

Abstract
Frustration from strong interdomain interactions can make misfolding a more severe problem in multidomain proteins than in single-domain proteins. On the basis of bioinformatic surveys, it has been suggested that lowering the sequence identity between neighboring domains is one of nature’s solutions to the multidomain misfolding problem. We investigate folding of multidomain proteins using the associative-memory, water-mediated, structure and energy model (AWSEM), a predictive coarse-grained protein force field. We find that reducing sequence identity not only decreases the formation of domain-swapped contacts but also decreases the formation of strong self-recognition contacts between β-strands with high hydrophobic content. The ensembles of misfolded structures that result from forming these amyloid-like interactions are energetically disfavored compared with the native state, but entropically favored. Therefore, these ensembles are more stable than the native ensemble under denaturing conditions, such as high temperature. Domain-swapped contacts compete with self-recognition contacts in forming various trapped states, and point mutations can shift the balance between the two types of interaction. We predict that multidomain proteins that lack these specific strong interdomain interactions should fold reliably.
Keywords: aggregation, funnel
Protein misfolding and productive protein folding bear a yin–yang relationship in the energy landscape theory of biomolecular self-organization (1). Only by comparing the strengths of the forces leading to proper structure to those that might, by chance, stabilize alternative structure can we quantitatively understand how proteins kinetically access their thermodynamically stable ordered states (1). In vivo and at low concentrations in vitro, unfolded small proteins avoid kinetic traps and generally find their way easily to their native state. Nevertheless, diseases caused by the misfolding of several specific proteins plague mankind (2, 3). Despite much effort, the patterns of interactions that allow pathological misfolding remain incompletely understood. Known pathological misfolding entails aggregation of specific proteins and thus the interactions of protein molecules with other copies of themselves. Energy landscape theory provides one natural explanation of this specificity in misfolding through the funneled nature of the monomeric protein energy landscape: Native-like interactions between different protein molecules like those found within a single protein are stronger than alternate nonnative interactions in the same molecule or interactions between peptide sequences chosen at random in the two molecules. Because of this intrinsic self-stickiness of foldable molecules, runaway domain swapping, in which native-like interactions are made between different copies of the same protein, provides a natural mechanism for aggregation (4–7). Indeed, transient protein aggregation during refolding at moderately high concentration does appear to be universal (8). Nevertheless this aggregation usually resolves itself eventually as the system comes to equilibrium, thus arguing that something more may be involved in natural pathological misfolding that seems permanent. One attractive idea is that there is an alternate “amyloid funnel” (9), which a protein may enter if the molecule has enough time to find it before completing its native folding. A funnel to the amyloid state has been thought to possibly be universal, because under appropriate denaturing conditions, it seems, even the most innocuous proteins can form amyloids (10). Alternatively, like the funnel for formation of a native structure, the amyloid funnel may be encoded in sequence signals (11, 12). We have been led to address these questions about the misfolding energy landscape in our effort to model a series of insightful experimental investigations on multidomain proteins (13, 14). Multidomain proteins are much more susceptible to misfolding than single-domain proteins because of the effective high local concentration of peptide binding partners. Experiments on artificial constructs in which related protein domains are fused together indicate that high sequence identity of the domains favors aggregation and that the initial interactions of the fused domains are critical to the aggregation process (13). At the same time, bioinformatic studies indeed show that neighboring domains in natural multidomain proteins have lower than expected sequence identity (15). These observations point to the importance of domain swapping as predicted by the minimal frustration principle from energy landscape theory (16). The present computational studies show that this is only part of the story, however. Indeed in our simulations additional sequence signals that allow some peptide fragments to recognize copies of themselves greatly increase the tendency to misfold. In silico, single mutations in these fragments can significantly reduce misfolding of a multidomain construct. It turns out that these sequence signals do not yield a globally unique structure but are able to act on rather high-entropy ensembles. They do not affect the ordered native state ensembles. These sequence signals allow alternate structures of self-recognizing peptide fragments to achieve minimally frustrated configurations that are locally competitive with the final native structure. Making these alternate structures, however, frustrates the formation of further tertiary structure in the protein so these misfolded states are globally higher in energy than the native state: Stable misfolding is encouraged by the high residual entropy of an ensemble of structures that can take advantage of a locally strong interaction. Misfolding thus occurs optimally under intermediate denaturing conditions. In this scenario, then, pathological aggregation remains consistent with a largely minimally frustrated and funneled energy landscape of each monomer. The residual frustration that encourages misfolding is not evident at the residue pair level but involves alternate pairings of hexamer fragments, allowing self-recognition in amyloid-like assemblies.
