and 5, necrotic lesion larger than 1.5 cm. As different lots of venom showed highly variable activities with respect to venom-induced lesion formation, different concentrations were required to induce visible lesion formation depending on the batch of venom used.
To induce hypothermia, mice were injected intraperitoneally with either bee (50 μg per mouse) or Western Diamondback (65 μg per mouse) venom and their temperature was followed by rectal thermometer.
Supplementary Material
Acknowledgments
We thank A. Unni and O. Colegio for critical review of the manuscript; F. Sutterwala and R. Flavell for advice and for providing the NLRC4-, caspase-1–, and NLRP3-deficient mice; and R. Flavell, F. Sutterwala, J. Bertin, and A. Coyle for the ASC-deficient mice. This study was supported by the Howard Hughes Medical Institute and National Institutes of Health Grants AI89771, DK071754, AI046688, and AI055502.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1221476110/-/DCSupplemental.
References
- 1.Fry BG, et al. The toxicogenomic multiverse: Convergent recruitment of proteins into animal venoms. Annu Rev Genomics Hum Genet. 2009;10:483–511. doi: 10.1146/annurev.genom.9.081307.164356. [DOI] [PubMed] [Google Scholar]
- 2.Vonk FJ, et al. Evolutionary origin and development of snake fangs. Nature. 2008;454(7204):630–633. doi: 10.1038/nature07178. [DOI] [PubMed] [Google Scholar]
- 3.Palm NW, Rosenstein RK, Medzhitov R. Allergic host defences. Nature. 2012;484(7395):465–472. doi: 10.1038/nature11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fry BG, Roelants K, Norman JA. Tentacles of venom: Toxic protein convergence in the Kingdom Animalia. J Mol Evol. 2009;68(4):311–321. doi: 10.1007/s00239-009-9223-8. [DOI] [PubMed] [Google Scholar]
- 5.Raghuraman H, Chattopadhyay A. Melittin: A membrane-active peptide with diverse functions. Biosci Rep. 2007;27(4-5):189–223. doi: 10.1007/s10540-006-9030-z. [DOI] [PubMed] [Google Scholar]
- 6.van den Bogaart G, Guzmán JV, Mika JT, Poolman B. On the mechanism of pore formation by melittin. J Biol Chem. 2008;283(49):33854–33857. doi: 10.1074/jbc.M805171200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kourie JI, Shorthouse AA. Properties of cytotoxic peptide-formed ion channels. Am J Physiol Cell Physiol. 2000;278(6):C1063–C1087. doi: 10.1152/ajpcell.2000.278.6.C1063. [DOI] [PubMed] [Google Scholar]
- 8.Kuhn-Nentwig L. Antimicrobial and cytolytic peptides of venomous arthropods. Cell Mol Life Sci. 2003;60(12):2651–2668. doi: 10.1007/s00018-003-3106-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayashi MA, et al. Cytotoxic effects of crotamine are mediated through lysosomal membrane permeabilization. Toxicon. 2008;52(3):508–517. doi: 10.1016/j.toxicon.2008.06.029. [DOI] [PubMed] [Google Scholar]
- 10.Oguiura N, Boni-Mitake M, Rádis-Baptista G. New view on crotamine, a small basic polypeptide myotoxin from South American rattlesnake venom. Toxicon. 2005;46(4):363–370. doi: 10.1016/j.toxicon.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Müller UR. Insect venoms. Chem Immunol Allergy. 2010;95:141–156. doi: 10.1159/000315948. [DOI] [PubMed] [Google Scholar]
- 12.León G, et al. Immune response towards snake venoms. Inflamm Allergy Drug Targets. 2011;10(5):381–398. doi: 10.2174/187152811797200605. [DOI] [PubMed] [Google Scholar]
- 13.Brown TC, Tankersley MS. The sting of the honeybee: An allergic perspective. Ann Allergy Asthma Immunol. 2011;107(6):463–470, quiz 471. doi: 10.1016/j.anai.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 14.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 15.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 16.Habermann E. Bee and wasp venoms. Science. 1972;177(4046):314–322. doi: 10.1126/science.177.4046.314. [DOI] [PubMed] [Google Scholar]
- 17.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 18.Iyer SS, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. 2009;106(48):20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King TP, Sobotka AK, Kochoumian L, Lichtenstein LM. Allergens of honey bee venom. Arch Biochem Biophys. 1976;172(2):661–671. doi: 10.1016/0003-9861(76)90121-1. [DOI] [PubMed] [Google Scholar]
- 20.Chen M, Wang H, Chen W, Meng G. Regulation of adaptive immunity by the NLRP3 inflammasome. Int Immunopharmacol. 2011;11(5):549–554. doi: 10.1016/j.intimp.2010.11.025. [DOI] [PubMed] [Google Scholar]
- 21.Kind LS, Ramaika C, Allaway E. Antigenic, adjuvant and permeability enhancing properties of melittin in mice. Allergy. 1981;36(3):155–160. doi: 10.1111/j.1398-9995.1981.tb01830.x. [DOI] [PubMed] [Google Scholar]
- 22.Metz M, et al. Mast cells can enhance resistance to snake and honeybee venoms. Science. 2006;313(5786):526–530. doi: 10.1126/science.1128877. [DOI] [PubMed] [Google Scholar]
- 23.Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell. 2006;126(6):1135–1145. doi: 10.1016/j.cell.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 24.Serhan CN. Resolution phase of inflammation: Novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 25.Akahoshi M, et al. Mast cell chymase reduces the toxicity of Gila monster venom, scorpion venom, and vasoactive intestinal polypeptide in mice. J Clin Invest. 2011;121(10):4180–4191. doi: 10.1172/JCI46139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Higginbotham RD. Mast cells and local resistance to Russell’s viper venom. J Immunol. 1965;95(5):867–875. [PubMed] [Google Scholar]
- 27.Higginbotham RD, Karnella S. The significance of the mast cell response to bee venom. J Immunol. 1971;106(1):233–240. [PubMed] [Google Scholar]
- 28.Profet M. The function of allergy: Immunological defense against toxins. Q Rev Biol. 1991;66(1):23–62. doi: 10.1086/417049. [DOI] [PubMed] [Google Scholar]
- 29.Schneider LA, Schlenner SM, Feyerabend TB, Wunderlin M, Rodewald HR. Molecular mechanism of mast cell mediated innate defense against endothelin and snake venom sarafotoxin. J Exp Med. 2007;204(11):2629–2639. doi: 10.1084/jem.20071262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kambe N, Nakamura Y, Saito M, Nishikomori R. The inflammasome, an innate immunity guardian, participates in skin urticarial reactions and contact hypersensitivity. Allergol Int. 2010;59(2):105–113. doi: 10.2332/allergolint.09-RAI-0160. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura Y, et al. Mast cells mediate neutrophil recruitment and vascular leakage through the NLRP3 inflammasome in histamine-independent urticaria. J Exp Med. 2009;206(5):1037–1046. doi: 10.1084/jem.20082179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dombrowski Y, et al. Honey bee (Apis mellifera) venom induces AIM2 inflammasome activation in human keratinocytes. Allergy. 2012;67(11):1400–1407. doi: 10.1111/all.12022. [DOI] [PubMed] [Google Scholar]
- 33.Kayagaki N, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 34.Brodsky IE, Monack D. NLR-mediated control of inflammasome assembly in the host response against bacterial pathogens. Semin Immunol. 2009;21(4):199–207. doi: 10.1016/j.smim.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 35.Brodsky IE, et al. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe. 2010;7(5):376–387. doi: 10.1016/j.chom.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.