

Abstract
Alzheimer’s disease (AD) is the most common cause of dementia worldwide. The pathogenesis of this neurodegenerative disease, currently without curative treatment, is associated with the accumulation of amyloid β (Aβ) in brain parenchyma and cerebral vasculature. AD patients are unable to clear this toxic peptide, leading to Aβ accumulation in their brains and, presumably, the pathology associated with this devastating disease. Compounds that stimulate the immune system to clear Aβ may therefore have great therapeutic potential in AD patients. Monophosphoryl lipid A (MPL) is an LPS-derived Toll-like receptor 4 agonist that exhibits unique immunomodulatory properties at doses that are nonpyrogenic. We show here that repeated systemic injections of MPL, but not LPS, significantly improved AD-related pathology in APPswe/PS1 mice. MPL treatment led to a significant reduction in Aβ load in the brain of these mice, as well as enhanced cognitive function. MPL induced a potent phagocytic response by microglia while triggering a moderate inflammatory reaction. Our data suggest that the Toll-like receptor 4 agonist MPL may be a treatment for AD.
Keywords: innate immunity, microglial cells, monocytes, phagocytosis, inflammation
Alzheimer’s disease (AD) is a neurodegenerative pathology characterized by the accumulation of amyloid beta (Aβ) and neurofibrillary tangles in the brain parenchyma (1). Inflammation, which occurs in parallel with the progression of the disease, is featured by the production of cytokines by activated microglia. The role of these cells in the pathogenesis of AD remains unclear and is an area of active investigation. Whereas chronic activation of microglial cells by Aβ can trigger the exaggerated release of cytokines and neurotoxic mediators that could be detrimental to neurons, microglia can also clear Aβ via increased phagocytosis and proteolytic degradation, which may be neuroprotective (2).
Toll-like receptors (TLRs) on the surface of microglial cells have been shown to bind Aβ, which triggers downstream intracellular signaling cascades (3, 4). Microglia deficient in TLR2, TLR4, or the coreceptor CD14 are not activated by Aβ and do not exhibit a phagocytic response (5). Transgenic AD mice lacking TLR4 have markedly elevated levels of diffuse and fibrillar Aβ (3). Furthermore, stimulation of microglial cells with TLR2-, TLR4-, or TLR9-specific agonists accelerates Aβ clearance both in vitro and in vivo (3, 6, 7).
Monophosphoryl lipid A (MPL) is a chemically detoxified lipid A moiety derived from Salmonella minnesota R595 LPS (8). This TLR4 ligand is at least 100-fold less pyrogenic than LPS yet maintains many of the immunomodulatory properties of LPS (9). Importantly, MPL is safe in humans and has been administered to millions of patients as a component of several vaccine formulations such as the Cervarix vaccine (10). We investigated herein the chronic use of the nonpyrogenic TLR4 agonist MPL and compared it with a strong TLR4 ligand (LPS) in a mouse model of AD.
Although the therapeutic potential of innate immune activation for AD is being evaluated in preclinical models, this concept has not been tested in humans. We propose that the age-related defects in immune cell function (11) commonly found in aging diseases such as AD (12) can be reconciled with a prophagocytic phenotype, yet mildly proinflammatory, which may lead to an improved clearance of Aβ.
Our data demonstrate that chronic, systemic administration of MPL ameliorates AD-like pathology by decreasing the cerebral Aβ load through the stimulation of the phagocytic capacity of innate immune cells.
Results
MPL Drives a Distinct TLR4 Stimulation from LPS.
MPL is derived from the LPS of the Gram-negative bacteria Salmonella minnesota R595 by three main chemical modifications: (i) elimination of the core oligosaccharide, (ii) hydrolysis of the 1-phosphate from the reducing end glucosamine, and (iii) removal of the acyl chain from the 3-position of the disaccharide (Fig. 1 A and B). The absence of the 1-phosphate on the MPL molecule was suggested to weaken the dimerization of TLR4/MD2 (myeloid differentiation factor-2) (13). This presumably induces a structural change in the TLR4 receptor complex that alters the recruitment of the adaptor proteins to the intracellular domain (13). Such a structural change may account for the distinct signaling properties of MPL, which predominantly activates the TLR4-TRAM (TRIF-related adaptor molecule)-TRIF (TIR-domain-containing adaptor protein inducing IFN-β) pathway over the more proinflammatory TLR4-MAL (MyD88-adaptor-like protein)-MyD88 (myeloid differentiation primary-response protein 88) signaling pathway (14). This differential use of intracellular adaptor proteins may be key to explaining the distinct effects observed after exposure of cells to MPL or LPS.
Fig. 1.