The computational approach we take is based on transferable energy functions that have been optimized to predict protein tertiary structures of monomers using simulated annealing (17). To do this annealing we leverage the associative-memory, water-mediated, structure and energy model (AWSEM)-MD software package that has been shown to accurately predict monomeric (18) and properly oligomeric protein structures (19). Although the energy function is bioinformatically based, it appears to have many elements of biophysical realism. Here we show the same energy function encodes also the local signals for misfolding, even though information on such misfolding was not used in training the algorithm. In addition to simulating the rapid annealing of several fused dimer constructs, we compute multidimensional free-energy profiles under varying thermodynamic conditions to understand the entropy/energy interplay on the landscape. We also quantify the statistics of the conformational states formed with both native structures and elements of misfolded structure. The self-recognition ensemble is energetically less stable than the native ensemble because the abnormally strong self-recognition interaction is highly local. The same energy function used for simulated annealing allows us to rapidly scan any sequence for fragments that are likely to participate in such amyloid-like structure formation. The results of this scan are consistent with other bioinformatic tools for identifying amyloidogenic sequences (11, 20–24).
Results and Discussion
Simulated Annealing of Fused Multidomain Proteins: Misfolding Correlates with the Sequence Identity Between Domains.
A couple of recent and insightful experimental studies of misfolding and aggregation have focused on the Ig domains of the vertebrate muscle protein titin (13, 14). One study showed that the rate of aggregation of the fused dimers is correlated with the sequence identity between the domains (13). Our simulation investigation parallels this experimental work. We investigated fused dimers in which the 27th Ig domain of human cardiac titin [TI I27; Protein Data Bank (PDB) ID 1TIU] is the first domain. Five proteins with varying sequence identity to I27 were chosen for the second domain, which is connected to the first domain via a four-residue glycine linker. In addition, we also simulated an SH3-SH3 fused dimer. Starting from totally extended conformations, 40 independent simulated annealing simulations were run for each fused protein. Simulated annealing searches for energetically stable structures by gradually reducing the temperature from slightly above the folding temperature, at which the efficiency of searching the conformational space is considered optimal, to the native temperature at which the native structure is stable. Our simulated annealing protocol (SI Text) is not an attempt to reproduce faithfully the experimental conditions (13, 14), but rather is used as a way of quickly searching the conformational landscape for states that could trap the protein and thereby inhibit or slow productive folding. At the end of a simulated annealing run, the protein typically adopts either the native conformation or a compact misfolded conformation such as the self-recognition state in the case of the I27-I27 fused dimer. Using the final structures from these simulations, we calculated the fraction of misfolded domains. Fig. 1 shows the fraction of misfolded domains vs. the sequence identity between the two domains. A domain is considered folded when the fraction of native contacts within the domain 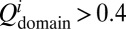 , i = 1, 2. As the sequence identity increases, the fraction of misfolded domains that result from simulated annealing increases, consistent with the observation that the sequence identity between neighboring domains in natural multidomain proteins is lower than would be expected by chance (13). However, what is the nature of these misfolded ensembles? What types of interactions are responsible for the misfolding?
, i = 1, 2. As the sequence identity increases, the fraction of misfolded domains that result from simulated annealing increases, consistent with the observation that the sequence identity between neighboring domains in natural multidomain proteins is lower than would be expected by chance (13). However, what is the nature of these misfolded ensembles? What types of interactions are responsible for the misfolding?
Fig. 1.

