

Abstract
The transmembrane (TM) and juxtamembrane (JM) regions of the ErbB family receptor tyrosine kinases connect the extracellular ligand-binding domain to the intracellular kinase domain. Evidence for the role of these regions in the mechanism of receptor dimerization and activation is provided by TM–JM peptides corresponding to the Neu (or rat ErbB2) receptor. Solid-state NMR and fluorescence spectroscopy show that there are tight interactions of the JM sequence with negatively charged lipids, including phosphatidylinositol 4,5-bisphosphate, in TM–JM peptides corresponding to the wild-type receptor sequence. We observe a release of the JM sequence from the negatively charged membrane surface using peptides containing an activating V664E mutation within the TM domain or in peptides engineered to form TM helix dimers with Val664 in the interface. These results provide the basis of a mechanism for coupling ligand binding to kinase activation in the full-length receptor.
Keywords: EGF receptor, HER2 receptor, PIP2
The receptor tyrosine kinases (RTKs) are a large family of membrane receptors that control cell growth, differentiation, and migration. These receptors have a three-domain architecture consisting of an extracellular ligand-binding domain, a single transmembrane helix, and an intracellular kinase domain. Receptor activation is triggered by ligand-mediated dimerization of receptor monomers or the structural rearrangement of inactive, preformed dimers (1, 2). Detailed information about the mechanisms of activation and regulation of RTKs has come largely from crystal structures of their extracellular and intracellular domains in inactive and active conformations. The activation mechanisms of the ErbB subfamily of RTKs are of particular interest because mutations and deletions that result in constitutive receptor activity have been identified in a number of human tumors (3, 4).
In the epidermal growth factor receptor (EGFR), a member of the ErbB subfamily, the structures of the extracellular domain with (5) and without (6) bound EGF revealed that the unliganded structure has a tethered conformation that undergoes a dramatic rearrangement upon ligand binding. On the intracellular side of the EGFR, crystal structures show that the activation loop associated with the active site in the kinase domain is in an open, active conformation before ligand binding (7) or locked in a Src tyrosine kinase/cyclin-dependent kinase-like inactive conformation (8). Activation is produced not via phosphorylation of the activation loop, as in other RTKs, but through the association of the intracellular kinase domains as an asymmetric dimer (8). An open question has involved how structural changes induced by ligand binding lead to the formation of an active, asymmetric dimer on the intracellular side of the receptor.
The single transmembrane (TM) helix and associated juxtamembrane (JM) sequences bridge the extracellular and intracellular domains and require a membrane environment to adopt their native structure. In general, ligand binding to the extracellular domain of the EGFR has the potential to change the proximity of the TM helices, their relative orientation, or both. To shed light on the role of the TM and JM regions in signaling across the membrane bilayer, we use two approaches to alter the proximity and orientation of the TM helices. First, we take advantage of a point mutation (V664E) in the TM sequence of the rat Neu (or ErbB2) receptor that leads to full oncogenic activation (9–11). The mutation was previously found to be sequence-specific, that is, substitution at positions 663 or 665 had no effect on receptor activity (12). In their original studies, Bargmann and Weinberg (12) raised the possibility that the mutation results in clustering of the receptor. Subsequent studies demonstrated that in the active receptor with the V664E point mutation, Glu664 mediates dimerization through hydrogen-bonding interactions (13) and that the sequence specificity is due to a tripeptide motif formed by Val663–Glu664–Gly665 (14). In this study, the V664E mutation allows us to modulate the interaction of the TM helices in a manner consistent with oncogenic receptor activation.
The proximity and orientation of the helices within the Neu TM helix dimer can also be controlled by attaching a known dimerization domain, such as the coiled coil domain of Put3, to the N terminus of peptides corresponding to the Neu TM–JM sequence. Put3 is a soluble transcriptional activator protein. Its C-terminal 28 residues associate in a stable left-handed coiled coil dimer and can induce dimer formation of attached TM sequences. Dimaio and coworkers (15) originally used the Put3 construct to engineer symmetric coiled coil orientations of E5, a 44-residue single-pass TM protein from bovine papillomavirus. We have subsequently shown that this approach is generally applicable to single-pass TM receptor proteins in the cytokine receptor family (16, 17). Fusion of the C terminus of the Put3 sequence to the N-terminal residue of the TM sequence of the Neu TM domain induces dimerization of the Neu TM domain in different orientations depending on the position of the fusion.
In this study, TM–JM peptides corresponding to the Neu (or rat ErbB2) receptor provide a way to conceptually link the crystal structures that have been determined of the extracellular and intracellular domains of ErbB family receptors. These peptides can be reconstituted into membrane bilayers of defined composition to provide a nativelike environment for proper folding. The TM sequence (residues 656–680) folds into a single membrane-spanning helix in bilayers and contains sequence motifs that mediate helix dimerization. We have previously shown that in Neu receptor TM peptides the V664E mutation leads to dimerization of TM helix (13, 18). The positively charged intracellular JM (residues 681–695) sequence binds strongly to negatively charged membranes. McLaughlin and coworkers (19, 20) have proposed that in the context of the full receptor this interaction may contribute to holding the kinase domain in an inactive conformation.