Distinct mechanisms mediating LPS- and MPL-induced TLR4 signaling. (A) Comparison of LPS from Salmonella minnesota R595 and (B) the major hexa-acyl monophoryl lipid A (MPL) chemically extracted from Salmonella minnesota R595. MPL is characterized by three main chemical modifications: (i) elimination of the core oligosaccharide, (ii) hydrolysis of the 1-phosphate from the reducing end glucosamine, and (iii) removal of the acyl chain from the 3-position of the disaccharide. (C) TLR4-specific activation of NF-κB and AP-1 in HEK293 cells expressing TLR4, MD-2, and CD14 following stimulation with MPL and LPS for 5 h (n = 3). In contrast, (D) no TLR2 activation is observed in HEK293-TLR2 cells from MPL when used in vitro at concentrations up to 2.5 µg/mL. The TLR2 ligand Pam3CSK4 was used as a positive control (n = 3). Using the same concentration of ligands (1 μg/mL), (E) degradation of IκBα in BV-2 microglia is delayed and milder following MPL treatment compared with LPS. BV-2 cells incubated with MPL exhibit significantly lower (F) ERK1/2 and (G) JNK1/2 phosphorylation compared with LPS. However, (H) p38 phosphorylation in MPL-stimulated BV-2 cells was not statistically different from that in LPS-treated cells (n = 4–6 for E–H). The lanes were run on the same gel but were noncontiguous. Data are expressed as the means ± SEM; **P < 0.01 vs. PBS.
To characterize the ligand–receptor interaction of MPL with the TLR4 receptor complex we used the HEK293 cell line transfected with TLR4, MD2, and CD14 genes, as well as an NF-κB and AP-1 reporter system. At the highest concentration of MPL tested (20 μg/mL), the activation of NF-κB and AP-1 was at a level comparable to a 200-fold lower concentration of LPS (0.1 μg/mL) (Fig. 1C). A neutralizing antibody directed against TLR4 inhibited the response to MPL. Incubating TLR2-transfected HEK293 cells with up to 2.5 μg/mL of MPL did not induce any activation of NF-κB and AP-1 (Fig. 1D), indicating that the activation was mediated only by TLR4 (because not more than 1 μg/mL MPL was used to treat cells in the following experiments). In accordance with the results observed in HEK293 cells, the degradation kinetics of the NF-κB inhibitor, IκBα, seemed both delayed and of a lower magnitude in MPL-treated microglia, even though the difference between MPL and LPS did not reach statistical significance (Fig. 1E).
In addition to NF-κB activation, MAPK cascades transduce signals from TLR activation, with effects on gene transcription and posttranscriptional modifications of cytoplasmic targets (15). We assessed the phosphorylation kinetics of three MAPKs (ERK, JNK, and p38) after stimulation of microglia with LPS and MPL. LPS induced strong phosphorylation of both ERK and JNK in microglia at 30 and 60 min poststimulation, whereas MPL had little effect (Fig. 1 F and G). LPS and MPL induced a similar level of p38 phosphorylation in microglial cells (Fig. 1H). This finding confirms a previous report (16) that both of these TLR4 ligands activate the p38 pathway to the same degree.
MPL Induces a Low Inflammatory Response by Microglia in Vitro.
Microglial cells are highly dynamic cells that can undergo rapid actin cytoskeleton remodeling to migrate or phagocytose foreign material. In vitro microglia activated by LPS exhibit a profound alteration in cell morphology, changing from a nonactivated amoeboid shape (Fig. 2A) to an elongated and multipolar morphology (Fig. 2B). MPL also stimulated cytoskeletal remodeling (Fig. 2C), but to a lower extent than LPS. Similar differences (MPL < LPS) were observed for cell migration (Fig. 2E and Fig. S1). To evaluate whether these morphological/motility changes correlated with innate immune activation, we compared the expression of cellular markers of microglia activation after treatment with either MPL or LPS. In contrast to LPS, MPL treatment did not induce the expression of cyclooxygenase-2 (COX-2) (Fig. 2F) or formation of nitrite (Fig. 2H), two classic markers of LPS-induced microglial activation. Whereas MPL did not trigger the production of cytokine-inducible NOS (iNOS) in microglia (Fig. 2I), treatment with either MPL or LPS led to similar decreases in Arginase 1 (Arg1) protein expression (Fig. 2J), a sensitive marker of alternative macrophage activation (17). This finding was unexpected because these two enzymes are generally inversely regulated (17). This reflects the multifaceted effects of the distinct TLR4 stimulation by MPL. Microglia treated with MPL induced both TNF-α mRNA (Fig. 2K) and protein (Fig. 2G) expression, although at lower levels compared with LPS. Both MPL and LPS induced similar levels of CCL2 mRNA expression (Fig. 2L). MPL and LPS induced equivalent levels of TLR2 gene expression after 2 h of incubation (Fig. 2M), but the level in MPL-treated cells was lower at 4 and 24 h (Fig. S2). These results demonstrate that MPL induces a low proinflammatory and transitory innate immune response in microglia while still inducing characteristic properties such as migration and cytoskeletal remodeling.