The fraction of misfolded domains found upon simulated annealing is plotted against the sequence identity between the two domains in the fused construct. Titin I27 was fused with five different proteins via a four-residue GLY linker to form two-domain proteins, as indicated in the plot. Forty annealing simulations were run for each fused protein from totally extended conformations. All proteins were studied with the same annealing schedule. A domain is considered folded when the fraction of native contacts within the domain 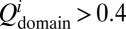 , i = 1, 2 at the end of all of the simulations. As the sequence identity increases, the fraction of misfolded domains increases accordingly.
, i = 1, 2 at the end of all of the simulations. As the sequence identity increases, the fraction of misfolded domains increases accordingly.
Self-Recognition Contacts and Domain-Swapped Contacts in the Misfolded Structures.
Interchain interactions are important for folding and misfolding when the protein concentration is high. In the case of fused dimer systems, the local concentration is always high—one domain has ample opportunity to interact with its covalently linked neighboring domain even when the concentration of molecules is low. Wright et al. have shown that, in the case of fused dimers of the Ig domains of titin, this initial interaction between the fused domains is the important step for determining aggregation rates (13). In these experiments, the aggregation rate was insensitive to the number of copies that were fused together for n ≥ 2. However, the nature of the misfolded structures remains unclear. Several a priori extreme possibilities exist—either specific interactions could drive misfolding or the misfolded structures might appear completely disordered and random. For evolved proteins, which satisfy the principle of minimal frustration, domain-swapped interactions are the most obvious candidate for specific interactions that drive misfolding. The counterparts of domain-swapped contacts in the monomer are native contacts that are in general stronger than other contacts and allow the protein to fold quickly and reliably. These same strong interactions can also drive oligomerization via domain swapping with nearby domains. Another candidate for misfolding, formation of self-recognition contacts, has been identified in studies of amyloid fibrils. Microcrystals of fibril-forming proteins reveal a “steric zipper” structure with two self-complementary β-sheets that form a spine of an amyloid fibril (25). These self-recognition contacts between two amyloidogenic segments in different molecules can be extremely strong but, unlike domain-swapping interactions, have no exact counterpart in the native structure and therefore can be involved only in misfolding. These segments are rich in hydrophobic amino acids, and bioinformatic studies indicate that evolution suppresses long stretches of hydrophobic amino acids (26). However, evolution has not completely eliminated them—these “amyloidogenic” segments appear to exist at a frequency of about one per protein (27) and are almost always buried in the natively folded structure. When the concentration of a particular protein is low, as is commonly the case in vivo, self-recognition of two buried segments is unlikely. Therefore, it is unsurprising that not every protein that has an amyloidogenic segment is involved in forming pathological fibrils or aggregates in vivo. Nonetheless, it is of great interest to study the role of these interactions in misfolding.
In Fig. 2, Left, we have shown a domain-swapped structure, one of the misfolded structures that resulted from simulated annealing of the fused SH3-SH3 construct. It is worth noting that a crystal structure of SH3 in domain-swapped form actually exists (PDB ID 1I07). AWSEM predicts the swapped structure, using only local structural information from the monomer of SH3 and a transferable tertiary contact potential. Lines are shown between residues forming tertiary contacts. Green lines indicate minimally frustrated interactions and red lines indicate highly frustrated interactions (28). Because these swapped interactions are between the same residue types as those in contact in the natively folded protein, they are minimally frustrated. The contact map in Fig. 2, Left is a 120 × 120 matrix, 58 residues from each SH3 and 4 residues from the linker. The lower left and upper right sections of the contact map show contacts within each monomer. The unsatisfied native contacts are shown in yellow, and the satisfied native contacts are shown in black. Both monomer structures have similar parts of the native structure formed. The nonnative contacts that are formed are shown in red. The upper left section shows contacts between the two monomers for the particular structure shown below the contact map. The swapped contacts that are formed are shown in black. These compose the majority of the formed intermonomer contacts in this case. Other nonnative intermonomer contacts exist mostly as a result of strong swapped contacts that are nearby in sequence. The swapping pattern is highly symmetrical, consistent with the symmetrical native structure of each monomer discussed previously. The lower right section of the contact map is a summary of the intermonomeric contacts formed in all 40 annealing simulations. The color scale indicates the frequency at which particular intermonomeric contacts occur in the final structures of the 40 annealing simulations. This contact map lacks symmetry—the swapped contacts in the upper left corner form more frequently than the contacts in the lower right corner. The more frequently formed swapped contacts involve the residue pairs that are closer to the linker region. Shorter sequence separation leads to a smaller entropy cost when these contacts are formed. This subset of swapped contacts forms in as many as 50% of the final structures.