Using two independent methods, solid-state NMR and fluorescence spectroscopy, we show that there are tight interactions of the JM sequence with negatively charged lipids in TM–JM peptides corresponding to the wild-type receptor sequence. We observe release of the JM sequence from the negatively charged membrane surface using peptides containing an activating V664E mutation within the TM domain or in peptides engineered to form TM helix dimers using the Put3 dimerization motif.
Results
V664E Mutation in Neu* TM–JM Peptides Stabilizes Dimers of the TM Domain.
We first show that the V664E mutation strengthens the association of the TM helices in the Neu* TM–JM peptides when reconstituted into membrane bilayers. Deuterium magic angle spinning (MAS) NMR spectroscopy provides a simple method to compare membrane association of TM peptides and establish differences in interfacial packing (21). The intensities of the spinning side bands in a deuterium MAS spectrum map out the deuterium line shape and are sensitive to molecular motion. Leucine, with a single deuterated methyl group at the end of its long side chain, is mobile in TM helices when oriented toward surrounding lipids and more constrained when packed within a helix interface and consequently can be used as a probe of the interface of interacting TM helices.
Fig. 1 presents deuterium MAS NMR spectra of peptides corresponding to the TM and intracellular JM sequences of the wild-type Neu and mutant Neu* receptors. Neu* TM–JM is a 47-residue peptide containing the V664E mutation. The peptides were synthesized with deuterated leucine at positions 668 or 670 (where the numbering corresponds to that of the full-length receptor). Leu668 is on the same face of the helix as Val664 or Glu664. In the structure of the Neu* TM dimer, Leu668 is packed in the dimer interface, whereas Leu670 is on the opposite face oriented toward the lipid acyl chains (18).
Fig. 1.

Deuterium NMR MAS spectroscopy of TM helix interactions. Neu TM–JM (Left) and Neu* TM–JM (Right) peptides containing deuterium-labeled Leu668 (A) and Leu670 (B) were reconstituted into DMPC:DMPG vesicles. Spectra were obtained at 25 °C and a MAS frequency of 5 kHz.
Fig. 1A presents deuterium MAS spectra of the Neu TM–JM and Neu* TM–JM peptides containing deuterated leucine at position 668. The spectra exhibit rotational side bands spaced at the MAS frequency of 5 kHz. Comparison of the side-band intensities between the right and left panels shows that the deuterium line shape is broader for Leu668 in the Neu* TM–JM peptide, indicative of motional restriction of the side chain, consistent with its position in the TM dimer interface. Fig. 1B shows deuterium MAS spectra of Neu TM–JM and Neu* TM–JM containing deuterated leucine at position 670. In contrast to Leu668, the deuterium line shapes are similar for Leu670 in both the Neu TM–JM and Neu* TM–JM peptides, and also similar to the narrow line shape for Leu668 in Neu TM–JM.
The deuterium line shapes and intensities support our previous structural studies that Leu668 is located within the interface of the Neu* TM–JM dimer (18), although they do not exclude the possibility that the helices are associating into higher-order oligomers. The comparison of deuterium line shapes also agrees with our previous comparisons of interhelical dipolar couplings showing that the V664E mutation shifts the monomer–dimer equilibrium toward the dimer state in Neu* (18). These observations indicate that the V664E mutation in the Neu* TM–JM peptide strengthens dimerization. We assume that the orientation and strengthened interaction of the TM helices in the Neu* TM–JM dimer (compared with Neu TM–JM) reflect the orientation and interaction of the TM helices in the activated full-length receptor.
Transmembrane Helix Interactions Influence the Interaction of the JM Domain with the Membrane Bilayer.
The intracellular JM sequence is positively charged and its binding with negatively charged membranes can be monitored using fluorescence spectroscopy. Fig. 2 presents fluorescence spectra of the isolated JM region of rat Neu (residues 682–696) labeled at the C terminus with Alexa568. Our measurements on the isolated JM sequence provide a comparison with previous experiments on isolated ErbB1 JM peptides (19) and with experiments below on the Neu and Neu* TM–JM sequences. The experiments were carried out in 1-palmitoyl-2-oleoyl-sn-phosphatidylcholine (POPC), 1-palmitoyl-2-oleoyl-sn-phosphatidylserine (POPS) membranes with increasing amounts of phosphatidylinositol 4,5-bisphosphate (PIP2). PIP2 is an integral player in the downstream events following the activation of RTKs and may be required for EGFR activation (22). Positively charged JM peptides sequester multivalent PIP2 when bound to membrane bilayers (19).
Fig. 2.

Fluorescence spectroscopy of JM–JM interactions. The influence of PIP2 on JM interactions was measured for the isolated JM domain and for the Neu TM–JM and Neu* TM–JM TM–JM peptides. (A) Interaction of the JM domain alone with PIP2 leads to JM–JM association and fluorescence quenching. Fluorescence of the Alexa568 label attached to the C terminus of the JM domain is quenched upon the addition of PIP2. (B) Titration of vesicles with PIP2 reconstituted with the TM–JM peptide of wild-type Neu TM–JM leads to a small increase in fluorescence and then a decrease in fluorescence. (C) Titration with PIP2 of vesicles reconstituted with the TM–JM peptide of Neu* TM–JM does not produce a change in fluorescence. The fluorescence measurements in A–C are representative of three independent reconstitutions.