Fig. 2.

MPL induces a low inflammatory response in microglia. BV-2 microglia were stimulated for 24 h with 1 μg/mL of LPS or MPL. F-actin (green) was stained with phalloidin to expose different cell morphologies: (A) nonactivated amoeboid shape, (B) LPS-activated elongated and multipolar morphology, and (C) an MPL-induced intermediate phenotype. (Scale bar, 100 μm.) (D) Total protrusion length was measured in a minimum of 70 cells per treatment group. (E) The migration of BV-2 cells was assessed using the scratch assay test (n = 3). (F) No induction of COX-2 was observed following MPL stimulation in contrast to LPS (n = 3–4). (G) A strong TNF-α secretion was seen in the media of LPS-incubated cells, whereas low levels were detected for MPL-treated cells (n = 6). (H and I) MPL did not generate nitrites or iNOS expression in comparison with the robust induction by LPS (n = 3–6). (J) Arginase 1 levels were similarly reduced following LPS or MPL stimulation (n = 3). (K) Two hours after cell stimulation, the transcriptional activation of TNF-α was lower in MPL-treated cells than in those treated with LPS. (L and M) Both MPL and LPS induced comparable levels of CCL2 and TLR2 mRNA at 2 h poststimulation (n = 3). The lanes were run on the same gel but were noncontiguous. Data are expressed as the means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (vs. PBS); &&P < 0.01, &&&P < 0.001 (vs. LPS).
MPL Induces a Strong Phagocytic Response.
Because MPL stimulated actin rearrangement and migration in microglia, we evaluated whether it could also modulate the phagocytic potential of these cells. Microglial cells treated with MPL showed a significant increase in their ability to phagocytize fluorescent beads (Fig. 3 A and B). We then investigated the ability of MPL-treated cells to internalize Aβ peptides. MPL-stimulated microglia exhibited a significant increase in the internalization of Aβ1–42 oligomers (Fig. 3C). Phagocytized Aβ oligomers colocalized with Lamp-2 in MPL-treated microglia, suggesting they were transported to lysosomes following phagocytosis (Fig. 3D and Fig. S3).
Fig. 3.

MPL stimulates phagocytosis in microglia and monocytes. BV-2 microglia were stimulated with 1 μg/mL of LPS or MPL for 18 h. (A) Both MPL and LPS stimulate the phagocytosis of fluorescent Escherichia coli beads by microglia (n = 3). (B) The intracellular localization of these beads (green) was validated by confocal microscopy. (Scale bar, 10 μm.) (C) Mean fluorescence intensity of internalized fluorescent Aβ oligomers was evaluated by flow cytometry and was increased after treatment with LPS or MPL. (D) Intracellular Aβ oligomers (red) were colocalizing with the lysosomal Lamp-2 proteins (green). (Scale bar, 10 μm.) (E) Distinct wild-type mice received a single LPS (20 μg) or MPL (50 μg) i.p. injection, and 24 h later 5 μg of fluorescent Aβ was injected via tail vein. Monocytes were isolated 2 h later and significantly more Aβ-containing cells were observed in MPL- and LPS-injected mice. (F) Scavenger receptor A (SR-A) mRNA was induced in microglia after 24 h of incubation with LPS or MPL. Data are expressed as the means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (vs. PBS); &P < 0.05, &&P < 0.01, &&&P < 0.001 (vs. LPS).
Monocytes are cells that would likely be targeted by peripheral injection of MPL. Interestingly, increased internalization of Aβ1–42 in vivo was also demonstrated in the blood of MPL-treated murine monocytes (Fig. 3E). The phagocytic response observed in MPL-treated microglia correlates with actin remodeling (Fig. 2 D and E) and induction of scavenger receptor A (SR-A) expression (Fig. 3F), both of which are crucial for internalization of extracellular material such as Aβ. On the surface of microglia, SR-As are important for adherence and phagocytosis of Aβ (18) but do not stimulate the production of reactive oxygen species (19). These results demonstrate that MPL induces a strong phagocytic response.
MPL Induces a Low Inflammatory Response While Triggering a Strong Monocytopoiesis in Mice.
To determine whether the low inflammatory response induced by MPL in vitro could be reproduced in vivo, we measured several cytokines and chemokines in the sera of wild-type C57BL/6 mice 2 and 6 h following a single i.p. injection of either MPL or LPS. MPL elevated the levels of most cytokines and chemokines that were measured, but the levels were substantially lower than what was observed in LPS-treated animals (Fig. 4). LPS induced considerably more TNF-α and IL-6 than MPL at 2 h postinjection (Fig. 4 A and B), and these cytokines were no longer detectable 6 h after MPL injection. MPL did not stimulate the release of IL-1β at the two time points examined (Fig. 4D). The levels of the chemokines CCL3, IP-10, CXCL-1, and CCL2 were all increased in MPL-treated mice but to lower concentrations than in the LPS group (Fig. 4 F–I).