Fig. 2.

Misfolded structures and their contact maps. (Left) A domain-swapped misfolded structure from simulated annealing simulations of the fused dimer SH3-SH3. (Right) A misfolded structure with a significant amount of self-recognition contacts from simulations of the fused dimer I27-I27. The different levels of frustration in the tertiary contacts as determined by the frustratometer analysis (28) are illustrated in the structures. In the structures, the two domains are in blue and yellow color, respectively. Minimally frustrated interactions are shown in green lines, and frustrated interactions are in red. The swapped contacts formed at the domain interface of SH3-SH3 are minimally frustrated, as expected from the principle of minimal frustration for native contacts. The self-recognition contacts formed at the domain interface of I27-I27 are also minimally frustrated, indicating that these contacts are stronger than random contacts. Within the set of four contact maps for each fused dimer, the lower left and upper right contact maps are for each domain, respectively. Formed native contacts are represented by black dots, formed nonnative contacts are in red, and the native contacts that are not formed are in yellow. The upper left contact map is the interdomain contact map. Black dots stand for formed swapped contacts. Red dots represent other types of interdomain contacts. In the case of I27-I27, we observe a different type of contact, the self-recognition contacts (in red), along the diagonal of the map. The lower left contact map is a summary contact map showing the total number of occurrences of each formed contact from the end structures of 40 annealing simulations. For SH3-SH3, the swapped contacts that are formed between the residues near the linker position appear more frequently than those that are more distantly connected through the sequence. For I27-I27, the self-recognition contacts formed between residue indexes 56 and 61 (HILILH) appear in almost all of the simulations.
In Fig. 2, Left, for the fused dimer I27-I27, we see that a very different type of misfolding occurs, which primarily involves self-recognition contacts. The summary contact map in the lower right section shows in red that a particular subset of self-recognition contacts is present in every final structure of the simulated annealing runs. As shown on the misfolded structure, these are stable and unfrustrated contacts formed by a parallel pairing of a strand of seven residues from monomer A with the same seven residues of monomer B. A calculation using the AWSEM-Amylometer (SI Text) identifies that these seven residues include a hexamer 56–61 (HILILH) that has the strongest self-recognition interaction among all hexamers in the sequence of I27. Furthermore, this self-recognition interaction is stronger than any possible native or other nonnative hexamer pair interaction in I27-I27 and is lower in energy than the threshold for amyloidogenicity as determined by the AWSEM-Amylometer.
Misfolded Ensembles Stabilized by Self-Recognition Interactions Are Entropically More Favored Than the Native.
Fig. 3 shows the energy and free-energy landscapes of the I27-I27 fused dimer along two reaction coordinates: a native reaction coordinate, the fraction of native contacts Q and a nonnative reaction coordinate, the sum of the number of self-recognition contacts Nself and the number of swapped contacts Nswap. The misfolded state I is energetically less stable than the native state N but entropically more favored. As shown in Fig. S1, below the folding temperature the native ensemble is the most populated state. As temperature increases, the misfolded ensemble becomes more and more highly populated. This type of metastable ensemble acts as a kinetic trap even below the folding temperature. When the fused dimer construct becomes trapped in this metastable state, large stretches of the structure are disordered and parts of the protein that are normally buried become exposed to solvent. This type of misfolding event could lead to aggregation when many fused dimers are present and is consistent with the experiments of Wright et al. (13).
Fig. 3.

Energy and free-energy surfaces for I27-I27 at its folding temperature. Nself and Nswap are the number of self-recognition contacts and the number of domain-swapped contacts, respectively. The trapped states I have higher energies than the native states N, as shown in the z axis, but have similar free energies to those of the native states, as shown by the color coding of the free energy, with scale indicated in the side bar. We see that the ensemble I states are entropically favored. As temperature increases, the intermediate ensemble will become more stable than the native ensemble.