The fluorescence emission band of Alexa568 is at 604 nm after excitation in its absorption band at 568 nm. As PIP2 is added, the fluorescence of the Alexa568 tag decreases (Fig. 2A). We attribute the decrease to quenching of the Alexa568 fluorescence due to clustering of JM peptides and close association of Alexa568 fluorophores in the plane of the membrane. The quenching can be reversed by the addition of the Ca2+ complex of calmodulin (Ca/CaM) and the direct interaction of PIP2 with the fluorophore can be measured using fluorescence resonance energy transfer (Figs. S1 and S2). These studies support previous fluorescence studies of McLaughlin et al. (19) using the JM sequence of the ErbB1 receptor containing an N-terminal acrylodan label. In these studies, it was found that (i) the 16-residue JM sequence of ErbB1 binds tightly to POPC:POPS bilayers, (ii) binding of the JM peptide can be reversed by the addition of Ca/CaM, and (iii) the positively charged JM sequence will attract PIP2 even in the presence of monovalent POPS. Together with the results in Fig. 2A, we conclude that a small amount of PIP2 with a net charge of −3 to −5 (23) leads to the association or clustering of the isolated JM domain when bound to membrane bilayers containing 23% monovalent POPS lipids. Whereas the clustering behavior of polybasic regions by PIP2 may be a general consequence of strong electrostatic interactions and relevant for PIP2-mediated processes (24), these results are relevant for the ErbB receptors, which are known to cluster (25) and appear to require PIP2 for activation (22).
We next asked whether the strengthened dimerization of the TM helix due to the V664E mutation influences the JM region of the peptide. Studies parallel to those described above were undertaken of JM–JM interactions in the context of the TM–JM peptides that are labeled with a fluorescent Alexa568 tag attached to their C terminus. We find that the JM–JM interactions in the Neu TM–JM and Neu* TM–JM peptides differ from peptides corresponding to the JM region alone and from each other. In Fig. 2B, titration with PIP2 of vesicles reconstituted with Neu TM–JM first leads to a small increase in fluorescence and then a small decrease in fluorescence. We attribute the small increase in fluorescence in Neu TM–JM to an increase in membrane binding of the JM domain and the subsequent decrease in fluorescence to the association of the JM regions of adjacent peptides on the membrane as PIP2 is added.
In Fig. 2C, titration of vesicles reconstituted with the Neu* TM–JM peptide does not produce a change in fluorescence. The results in Fig. 2 are reproducible (n = 3) and indicate that the strengthened dimerization of the TM helix due to the V664E mutation influences the JM region of the peptide. Two possible explanations for the absence of fluorescence changes upon the addition of PIP2 are that (i) the JM region in the Neu* TM–JM dimer is associated with the membrane, but does not interact with the JM region of the opposing monomer in the dimer structure, or (ii) the JM region is not associated with the membrane.
Transmembrane Helix Interactions Influence the Dynamics of the JM Domain.
To address whether the absence of a change of fluorescence emission of the Neu* TM–JM dimer upon the addition of PIP2 is due to the dissociation of the JM domain from the membrane, we measured the dynamics of the JM region by NMR spectroscopy. If the JM domain is released from the membrane in the Neu* TM–JM dimer, it would be expected to increase in mobility.
Solid-state NMR spectra are sensitive to molecular motions over a wide range of time scales. NMR methods can be used to enhance signals from flexible regions undergoing rapid isotropic motion or rigid regions whose motions are slow compared with the MAS frequencies used to obtain high-resolution spectra. In this section, we compare the signal intensities of 13C-labeled Tyr690 and Arg693 located in the middle of the JM sequence using two different NMR techniques. First, MAS spectra of Neu TM–JM and Neu* TM–JM were obtained using direct polarization (DP) by applying a single 13C excitation pulse. The signal intensities are not sensitive to molecular motion under DP. Second, spectra were obtained using the INEPT (insensitive nuclei enhancement by polarization transfer) pulse sequence, where polarization is transferred from 1H to 13C through J-couplings. The INEPT sequence enhances sites that are mobile with correlation times of < 0.01 μs (26) (i.e., in solutions in which anisotropic interactions such as dipolar couplings are averaged). In a similar study, Ladizhansky and coworkers (27) used the INEPT pulse sequence to distinguish regions of the positively charged myelin basic protein that are not associated with negatively charged lipid bilayers from those regions that are membrane-associated.