Fig. 4.

Mild activation of the immune system in response to systemic MPL injection in mice. (A–I) Cytokine and chemokine profile in sera of wild-type mice 2 and 6 h after an i.p. injection of LPS (20 μg) or MPL (50 μg). Results are shown in relative units (RU), pg/mL or ng/mL (n = 4–5). The bars represent mean ± SEM. (J) Representative TLR2 mRNA in situ hybridization in brains of mice injected i.p. with PBS, MPL (50 μg), or LPS (3 μg). Twenty-four hours after the injection, LPS provoked a strong TLR2 induction in the circumventricular organs, choroid plexus, and brain parenchyma, whereas MPL triggered weak expression, mainly in circumventricular organs. (Scale bar, 500 μm.) *P < 0.05.
We also examined the inflammatory response in the brains of these mice by analyzing TLR2 and TNF-α mRNA expression, two sensitive markers of microglia activation (20). LPS induced robust TLR2 and TNF-α expression in circumventricular organs (CVOs), choroid plexus, and throughout the brain parenchyma, whereas MPL triggered low expression mainly in CVOs (Fig. 4J and Fig. S4). Interestingly, whereas i.p. injection of either MPL or LPS stimulated similar expansion of blood monocytes (Fig. 5), MPL induced a milder proinflammatory cytokine response compared with LPS.
Fig. 5.
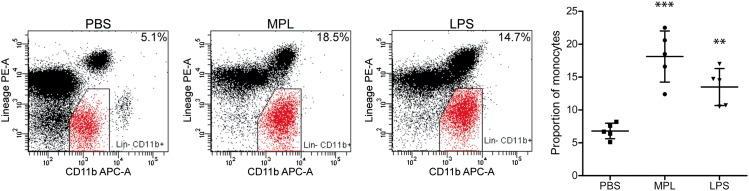
MPL triggers the expansion of blood monocytes in mice. Wild-type mice received a single i.p. injection of PBS, MPL (50 μg), or LPS (20 μg) and their blood was analyzed 24 h later by flow cytometry. Cells were considered to be monocytes by their expression of CD11b and the lack of lineage (Lin−) markers: CD3−, NK1.1−, Ly6G−, and B220− (n = 5). Data are expressed as the percentage of total CD45+ leukocytes excluding debris. The bars represent mean ± SD; **P < 0.01, ***P < 0.001 (vs. PBS).
MPL Treatment Improves AD-Related Pathology of APPswe/PS1 Mice.
To investigate whether treatment with MPL might affect AD-related pathology in vivo, MPL, LPS, or PBS was administered weekly by i.p. injection to APPswe/PS1 mice for 12 consecutive weeks beginning when the mice were 3 mo old. Cognitive function and Aβ deposition were assessed for each mouse 2 and 3 wk after the final injection, respectively. Using a T water maze behavioral test to assess hippocampus-based spatial learning and memory, experiments were conducted to determine whether clearance of Aβ correlated with improved cognitive functions in these mice. Compared with the PBS-treated control group, the MPL-treated APPswe/PS1 mice showed significant improvement in cognitive functions (Fig. 6I). LPS treatment, however, did not lead to significant improvement in the cognitive performances of APPswe/PS1 mice.
Fig. 6.

MPL treatment reduces Aβ levels and cognitive deficits in APPswe/PS1 mice. MPL (50 μg), LPS (3 μg), or PBS was administered once a week by i.p. injection in 3-mo-old APPswe/PS1 mice for 12 consecutive weeks. (A–C) Representative Aβ immunoreactivity in cortex and hippocampus is shown in brain sections of APPswe/PS1 mice injected with PBS, MPL, or LPS. (Scale bar, 500 μm.) (D and E) Compared with the PBS control group, the number and the area of Aβ plaques are significantly reduced in the cortex of MPL-treated mice (n = 9). (F) Extracellular Aβ monomers in mouse brains were quantified by Western blot analysis. (G) MPL-treated mice had significantly fewer monomers compared with controls, whereas the LPS group remained essentially unchanged (n = 8). (H) Analysis of CD45+ brain cells in 10-mo-old APPswe/PS1 after five consecutive daily i.p. injections of MPL (25 μg). Flow cytometry analysis of CD45+ brain cells that were immunoreactive for intracellular Aβ (using monoclonal anti-Aβ 6E10 antibody) revealed a significant increase in MPL-injected mice compared with the PBS control group (n = 4–5). (I) The hippocampus-based spatial learning and memory of APPswe/PS1 mice was evaluated in the T water maze behavioral test. APPswe/PS1 mice treated with MPL had a significant improvement of their cognitive functions as shown by their lower number of trials to reach the criterion in the reversal phase of the test (n = 9–19). Each point represents a single mouse and the horizontal bars are the mean for each group. *P < 0.05, **P < 0.01 (vs. PBS).