Point Mutations and Their Effects in I27-I27 and SH3-SH3.
Removal and addition of strong self-recognition interactions by point mutations can change the degree of misfolding significantly. The foldability of the fused dimers correlates well with the sequence identity between the two domains as shown in Fig. 1. As the sequence identity of neighboring domains decreases, the fused protein folds correctly more often. In simulated annealing of I27-I27, strong self-recognition contacts often result in misfolding. Can we mutate the sequence to eliminate these strong interdomain interactions so that the protein folds more often? We constructed a double mutant of I27, V11D and I59E. The resulting I27*-I27* has 100% sequence identity between the two domains, but two of the strongest interdomain hexamer pairs that were found most frequently in previous misfolded structures in I27-I27 simulated annealing have been greatly weakened. The AWSEM-Amylometer was used to find the appropriate mutations that would reduce the strength of these self-recognition interactions the most. Despite having 100% sequence identity, I27*-I27* folds three times more often than the I27-I27 system and folds as often as fused I27-I31 (57% sequence identity).
For SH3-SH3, which folds well, we tested to see whether conversely a single-point mutation that introduces a strongly self-recognizing hexamer can induce misfolding in the simulated annealing. Again, the AWSEM-Amylometer was used to suggest an appropriate mutation. After introducing the E27I mutation, the fraction of misfolded domains goes up from 18% to 79%, using the identical simulation protocol that was used before. These two mutation studies suggest that one of the important effects of lowering the sequence identity between neighboring domains is to decrease the probability of strongly self-recognizing segments occurring, which in turn lowers the chance of misfolding via interdomain interactions.
Competing Roles of Self-Recognition Contacts and Swapped Contacts.
As shown in Fig. 3, a combination of self-recognition contacts and swapped contacts contributes to the stabilization of misfolded ensembles. Some self-recognition contacts are particularly strong. Swapped contacts are also strong as would be anticipated by the principle of minimal frustration. By making point mutations, we can change the relative strength of both interactions and study their competing roles in forming various misfolded structures. As shown in Fig. 4, the I27-I27 fused dimer, where strong self-recognition interactions are present, favors misfolded structures with these interactions satisfied. Forming these contacts in turn inhibits misfolded structures with many swapped contacts from forming. When point mutations were made to reduce the stability of these hexamer pairs, the structures with a significant number of swapped contacts appear more often. The converse is true for SH3-SH3. The point mutation E27I introduces a strong hexamer pair, promoting the formation of misfolded structures with the hexamer pair forming instead of forming domain-swapped structures. Both types of contacts can be strong, and depending on the specific sequence and simulation conditions, one or both can contribute to misfolding.
Fig. 4.

Competing roles of the swapped contacts and self-recognition contacts in the “wild-type” (WT) fused dimer I27-I27, SH3-SH3, and their mutants (MT) I27*-I27* and SH3*-SH3*. Forty annealing simulations were carried out for all dimers. 〈Ncontacts〉 is the number of contacts averaged over all 40 simulations. Nintercontact is the number of interdomain contacts. The interdomain contacts include the swapped contacts, self-recognition contacts, and other types of interdomain contacts. (A) Single-point mutation introduces a pair of strong self-recognition contacts in SH3*-SH3*, suppressing the formation of swapped contacts. Significant misfolding with formation of self-recognition contacts occurs after the mutation. (B) Two point mutations eliminated two pairs of the strongest self-recognition contacts in I27; therefore the swapped contacts play a more dominant role.