Fig. 3 presents 1D solid-state NMR spectra obtained with MAS with DP and INEPT. The measurements were made on the same samples of Neu TM–JM and Neu* TM–JM reconstituted into dimyristoylphosphatidylcholine:dimyristoylphosphatidylglycerol (DMPC:DMPG) vesicles. The measurements are without PIP2 incorporated into the membranes. 1D 13C-DP spectra of Neu TM-JM and Neu* TM-JM are shown in Fig. 3A. The aliphatic resonances originating from natural abundance 13C of the lipids and 13C labels on Tyr690 and Arg693 are observed between 0–70 ppm. The resolved resonances of Tyr690 and Arg693 in the JM region are labeled Y and R, respectively. The aromatic resonances of Tyr690 occur between 110 and 160 ppm. The guanidinium Cζ resonance of Arg693 is observed at ∼160 ppm. The backbone carbonyl resonances are at ∼175 ppm. All of the 13C resonances that are expected in the TM–JM peptides are observed because the DP experiment is not sensitive to peptide motion.
Fig. 3.

Solid-state 13C MAS NMR of JM domain dynamics. Solid-state NMR spectra were obtained of the Neu TM–JM and Neu* TM–JM peptides containing U-13C-labeled Tyr690 and Arg693 in the JM region of the Neu TM–JM peptides at 37 °C. Spectra were obtained with direct polarization (DP) (A) and the INEPT sequence (B). The peaks from Tyr690 and Arg693 in the JM region are labeled. The CP and INEPT sequences highlight the rigid and mobile regions, respectively.
Fig. 3B presents 1D 13C-INEPT spectra of Neu TM–JM and Neu* TM–JM. There is a distinct difference in the spectra of the Neu and Neu* peptides. For Neu* TM–JM, peaks from the protonated Cɛ and Cδ resonances of aromatic ring of Tyr690 are observed at ∼118 and 133 ppm, respectively. The 13C resonances of the unprotonated Tyr690 Cγ, Tyr690 Cζ, and Arg693 Cζ carbons are not observed because they lack a directly bonded proton. The observation of Tyr690 resonances in the INEPT spectrum of Neu* TM–JM provides direct evidence that the JM domain is mobile, consistent with the dissociation of the JM domain from the membrane surface. In contrast, the absence of the Tyr690 resonances in the INEPT spectrum of Neu TM–JM is consistent with lack of isotropic motion due to membrane binding.
Engineering Active and Inactive Receptor Dimer Orientations in the Neu Receptor.
Comparisons of the Neu TM–JM and Neu* TM–JM peptides show how receptor dimerization influences binding of the JM domain to the membrane bilayer. In a preformed dimer, coupling of the TM domain to the JM sequence may occur by influencing the relative proximity of the TM helices and/or their orientation. In this section, we test whether the orientation of the TM helix modulates association of the JM sequence with negatively charged membrane bilayers.
The orientation of the helices within the Neu TM helix dimer can be controlled by attaching a known soluble dimerization domain to the N terminus of the TM–JM peptide. Fusion of the C terminus of the Put3 sequence to the N-terminal residue of the TM sequence of the Neu TM domain induces dimerization of the Neu TM domain in different orientations depending on the position of the fusion (Fig. S3). The positions of the heptad repeats characteristic of left-handed coiled coils are denoted a–g. The predicted interfaces of four Put3–Neu fusion protein constructs are shown in the helical wheel diagrams in Fig. 4, where positions a and d form the interface in coiled coils. The Put3-Neu4 construct with Val664 and Leu668 in the interface corresponds to Neu* TM–JM.
Fig. 4.

Dependence of JM–membrane interactions using engineered Put3–Neu TM–JM constructs. Sequences (A) and helical wheel diagrams (B–E) are shown of the four Put3–Neu TM–JM constructs. Deletion of residues at the junction between the Put3 sequence and the Neu TM–JM sequence leads to four different orientations of the helices in the engineered dimers. The a and d positions correspond to the helix interface. (F) Tryptophan fluorescence of the four Put3–Neu TM–JM constructs in POPC:POPS vesicles. (G) Tryptophan fluorescence of the four Put3–Neu TM–JM constructs in POPC:POPS vesicles with a molar ratio of PIP2 to peptide of 1:1.
We chemically synthesized peptides listed in Fig. 4A. Each of the four Put3–Neu TM–JM peptides contains a single tryptophan at the C terminus of the JM domain. These peptides were reconstituted into POPC:POPS (10:3) lipid bilayers with and without added PIP2 for fluorescence measurements. Binding and insertion of tryptophan into hydrophobic membranes results in a blue shift and intensity increase of the fluorescence emission band. Fig. 4 F and G present fluorescence spectra of Put3–Neu1–4 in the region of the tryptophan emission band. The fluorescence intensity from the tryptophan incorporated at the C terminus was significantly reduced for the Put3–Neu4 peptide. More importantly, compared with other sequences, we observed a red shift for the spectrum of the Put3–Neu4 peptide. We could also observe small differences in the fluorescence intensities from Put3–Neu1, 2, and 3 peptides. In POPC:POPS membranes without added PIP2, these differences in the intensity are not accompanied with a spectral shift. In membranes with PIP2, there are slight red shifts between Put3–Neu3, Put3–Neu2, and Put3–Neu1.
These observations suggest that the C-terminal tryptophan of the Put3–Neu4 dimer is in a hydrophilic environment as a result of release of the JM domain from the membrane. This observation is consistent with the results described in the previous two sections using fluorescence and solid-state NMR spectroscopy and shows that the interaction of the JM region to lipid bilayers depends on the orientation of the TM helices.