Administration of MPL caused a significant reduction in the number and size of Aβ deposits, as well as the quantity of soluble Aβ in the brain (P = 0.0020, 0.0018, and 0.0027, respectively, versus PBS control group; Fig. 6 A–G). Compared with controls, the size and number of Aβ plaques were considerably greater in LPS-treated animals, whereas the level of soluble Aβ monomers was equivalent. The percentage of CD45+ brain cells that contained Aβ after i.p. injections of MPL was determined to investigate whether MPL treatment led to lower Aβ levels by promoting phagocytosis of Aβ. The brains of mice treated with MPL had significantly higher percentages of Aβ-positive cells than those of the control group (approximately five times higher; Fig. 6H). These data showed that MPL treatment can stimulate the clearance of Aβ and improve cognitive function of APPswe/PS1 mice. To understand whether an increase in the infiltration of bone marrow-derived microglia could be responsible for these beneficial effects, we generated APPswe/PS1 chimeric mice in which bone marrow cells were GFP+/−. The number of these cells, mostly found in perivascular spaces and the choroid plexus, was not increased following 12 weekly injections of MPL in APPswe/PS1 mice (Fig. S5).
Repeated Weekly Injections of MPL in APPswe/PS1 Mice Generate a Transient Innate Immune Response and Do Not Create an Immune Tolerance.
We examined the pathways previously explored (following an acute injection of MPL) to assess the effects of the chronic treatment regimen used in APPswe/PS1 mice. Both TLR2 and TNF-α gene expression in the CVOs vanished 3 wk after the last injection of MPL (Fig. S6). In chronically LPS-challenged mice, we could not observe TNF-α mRNA in the brain, and we detected a very weak or no TLR2 signal. In the periphery, monocytes were at normal levels 6 d after the last injection (Fig. S7B). Blood TNF-α, IL-6, IL-10, CCL2, IFN-γ, and IL-12p70 were all at baseline concentrations 3 wk after the last injection and not statistically different among treatment groups (Fig. S8). To evaluate whether repeated weekly injections of MPL induced immune tolerance in APPswe/PS1 mice, we measured its ability to trigger TNF-α and CCL2 release (Fig. S9) as well as expansion of monocytes (Fig. S7A) in blood after 12 weekly injections. Essentially, MPL had effects similar to a single acute injection (Figs. 4 and 5). This is in line with evidence showing that endotoxin tolerance is not maintained more than 5 d (21). These data demonstrate that repeated weekly MPL injections do not produce a sustained state of immune activation and that the ligand remains able to activate TLR4 similarly to a single acute injection.
Discussion
The data reported here show that the TLR4 ligand MPL induces phagocytosis of Aβ by mononuclear phagocytes yet elicits an inflammatory response that is considerably lower than generated by LPS. Chronic systemic administration of MPL significantly improved AD-related pathology in treated APPswe/PS1 mice, evidenced by reduced levels of Aβ in the brain and improved cognitive function.
Although a strong inflammatory response is potentially harmful, many studies highlighting the efficacy of finely tuned innate immune activation for treatment of AD in transgenic mice have been published (2, 22). For instance, induction of the proinflammatory cytokines IL-1β, TNF-α, IL-6, and IFN-γ correlated with reduced Aβ deposition in AD mouse models (23–26). Recently, TNF-α receptor ablation was shown to enhance AD pathology in transgenic mice, likely because of impaired Aβ phagocytosis (27). Additionally, activation of microglia has been reported to induce lysosome acidification and increased degradation of Aβ (28). In this regard, MPL seems to induce a suitable degree of innate immune stimulation that reduces the accumulation of Aβ and improves spatial memory in APPswe/PS1 mice.
Stimulation of microglia and monocytes with MPL induced increased phagocytosis of Aβ by these cells. Whereas it is widely accepted that p38, ERK, JNK, and NF-κB all induce TLR-mediated cytokine production, p38 has been shown to up-regulate scavenger receptor expression and induction of phagocytic activity (29, 30). Previous studies have also shown that the Aβ phagocytic activity involving cell surface receptors such as SR-A is altered in AD and aged mice (31, 32). Although MPL does not induce ERK or JNK and stimulates NF-κB to a lesser extent than LPS, it does activate p38 strongly and drives the expression of SR-A in microglial cells. These findings suggest that MPL selectively stimulates p38 and promotes Aβ uptake while avoiding the extensive production of potentially harmful proinflammatory mediators induced by other signaling pathways.