The single-molecule experiments of Borgia et al. (14) reveal some misfolding of I27-I27 while refolding under native conditions. Under these conditions only a small amount of misfolded proteins is formed. Using a symmetrized Go model Borgia et al. were able to propose several candidate misfolded (domain-swapped) structures and found that the FRET distances that they measured were consistent with these structures, although they were not able to distinguish between them. Interestingly, one of the domain-swapped structures that they observed in simulation was also found in our simulated annealing simulations of I27*-I27*, but was not found by us for I27-I27. In our simulations of I27-I27, the self-recognition hexamer pairing is so strong that it suppresses the formation of a fully domain-swapped structure. In I27*-I27*, the weakening of the self-recognition interactions allows domain-swapped interactions to predominate. To compare our structures with the experimental results of Borgia et al., we took the self-recognition ensemble of misfolded structures from the I27-I27 simulations and measured the distance between the residues that were labeled in the experiments, residues 3 and 83. The distribution of distances peaks around 2.3 nm, as shown in Fig. S2, very close to 2.0 nm, the distance in the domain-swapped structure proposed in their paper. For further experiments to clarify the detail of the misfolded structure, we suggest that two FRET labels be put near the strongly self-recognizing hexamer of each monomer. If the self-recognition contacts are in fact formed as predicted by the rapid simulated annealing of the present model and also remain stable under experimental conditions, the transfer efficiency should be high and strongly peaked for the newly labeled misfolded construct.
Folding and Misfolding from Single Domain to Multidomain: Implications for Aggregation.
In single-domain protein folding, the native interactions are in competition with nonnative intrachain interactions. Evolution has made these nonnative intrachain interactions weaker than the native ones so that proteins fold correctly on biological timescales. The misfolding problem gets more severe in the case of multidomain proteins or proteins at high concentration because of the possibility of forming strong interdomain contacts. Nearly all protein sequences have at least one amyloidogenic segment (27). Due to the strong interactions between them, multiple copies of these segments in proximity could cause the protein or proteins to adopt transiently stable trapped conformations that trigger further aggregation. For I27, as shown in Fig. 5A, inclusion of interdomain interactions in the AWSEM-Amylometer calculation reveals a specific self-recognizing hexamer interaction that is amyloidogenic according to our empirically determined threshold. It is important to remember that this interaction is simply not possible in the case of the monomer itself because only a single copy of the hexamer exists in a monomer. When simulated annealing runs were performed on the I27-I27 fused dimer, it was indeed this particular interaction that was the dominant cause for misfolding, as discussed previously. On the other hand, SH3-SH3 folds as well as its monomer because all of the hexamers in SH3 are only weakly self-recognizing. Our folding and misfolding results on the I27 and SH3 fused dimers are consistent with the experimental aggregation studies that show that the I27-I27 fused dimer aggregates but SH3-SH3 does not (13).
Fig. 5.

Comparisons of stability (energy per residue) among various zero-temperature structures and ensembles of thermally sampled structures of I27 (A and C) and SH3 (B). The stability for the native monomeric structure (green vertical line) is calculated from the AWSEM energy function. The strongest nonnative hexamer pairing possible in the monomer (magenta bar) is significantly less stable than the native structure, indicating that misfolding by inappropriate pairing of strands will be unlikely during the folding of the monomer for both I27 and SH3. In A and B, the blue bars represent the distribution of the stability of all of the self-recognition hexamer pairs, calculated from the AWSEM-Amylometer (SI Text). If the stability of the strongest self hexamer pair is competitive with the native structure, as in the case of I27-I27, the particular self pair becomes responsible for the misfolding of the fused protein in our simulation and potentially, would trigger further aggregation in solution. For SH3-SH3, B predicts that fused protein should fold as well as the monomer in the simulation, because all self hexamer pairings are weaker than the most stable nonnative hexamer pairing in the monomer. In C, the stability distributions of various ensembles of structures collected from the simulations of I27-I27 are shown. The native ensemble (green) is energetically more stable than both the domain-swapped ensemble (blue) and the self-recognition ensemble (red). Nevertheless, the local interactions between the self-pairing hexamers from the self-recognition ensembles, shown in cyan, are even stronger than typical energies in the native folded ensemble.