Discussion
We describe studies using peptides corresponding to the TM and JM sequences of the Neu receptor that provide an explanation for how activation of the intracellular kinase domain can be regulated by the proximity and orientation of the TM helices. The TM–JM constructs bridge the structures of the extracellular ligand-binding domain and the intracellular kinase domain. They complement the crystal structures obtained of these domains and provide insights into the role of the membrane environment in regulating signal transduction. Our key finding is that the JM sequence is released from the negatively charged membrane surface when the TM helices are placed in an orientation reflecting the active state of the receptor.
The idea of rotational coupling of the TM helices in activation of the ErbB-family receptors was first raised in studies of the Neu receptor containing the V664E mutation. Stern and coworkers (14, 28) found that only a single position of the V663–E664–G665 dimerization motif results in activation. Shifting the position of this motif by one residue generated an inactive receptor. Bell et al. (29) extended these studies by demonstrating that there is a periodic activation of the Neu and PDGF-β receptors as the dimerization motif is shifted across the TM domain.
More recently, Pike and coworkers (30, 31) have shown that the intracellular JM domain is allosterically coupled to the ligand-binding site. They found that positive linkage and negative cooperativity in EGF binding requires the presence of the intracellular JM domain. EGF has higher affinity for the first site on the EGFR dimer relative to the receptor monomer (positive linkage) as well as for the second site on the receptor dimer (negative cooperativity). They concluded that there is inside-out signaling by the EGF receptor and suggested that the position of the JM domain controls the rotation or tilt of the TM helix, which in turn influences the structure and interactions of the extracellular domain.
There is substantial evidence that isolated TM helices corresponding to the ErbB receptors dimerize in membrane bilayers (32–34). The recent solution NMR structure of the ErbB2 TM domain dimer shows that there is a preferential TM helix interface in the wild-type receptor (35) that coincides with the interface previously described in the active (V664E) mutant of the Neu receptor TM dimer (18). These results argue that TM dimerization is associated with a single interface (corresponding to the active receptor) and there is a shift in the monomer–dimer equilibrium toward the dimer with the V664E mutation.
For the full-length receptor, the concept that the TM helices do not interact in the inactive state is supported by two studies showing that uncoupling of the extracellular domain from the TM domain induces receptor activation. In one case, a flexible linker is inserted between the extracellular domain and the TM domain, and in the second, the entire extracellular domain is truncated. Both changes result in ligand-independent receptor activation (10, 36). An explanation consistent with the studies showing that the isolated TM helices have a propensity to dimerize is that the extracellular domain of the inactive receptor constrains the proximity or orientation of the TM helices. When these constraints are removed, the helices dimerize in an activating orientation. Jura et al. (37) suggested that ligand binding may simply change the position of the C-terminal ends of the extracellular domains and bring the TM helices into close proximity. In fact, there does not seem to be a specific TM helix interface that is required for activation (38, 39), despite the observation of a sequence motif in the TM domain that is roughly conserved across the RTK family (40). In the wild-type full-length receptor, ligand binding may simply change the relative orientation and proximity of the helices without inducing helix dimerization.
Fig. 5 presents a cartoon that places our studies on isolated TM–JM peptides into the context of a possible activation mechanism of the full-length receptor. The inactive receptor can exist either as a monomer or preformed dimer. We find that PIP2 binds to the positively charged cluster in the JM region of the Neu receptor and mediates JM–JM association. PIP2-mediated association of the JM regions would stabilize the inactive dimer. Deletion of the cytoplasmic domain shifts the monomer-dimer equilibrium toward the monomer (41). Interestingly, an increase in preformed dimers is observed at the cell periphery where there is an increase in PIP2 generation (41). Such an increase in preformed dimers (with a high-affinity ligand binding site) may correlate with an increase in EGFR activity observed with increasing PIP2 (22).
Fig. 5.
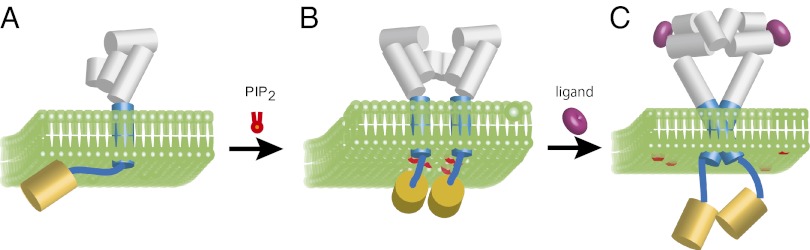
Model of membrane release of the intracellular JM domain upon ligand binding. (A) In the monomer of the Neu receptor, the positively charged JM sequence associates with the negatively charged cytoplasmic surface of the plasma membrane. The structures of the extracellular domain of the EGFR with (5) and without (6) bound EGF revealed that the unliganded structure has a tethered conformation that undergoes a dramatic rearrangement upon ligand binding. (B) In the absence of ligand, receptors in the ErbB receptor family are able to form inactive dimers. Dimerization seems to be mediated by interactions in both the extracellular and cytoplasmic domains, because deletion of both results in an increase in EGFR dimers (10, 41). We suggest that dimerization is mediated in part by JM–JM interactions through PIP2. (C) Ligand binding results in a conformational change of the extracellular domain, which is coupled to a change in the orientation of the TM helices. The change in helix orientation releases the JM domain from the membrane and allows asymmetric association of the kinase domain. Tethering the JM domain to the membrane by engineering palmitoylation sites removes positive linkage and negative cooperativity observed with ligand binding (30, 31).