There are several possibilities to explain how peripheral administration of MPL reduces Aβ levels in the brain and restricts cognitive deficits in APPswe/PS1 mice. First, MPL-activated, highly Aβ phagocytic myeloid cells in the periphery could act as a sink for Aβ in the blood. This sink effect could contribute to reduce the level of Aβ in the brain via equilibrium-driven redistribution of Aβ to the periphery, as observed in Aβ-specific immunotherapy (33). This is in line with the increase in Aβ phagocytosis by blood monocytes stimulated with MPL. However, the increased proportion of CD45+ cells containing Aβ found in the brain of MPL-treated mice suggests that peripheral clearance is not the exclusive mechanism involved in eliminating Aβ. In this regard, it is noteworthy that CD45+ microglial cells have been shown to promote the clearance of oligomeric Aβ (34). A compatible mode of action could be the stimulation of microglia or macrophages located in the CVOs, which lay outside the blood–brain barrier (BBB). Once activated, these cells can mediate progressive release of cytokines across the BBB and into the brain parenchyma (20). Finally, systemic administration of MPL may promote mobilization of microglial precursors from bone marrow, which move into the brain perivascular spaces and clear Aβ via phagocytosis (35, 36).
Although LPS was found to induce Aβ phagocytosis better than MPL in microglia and monocytes, chronic systemic LPS administration in APPswe/PS1 mice exacerbated the Aβ plaque load. This result is consistent with published studies showing that acute injection of LPS activates microglia and increases Aβ phagocytosis (6, 37), whereas chronic exposure to LPS leads to higher deposition of Aβ (38, 39). Excessive and sustained inflammation produced by chronic LPS stimulation is thought to be the main cause for this phenomenon (38–40). Indeed, multiple lines of evidence link inflammatory mediators to increased amyloidogenesis by different mechanisms, including enhanced transcription (41) and translation (42) of the APP gene and JNK-dependent stimulation of gamma-secretase enzymatic activity (43). The findings that MPL induces increased Aβ phagocytosis, but only modest inflammation, could explain the highly efficient reduction of Aβ levels in vivo by this TLR4 ligand compared with LPS.
Mawuenyega et al. (44) recently demonstrated that AD is characterized by an impairment in the clearance of Aβ, rather than increased production. Notably, the reduction of factors associated with hematopoiesis and phagocytosis has been associated with development of AD (12). Moreover, it has been shown that myeloid cells from patients with AD and mild cognitive impairment phagocytose Aβ less efficiently than cells from normal controls (31). Therapeutics promoting the elimination of Aβ by scavenger cells such as monocytes, macrophages, or microglia could be valuable tools in the fight against AD. We demonstrate here that TLR4 stimulation with the detoxified ligand MPL significantly improved AD-related pathology. Although the safety of the MPL treatment regimen used here has not been confirmed in humans, this compound has been administered to hundreds of thousands of humans as an adjuvant in different vaccines (9) and is currently used as a component of a marketed human vaccine (Cervarix). Based on our findings, we propose that MPL holds great promise as a safe and effective treatment for this neurodegenerative disease.
Materials and Methods
Materials and methods are described in detail in SI Materials and Methods.
Ligand Preparation.
The MPL immunostimulant was produced at GlaxoSmithKline from the LPS of S. minnesota R595 following alkaline procedures, which have been described previously (45, 46).
NF-κB/AP-1 Activation Assay.
Transfected HEK 293 cells with the expression vectors encoding human TLR4, MD-2, and CD14 were further stably transfected with the NF-κB reporter vector pNifty-2 secreted alkaline phosphatase. Cells were stimulated for 5 h in FCS-free medium containing different concentrations of MPL and LPS or PBS.
Quantification of Cytokine and Chemokine Levels in Mouse Sera.
Blood (sera) cytokine and chemokine levels were measured in C57BL/6 mice using ELISA or a Luminex mouse cytokine-chemokine kit after the i.p. injection of either LPS (20 μg) or MPL (50 μg) at 2 or 6 h postinjection.
TLR2 in Situ Hybridization.
In situ hybridization histochemical localization of TLR2 on brain slices was performed using 35S-labeled cRNA probes according to a protocol described previously (47).
FACS Analysis on Mouse Whole Blood and Aβ1–42 Uptake by Monocytes.
C57BL/6 mice received an i.p. injection of PBS, MPL (50 µg), or LPS (20 µg). Twenty-four hours after treatment, mice received 5 µg of fluorescent HiLyte Fluor 488-labeled Aβ1–42 via a tail vein injection and peripheral blood was harvested 2 h later. Leukocytes were stained and acquired on a flow cytometer.
Transgenic Mouse Lines and Treatment.
All protocols were conducted according to the Canadian Council on Animal Care guidelines, as administered by the Laval University Animal Welfare Committee. MPL (50 μg), LPS (3 μg), or PBS was administered once a week by i.p. injection in 3-mo-old APPswe/PS1 mice for 12 consecutive weeks.