Self-recognizing segments tend to be short, typically between five and seven residues long, and so the interactions that stabilize the resulting misfolded structures are highly localized in sequence. This short sequence length is about that of a Kuhn statistical segment of a polypeptide (29) so the entropy loss in such self-recognition is comparable to that of bringing a single pair of residues together. This allows self-recognition to occur locally. Therefore, as shown in Fig. 5C, even though the local self interactions between the hexamers are much stronger than the average native interactions, the self-recognition ensemble is energetically less stable than the native ensemble. Swapped contacts are not as strong as the strongest self-recognition contacts but are present diffusely throughout the sequence. Forming domain-swapped structures requires the cooperation of contacts throughout the sequence but results in structures that are closer in energy to the natively folded structure. Both of these types of contacts can lead to oligomerization, but the resulting misfolded structures have different thermodynamic contributions to their stability. To what extent these two types of interaction contribute to the different stages of pathological aggregate formation in vivo remains an open question, but the coincidence of detecting amyloid diseases and occurrence of fevers in patients is intriguing (30).
Conclusion
We investigated misfolding in fused dimer constructs. Our results are consistent with the experiments of Borgia et al. (14) who have shown that aggregation rates are higher for fused dimers with high sequence identity between the domains and that the interaction between the two domains of the same molecule is the rate-determining step in this process. Nature’s strategy of lowering the sequence identity between nearby domains can now be understood as reducing both the number of strong domain-swapped contacts and the number of self-recognition contacts, in turn lowering the chance of misfolding. Our misfolded structure ensembles, both domain swapped and self-recognized, are also consistent with the FRET distances obtained by the single-molecule studies of Borgia et al. (14). Both ensembles are energetically less favored than the native ensemble, and the self-recognized structures are particularly favored by entropy owing to their using strong local interactions but having otherwise largely unfolded structure. The unfolded nature of the self-recognizing structures would seem to make them particularly prone to triggering aggregation, although this has not been directly studied here. How multiple copies of self-recognizing structures interact and order is currently under investigation. Finally, our study and others suggest that aggregation propensity under mildly denaturing conditions is sequence specific, and self-recognition in particular is highly local. This bodes well for future therapeutic intervention efforts and provides an optimistic contrast to the commonly held notion that the amyloid state is a low-energy “black hole” state of all proteins.
Supplementary Material
Acknowledgments
We thank Jane Clarke and Mikael Oliveberg for stimulating discussions. We also thank Yaakov Levy and Mikael Oliveberg for a critical reading of the manuscript. W.Z. and N.P.S. were supported by Grants R01 GM44557 and P01 GM071862 from the National Institute of General Medical Sciences. Additional support was also provided by the D. R. Bullard-Welch Chair at Rice University.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1222130110/-/DCSupplemental.
References
- 1.Wolynes PG. Energy landscapes and solved protein-folding problems. Philos Trans R Soc A. 2005;363:453–467. doi: 10.1098/rsta.2004.1502. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426(6968):900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 3.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 4.Silow M, Tan Y-J, Fersht AR, Oliveberg M. Formation of short-lived protein aggregates directly from the coil in two-state folding. Biochemistry. 1999;38(40):13006–13012. doi: 10.1021/bi9909997. [DOI] [PubMed] [Google Scholar]
- 5.Yang S, et al. Domain swapping is a consequence of minimal frustration. Proc Natl Acad Sci USA. 2004;101(38):13786–13791. doi: 10.1073/pnas.0403724101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sambashivan S, Liu Y, Sawaya MR, Gingery M, Eisenberg D. Amyloid-like fibrils of ribonuclease A with three-dimensional domain-swapped and native-like structure. Nature. 2005;437(7056):266–269. doi: 10.1038/nature03916. [DOI] [PubMed] [Google Scholar]