Ligand binding induces a conformational change in the receptor and association of the TM helices in a specific orientation. The mechanism by which the relative orientation of the TM domains releases the JM sequence from the membrane surface is not known but likely involves decreasing its electrostatic interaction with negatively charged membrane surface (42).
Finally, in the ErbB family of receptors, the proposed change in exposure of the JM domain provides an additional mechanism for regulation through interactions with cytosolic proteins. The JM sequence contains a number of sequence motifs that serve to modulate receptor activity through interactions with intracellular proteins. These include basolateral targeting signals, a lysosomal sorting motif, and binding sites for phosphoinositide kinases and for calmodulin (43, 44). Fig. 5 suggests that these motifs may not be accessible to intracellular proteins until the JM domain is released from the membrane.
Materials and Methods
Materials.
13C-labeled amino acids were purchased from Cambridge Isotope Laboratories. DMPC, DMPG, POPC, and POPS were obtained from Avanti Polar Lipids as lyophilized powders and used without further purification. Alexa Fluor 568 C5-maleimide was purchased from Invitrogen.
Peptide Synthesis and Purification.
Peptides corresponding to the TM and JM regions of the Neu receptor (650–696) were synthesized by solid-phase methods with the following sequence: EQRASPVTFIIATVV664GVLL668FL670ILVVVVGILIKRRRQKIRKYT691MRLL-NH2. The C terminus was amidated. The synthetic peptides were purified by reverse-phase HPLC on a C4 column with a gradient of formic acid/1-propanol (4:1) over formic acid/water (2:3). The purity was confirmed with MALDI mass spectrometry and analytical reverse phase HPLC. For fluorescent-labeled peptides, Alexa Fluor 568 C5-maleimide was introduced to the sulfide group on cysteine at the C terminus of the TM–JM peptide by mixing the peptide and the fluorescence derivative in dimethylformamide under basic conditions.
Reconstitution of Peptides into Membrane Bilayers.
The Neu receptor peptides were cosolubilized with lipid and octyl-β-glucoside in trifluoroethanol. For NMR experiments, the peptide:lipid molar ratio was 1:50 and the molar ratio between DMPC and DMPG or between POPC and POPS was 10:3. PIP2 was not used in these experiments. For fluorescence experiments, the peptide-to-lipid ratio ranged from 1:100–1:5,000 and the lipid concentration was 200–250 μM in Mops buffer (10 mM Mops and 0.1 M KCl, pH 7.0). The molar ratio between POPC and POPS was 10:3. The solution was incubated for 90 min at 37 °C, after which the solvents were removed under a stream of argon gas and then under vacuum. Mes buffer (50 mM Mes, 50 mM NaCl, and 5 mM DTT, pH 6.2) was added to the solid from the previous step and mixed at 37 °C for 6 h. The octyl-β-glucoside was removed by dialysis. The method for reconstitution parallels our previous studies using IR and NMR spectroscopy to incorporate the Neu and Neu* TM peptides into membrane bilayers as TM dimers as assayed by the observed IR dichroic ratio of the amide I band and interhelical dipolar couplings (18). For NMR experiments, the reconstituted membranes were pelleted and loaded into NMR rotors.
Fluorescence Spectroscopy.
Fluorescence experiments were carried out on an Hitachi F-2500 fluorescence spectrophotometer or an Horiba Jobin Yvon FL-3 22 fluorimeter. After the reconstitution, we formed vesicles by extrusion of multilamellar vesicles through 200-nm polycarbonate filters. For experiments with PIP2, the PIP2 was introduced into the membranes by addition of PIP2 micelles to the vesicle solution, and the fluorescence measurements were made within 1 h to minimize PIP2 hydrolysis. The PIP2 concentration ranged from 0.05 μM to 4 μM, or PIP2-to-peptide ratios of 1:50–2:1.
Solid-State NMR Spectroscopy.
Solid state NMR MAS experiments were performed on Varian Infinity-plus 500, 600, and 700 spectrometers using triple-resonance MAS probes with 3.2- and 4.0-mm rotors. For deuterium-observe experiments, single pulse excitation was used with a 5-μs 90° pulse length, followed by a 10-μs delay before data acquisition. The probe temperature was maintained at 25 °C. For 13C observe experiments, the MAS frequency was maintained at 12 kHz. Two-phase modulated decoupling was used during the acquisition period, except for the direct polarization experiment, in which continuous-wave decoupling was used. The decoupling field strength was typically 70 kHz. 13C chemical shifts were referenced to tetramethylsilane. The probe temperature was maintained at 37 °C. For INEPT experiments, delays of 1.2 and 1.0 ms were used.