Aβ Plaque Immunofluorescence and Stereological Analysis.
To stain Aβ plaques, free-floating sections were immunolabeled with the monoclonal anti-Aβ (6E10). The number of plaques and the area occupied by all Aβ-labeled plaques were determined by stereological analysis as described previously (35).
Western Blot Analysis of Brain Samples.
Monomeric Aβ and β-actin were detected by immunoblotting with the monoclonal antibodies anti-Aβ (6E10) and anti-β-actin (13E5).
Isolation of Brain Leukocytes and FACS Analysis.
Brain leukocytes were isolated by Percoll gradient, stained with anti–CD45-PE and anti-Aβ (6E10), and analyzed by flow cytometry.
Left–Right Discrimination Learning.
Mice were placed at the stem of a water-filled T maze and choose to swim either left or right until they found the submerged platform and escaped to it. The reversal learning phase was then conducted 48 h later. During this phase, the same protocol was repeated except that the mice were trained to find the new location of the escape platform on the side opposite that on which they had learned during the acquisition phase.
Additional Information.
The experiments conducted with BV-2 cells are described in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Martine Lessard, Marie-Pier Girouard, and Marie-Michèle Plante for technical help. GlaxoSmithKline Vaccines and the Canadian Institutes in Health Research (CIHR) supported this research. J.-P.M. is supported by a doctoral Studentship from the CIHR, M.H. by a postdoctoral fellowship from the Natural Sciences and Engineering Research Council, and P.T. by a master scholarship from Bourse de Recherche en Milieu Pratique Innovation (Fonds Québécois de la Recherche sur la Nature et les Technologies–Conseil de Recherches en Sciences Naturelles et en Génie). S.R. holds a Canadian Research Chair in Neuroimmunology.
Footnotes
Conflict of interest statement: M.H., P.T.-J., A.-M.L., R.J., C.C., V.B., R.P., A.P., and D.L. are employees of GlaxoSmithKline Vaccines. This research was supported in part by GlaxoSmithKline Vaccines.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1215165110/-/DCSupplemental.
References
- 1.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362(4):329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 2.Wyss-Coray T. Inflammation in Alzheimer disease: Driving force, bystander or beneficial response? Nat Med. 2006;12(9):1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 3.Tahara K, et al. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129(Pt 11):3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer’s disease. Neurobiol Dis. 2010;37(3):503–509. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar Abeta-stimulated microglial activation. J Neurosci. 2009;29(38):11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herber DL, et al. Microglial activation is required for Abeta clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J Neuroimmune Pharmacol. 2007;2(2):222–231. doi: 10.1007/s11481-007-9069-z. [DOI] [PubMed] [Google Scholar]
- 7.Scholtzova H, et al. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J Neurosci. 2009;29(6):1846–1854. doi: 10.1523/JNEUROSCI.5715-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casella CR, Mitchell TC. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell Mol Life Sci. 2008;65(20):3231–3240. doi: 10.1007/s00018-008-8228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cluff CW. Monophosphoryl lipid A (MPL) as an adjuvant for anti-cancer vaccines: Clinical results. Adv Exp Med Biol. 2010;667:111–123. doi: 10.1007/978-1-4419-1603-7_10. [DOI] [PubMed] [Google Scholar]
- 10.Garçon N, Van Mechelen M. Recent clinical experience with vaccines using MPL- and QS-21-containing adjuvant systems. Expert Rev Vaccines. 2011;10(4):471–486. doi: 10.1586/erv.11.29. [DOI] [PubMed] [Google Scholar]
- 11.Shaw AC, Joshi S, Greenwood H, Panda A, Lord JM. Aging of the innate immune system. Curr Opin Immunol. 2010;22(4):507–513. doi: 10.1016/j.coi.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ray S, et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007;13(11):1359–1362. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 13.Park BS, et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458(7242):1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 14.Mata-Haro V, et al. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316(5831):1628–1632. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 15.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 16.Cekic C, et al. Selective activation of the p38 MAPK pathway by synthetic monophosphoryl lipid A. J Biol Chem. 2009;284(46):31982–31991. doi: 10.1074/jbc.M109.046383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Gayar S, Thüring-Nahler H, Pfeilschifter J, Röllinghoff M, Bogdan C. Translational control of inducible nitric oxide synthase by IL-13 and arginine availability in inflammatory macrophages. J Immunol. 2003;171(9):4561–4568. doi: 10.4049/jimmunol.171.9.4561. [DOI] [PubMed] [Google Scholar]
- 18.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17(3):553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 19.Husemann J, Loike JD, Kodama T, Silverstein SC. Scavenger receptor class B type I (SR-BI) mediates adhesion of neonatal murine microglia to fibrillar beta-amyloid. J Neuroimmunol. 2001;114(1-2):142–150. doi: 10.1016/s0165-5728(01)00239-9. [DOI] [PubMed] [Google Scholar]