- 7.Bennett MJ, Sawaya MR, Eisenberg D. Deposition diseases and 3D domain swapping. Structure. 2006;14(5):811–824. doi: 10.1016/j.str.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 8.Oliveberg M. Alternative explanations for “Multistate” kinetics in protein folding: Transient aggregation and changing transition-state ensembles. Acc Chem Res. 1998;31:765–772. [Google Scholar]
- 9.Otzen DE, Kristensen O, Oliveberg M. Designed protein tetramer zipped together with a hydrophobic Alzheimer homology: A structural clue to amyloid assembly. Proc Natl Acad Sci USA. 2000;97(18):9907–9912. doi: 10.1073/pnas.160086297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacPhee CE, Dobson CM. Formation of mixed fibrils demonstrates the generic nature and potential utility of amyloid nanostructures. J Am Chem Soc. 2000;122:12707–12713. [Google Scholar]
- 11.Fernandez-Escamilla A-M, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22(10):1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- 12.Tartaglia GG, et al. Prediction of aggregation-prone regions in structured proteins. J Mol Biol. 2008;380(2):425–436. doi: 10.1016/j.jmb.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 13.Wright CF, Teichmann SA, Clarke J, Dobson CM. The importance of sequence diversity in the aggregation and evolution of proteins. Nature. 2005;438(7069):878–881. doi: 10.1038/nature04195. [DOI] [PubMed] [Google Scholar]
- 14.Borgia MB, et al. Single-molecule fluorescence reveals sequence-specific misfolding in multidomain proteins. Nature. 2011;474(7353):662–665. doi: 10.1038/nature10099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han J-H, Batey S, Nickson AA, Teichmann SA, Clarke J. The folding and evolution of multidomain proteins. Nat Rev Mol Cell Biol. 2007;8(4):319–330. doi: 10.1038/nrm2144. [DOI] [PubMed] [Google Scholar]
- 16.Bryngelson JD, Wolynes PG. Spin glasses and the statistical mechanics of protein folding. Proc Natl Acad Sci USA. 1987;84(21):7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papoian GA, Ulander J, Eastwood MP, Luthey-Schulten Z, Wolynes PG. Water in protein structure prediction. Proc Natl Acad Sci USA. 2004;101(10):3352–3357. doi: 10.1073/pnas.0307851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davtyan A, et al. AWSEM-MD. Protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J Phys Chem B. 2012;116(29):8494–8503. doi: 10.1021/jp212541y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng W, Schafer NP, Davtyan A, Papoian GA, Wolynes PG. Predictive energy landscapes for protein-protein association. Proc Natl Acad Sci USA. 2012;109(47):19244–19249. doi: 10.1073/pnas.1216215109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frousios KK, Iconomidou VA, Karletidi C-M, Hamodrakas SJ. Amyloidogenic determinants are usually not buried. BMC Struct Biol. 2009;9:44. doi: 10.1186/1472-6807-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galzitskaya OV, Garbuzynskiy SO, Lobanov MY. Prediction of amyloidogenic and disordered regions in protein chains. PLoS Comput Biol. 2006;2(12):e177. doi: 10.1371/journal.pcbi.0020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamodrakas SJ, Liappa C, Iconomidou VA. Consensus prediction of amyloidogenic determinants in amyloid fibril-forming proteins. Int J Biol Macromol. 2007;41(3):295–300. doi: 10.1016/j.ijbiomac.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 23.López de la Paz M, Serrano L. Sequence determinants of amyloid fibril formation. Proc Natl Acad Sci USA. 2004;101(1):87–92. doi: 10.1073/pnas.2634884100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z, Chen H, Lai L. Identification of amyloid fibril-forming segments based on structure and residue-based statistical potential. Bioinformatics. 2007;23(17):2218–2225. doi: 10.1093/bioinformatics/btm325. [DOI] [PubMed] [Google Scholar]
- 25.Sawaya MR, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447(7143):453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz R, Istrail S, King J. Frequencies of amino acid strings in globular protein sequences indicate suppression of blocks of consecutive hydrophobic residues. Protein Sci. 2001;10(5):1023–1031. doi: 10.1110/ps.33201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci USA. 2010;107(8):3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferreiro DU, Hegler JA, Komives EA, Wolynes PG. Localizing frustration in native proteins and protein assemblies. Proc Natl Acad Sci USA. 2007;104(50):19819–19824. doi: 10.1073/pnas.0709915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flory P, et al. Statistical mechanics of chain molecules. Biopolymers. 2004;8:699–700. [Google Scholar]
- 30.Wolynes PG, Eaton W. The physics of protein folding. Phys World. 1999;12:39–44. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.