Supplementary Material
Acknowledgments
We thank R. Yamaguchi for help with peptide purification and Stuart McLaughlin and Martine Ziliox for critical reading of the manuscript. This work was supported by a grant from the Naito Foundation (to T.S.) and by Grant GM 46732 from the National Institutes of Health (to S.O.S.). C.M. was a research fellow of the Japan Society for the Promotion of Science.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1215207110/-/DCSupplemental.
References
- 1.Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 2.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 3.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 4.Holbro T, Civenni G, Hynes NE. The ErbB receptors and their role in cancer progression. Exp Cell Res. 2003;284(1):99–110. doi: 10.1016/s0014-4827(02)00099-x. [DOI] [PubMed] [Google Scholar]
- 5.Ogiso H, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110(6):775–787. doi: 10.1016/s0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 6.Ferguson KM, et al. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003;11(2):507–517. doi: 10.1016/s1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 7.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277(48):46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125(6):1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 9.Bargmann CI, Hung MC, Weinberg RA. Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell. 1986;45(5):649–657. doi: 10.1016/0092-8674(86)90779-8. [DOI] [PubMed] [Google Scholar]
- 10.Bargmann CI, Weinberg RA. Increased tyrosine kinase activity associated with the protein encoded by the activated neu oncogene. Proc Natl Acad Sci USA. 1988;85(15):5394–5398. doi: 10.1073/pnas.85.15.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bargmann CI, Hung MC, Weinberg RA. The neu oncogene encodes an epidermal growth factor receptor-related protein. Nature. 1986;319(6050):226–230. doi: 10.1038/319226a0. [DOI] [PubMed] [Google Scholar]
- 12.Bargmann CI, Weinberg RA. Oncogenic activation of the neu-encoded receptor protein by point mutation and deletion. EMBO J. 1988;7(7):2043–2052. doi: 10.1002/j.1460-2075.1988.tb03044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith SO, Smith CS, Bormann BJ. Strong hydrogen bonding interactions involving a buried glutamic acid in the transmembrane sequence of the neu/erbB-2 receptor. Nat Struct Biol. 1996;3(3):252–258. doi: 10.1038/nsb0396-252. [DOI] [PubMed] [Google Scholar]
- 14.Burke CL, Lemmon MA, Coren BA, Engelman DM, Stern DF. Dimerization of the p185neu transmembrane domain is necessary but not sufficient for transformation. Oncogene. 1997;14(6):687–696. doi: 10.1038/sj.onc.1200873. [DOI] [PubMed] [Google Scholar]
- 15.Mattoon D, Gupta K, Doyon J, Loll PJ, DiMaio D. Identification of the transmembrane dimer interface of the bovine papillomavirus E5 protein. Oncogene. 2001;20(29):3824–3834. doi: 10.1038/sj.onc.1204523. [DOI] [PubMed] [Google Scholar]
- 16.Seubert N, et al. Active and inactive orientations of the transmembrane and cytosolic domains of the erythropoietin receptor dimer. Mol Cell. 2003;12(5):1239–1250. doi: 10.1016/s1097-2765(03)00389-7. [DOI] [PubMed] [Google Scholar]
- 17.Staerk J, et al. Orientation-specific signalling by thrombopoietin receptor dimers. EMBO J. 2011;30(21):4398–4413. doi: 10.1038/emboj.2011.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith SO, et al. Transmembrane interactions in the activation of the Neu receptor tyrosine kinase. Biochemistry. 2002;41(30):9321–9332. doi: 10.1021/bi012117l. [DOI] [PubMed] [Google Scholar]
- 19.McLaughlin S, Smith SO, Hayman MJ, Murray D. An electrostatic engine model for autoinhibition and activation of the epidermal growth factor receptor (EGFR/ErbB) family. J Gen Physiol. 2005;126(1):41–53. doi: 10.1085/jgp.200509274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sengupta P, et al. EGFR juxtamembrane domain, membranes, and calmodulin: Kinetics of their interaction. Biophys J. 2009;96(12):4887–4895. doi: 10.1016/j.bpj.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W, Crocker E, Constantinescu SN, Smith SO. Helix packing and orientation in the transmembrane dimer of gp55-P of the spleen focus forming virus. Biophys J. 2005;89(2):1194–1202. doi: 10.1529/biophysj.104.057844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michailidis IE, et al. Phosphatidylinositol-4,5-bisphosphate regulates epidermal growth factor receptor activation. Pflugers Archiv- Eur J Phys. 2011;461(3):387–397. doi: 10.1007/s00424-010-0904-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLaughlin S, Wang JY, Gambhir A, Murray D. PIP(2) and proteins: Interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 24.van den Bogaart G, et al. Membrane protein sequestering by ionic protein-lipid interactions. Nature. 2011;479(7374):552–555. doi: 10.1038/nature10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clayton AHA, Tavarnesi ML, Johns TG. Unligated epidermal growth factor receptor forms higher order oligomers within microclusters on A431 cells that are sensitive to tyrosine kinase inhibitor binding. Biochemistry. 2007;46(15):4589–4597. doi: 10.1021/bi700002b. [DOI] [PubMed] [Google Scholar]