- 20.Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9(6):429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- 21.del Fresno C, et al. Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: Demonstration in isolated monocytes from cystic fibrosis patients. J Immunol. 2009;182(10):6494–6507. doi: 10.4049/jimmunol.0803350. [DOI] [PubMed] [Google Scholar]
- 22.Yong VW, Rivest S. Taking advantage of the systemic immune system to cure brain diseases. Neuron. 2009;64(1):55–60. doi: 10.1016/j.neuron.2009.09.035. [DOI] [PubMed] [Google Scholar]
- 23.Chakrabarty P, Herring A, Ceballos-Diaz C, Das P, Golde TE. Hippocampal expression of murine TNFα results in attenuation of amyloid deposition in vivo. Mol Neurodegener. 2011;6:16. doi: 10.1186/1750-1326-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chakrabarty P, et al. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J Immunol. 2010;184(9):5333–5343. doi: 10.4049/jimmunol.0903382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakrabarty P, et al. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: Evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010;24(2):548–559. doi: 10.1096/fj.09-141754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaftel SS, et al. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 2007;117(6):1595–1604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montgomery SL, et al. Ablation of TNF-RI/RII expression in Alzheimer’s disease mice leads to an unexpected enhancement of pathology: Implications for chronic pan-TNF-α suppressive therapeutic strategies in the brain. Am J Pathol. 2011;179(4):2053–2070. doi: 10.1016/j.ajpath.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majumdar A, et al. Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Mol Biol Cell. 2007;18(4):1490–1496. doi: 10.1091/mbc.E06-10-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doyle SE, et al. Toll-like receptors induce a phagocytic gene program through p38. J Exp Med. 2004;199(1):81–90. doi: 10.1084/jem.20031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong L, Ge B-X. MyD88-independent activation of a novel actin-Cdc42/Rac pathway is required for Toll-like receptor-stimulated phagocytosis. Cell Res. 2008;18(7):745–755. doi: 10.1038/cr.2008.65. [DOI] [PubMed] [Google Scholar]
- 31.Fiala M, Veerhuis R. Biomarkers of inflammation and amyloid-beta phagocytosis in patients at risk of Alzheimer disease. Exp Gerontol. 2010;45(1):57–63. doi: 10.1016/j.exger.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28(33):8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2009;8(1):16–30. doi: 10.2174/187152709787601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Y, et al. CD45 deficiency drives amyloid-β peptide oligomers and neuronal loss in Alzheimer’s disease mice. J Neurosci. 2011;31(4):1355–1365. doi: 10.1523/JNEUROSCI.3268-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simard AR, Soulet D, Gowing G, Julien J-P, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49(4):489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 36.Mildner A, et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J Neurosci. 2011;31(31):11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herber DL, et al. Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. 2004;190(1):245–253. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 38.Sheng JG, et al. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis. 2003;14(1):133–145. doi: 10.1016/s0969-9961(03)00069-x. [DOI] [PubMed] [Google Scholar]
- 39.Lee JW, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37. doi: 10.1186/1742-2094-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blasko I, Marx F, Steiner E, Hartmann T, Grubeck-Loebenstein B. TNFalpha plus IFNgamma induce the production of Alzheimer beta-amyloid peptides and decrease the secretion of APPs. FASEB J. 1999;13(1):63–68. doi: 10.1096/fasebj.13.1.63. [DOI] [PubMed] [Google Scholar]
- 41.Trejo J, et al. A direct role for protein kinase C and the transcription factor Jun/AP-1 in the regulation of the Alzheimer’s beta-amyloid precursor protein gene. J Biol Chem. 1994;269(34):21682–21690. [PubMed] [Google Scholar]
- 42.Rogers JT, et al. Translation of the alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5′-untranslated region sequences. J Biol Chem. 1999;274(10):6421–6431. doi: 10.1074/jbc.274.10.6421. [DOI] [PubMed] [Google Scholar]
- 43.Liao Y-F, Wang B-J, Cheng H-T, Kuo L-H, Wolfe MS. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279(47):49523–49532. doi: 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- 44.Mawuenyega KG, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330(6012):1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baldridge JR, Crane RT. Monophosphoryl lipid A (MPL) formulations for the next generation of vaccines. Methods. 1999;19(1):103–107. doi: 10.1006/meth.1999.0834. [DOI] [PubMed] [Google Scholar]
- 46.Ulrich JT, Myers KR. Monophosphoryl lipid A as an adjuvant: Past experiences and new directions. Pharm Biotechnol. 1995;6:495–524. [PubMed] [Google Scholar]
- 47.Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1beta in mediating NF-kappaB activity and COX-2 transcription in cells of the blood-brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci. 1999;19(24):10923–10930. doi: 10.1523/JNEUROSCI.19-24-10923.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.