- 26.Nowacka A, Mohr PC, Norrman J, Martin RW, Topgaard D. Polarization transfer solid-state NMR for studying surfactant phase behavior. Langmuir. 2010;26(22):16848–16856. doi: 10.1021/la102935t. [DOI] [PubMed] [Google Scholar]
- 27.Zhong L, Bamm VV, Ahmed MAM, Harauz G, Ladizhansky V. Solid-state NMR spectroscopy of 18.5 kDa myelin basic protein reconstituted with lipid vesicles: Spectroscopic characterisation and spectral assignments of solvent-exposed protein fragments. Biochim Biophys. Acta-Biomembr. 2007;1768(12):3193–3205. doi: 10.1016/j.bbamem.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cao H, Bangalore L, Bormann BJ, Stern DF. A subdomain in the transmembrane domain is necessary for p185neu* activation. EMBO J. 1992;11(3):923–932. doi: 10.1002/j.1460-2075.1992.tb05131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bell CA, et al. Rotational coupling of the transmembrane and kinase domains of the Neu receptor tyrosine kinase. Mol Biol Cell. 2000;11(10):3589–3599. doi: 10.1091/mbc.11.10.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macdonald-Obermann JL, Pike LJ. Palmitoylation of the EGF receptor impairs signal transduction and abolishes high-affinity ligand binding. Biochemistry. 2009;48(11):2505–2513. doi: 10.1021/bi802249x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macdonald-Obermann JL, Pike LJ. The intracellular juxtamembrane domain of the epidermal growth factor (EGF) receptor is responsible for the allosteric regulation of EGF binding. J Biol Chem. 2009;284(20):13570–13576. doi: 10.1074/jbc.M109.001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mendrola JM, Berger MB, King MC, Lemmon MA. The single transmembrane domains of ErbB receptors self-associate in cell membranes. J Biol Chem. 2002;277(7):4704–4712. doi: 10.1074/jbc.M108681200. [DOI] [PubMed] [Google Scholar]
- 33.Duneau JP, Vegh AP, Sturgis JN. A dimerization hierarchy in the transmembrane domains of the HER receptor family. Biochemistry. 2007;46(7):2010–2019. doi: 10.1021/bi061436f. [DOI] [PubMed] [Google Scholar]
- 34.Beevers AJ, Damianoglou A, Oates J, Rodger A, Dixon AM. Sequence-dependent oligomerization of the Neu transmembrane domain suggests inhibition of “conformational switching” by an oncogenic mutant. Biochemistry. 2010;49(13):2811–2820. doi: 10.1021/bi902087v. [DOI] [PubMed] [Google Scholar]
- 35.Bocharov EV, et al. Spatial structure of the dimeric transmembrane domain of the growth factor receptor ErbB2 presumably corresponding to the receptor active state. J Biol Chem. 2008;283(11):6950–6956. doi: 10.1074/jbc.M709202200. [DOI] [PubMed] [Google Scholar]
- 36.Sorokin A. Activation of the EGF receptor by insertional mutations in its juxtamembrane regions. Oncogene. 1995;11(8):1531–1540. [PubMed] [Google Scholar]
- 37.Jura N, et al. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell. 2009;137(7):1293–1307. doi: 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kashles O, et al. Ligand-induced stimulation of epidermal growth factor receptor mutants with altered transmembrane regions. Proc Natl Acad Sci USA. 1988;85(24):9567–9571. doi: 10.1073/pnas.85.24.9567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carpenter CD, et al. Structural analysis of the transmembrane domain of the epidermal growth factor receptor. J Biol Chem. 1991;266(9):5750–5755. [PubMed] [Google Scholar]
- 40.Sternberg MJ, Gullick WJ. A sequence motif in the transmembrane region of growth factor receptors with tyrosine kinase activity mediates dimerization. Protein Eng. 1990;3(4):245–248. doi: 10.1093/protein/3.4.245. [DOI] [PubMed] [Google Scholar]
- 41.Chung I, et al. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature. 2010;464(7289):783–787. doi: 10.1038/nature08827. [DOI] [PubMed] [Google Scholar]
- 42.Sato T, Pallavi P, Golebiewska U, McLaughlin S, Smith SO. Structure of the membrane reconstituted transmembrane-juxtamembrane peptide EGFR(622-660) and its interaction with Ca2+/calmodulin. Biochemistry. 2006;45(42):12704–12714. doi: 10.1021/bi061264m. [DOI] [PubMed] [Google Scholar]
- 43.Hake MJ, Choowongkomon K, Kostenko O, Carlin CR, Sönnichsen FD. Specificity determinants of a novel Nck interaction with the juxtamembrane domain of the epidermal growth factor receptor. Biochemistry. 2008;47(10):3096–3108. doi: 10.1021/bi701549a. [DOI] [PubMed] [Google Scholar]
- 44.Kil SJ, Carlin C. EGF receptor residues leu679, leu680 mediate selective sorting of ligand-receptor complexes in early endosomal compartments. J Cell Physiol. 2000;185(1):47–60. doi: 10.1002/1097-4652(200010)185:1<47::AID-JCP4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.