

Abstract
One of the most difficult problems in modern genomics is the assembly of full-length chromosomes using next generation sequencing (NGS) data. To address this problem, we developed “reference-assisted chromosome assembly” (RACA), an algorithm to reliably order and orient sequence scaffolds generated by NGS and assemblers into longer chromosomal fragments using comparative genome information and paired-end reads. Evaluation of results using simulated and real genome assemblies indicates that our approach can substantially improve genomes generated by a wide variety of de novo assemblers if a good reference assembly of a closely related species and outgroup genomes are available. We used RACA to reconstruct 60 Tibetan antelope (Pantholops hodgsonii) chromosome fragments from 1,434 SOAPdenovo sequence scaffolds, of which 16 chromosome fragments were homologous to complete cattle chromosomes. Experimental validation by PCR showed that predictions made by RACA are highly accurate. Our results indicate that RACA will significantly facilitate the study of chromosome evolution and genome rearrangements for the large number of genomes being sequenced by NGS that do not have a genetic or physical map.
Keywords: computational genomics, comparative genomics, bioinformatics, chromosome breakpoints, mammals
Whole genome sequencing of vertebrate species has greatly advanced comparative genomics and provided novel insights into animal biology (1–3). One of the most important advantages of having whole genome sequences is the capacity to understand the evolutionary history of genome organization and structural variation caused by chromosome rearrangements (4–7). However, the number of animal genomes sequenced by next generation sequencing (NGS) is rapidly outpacing the number of genomes with physical or genetic maps for anchoring the assemblies to chromosomes, which is necessary for elucidating the biological consequences of chromosome rearrangements and for shedding new light on the molecular signatures of human variation and disease mechanisms (8–10). More large-scale genome sequencing projects are planned using NGS technologies, including the Genome 10K (G10K) and the 5K Insect Genome (i5K) initiatives (11, 12). Thus, improved methods are required for the assembly of chromosome-scale DNA fragments from NGS data.
Since the emergence of NGS technology, several groups have developed de novo assemblers based on NGS data, such as ABySS (13), ALLPATH-LG (14), SOAPdenovo (15), and Velvet (16). However, the limitation of NGS read length makes it extremely difficult to assemble the reads into chromosomes for large genomes. This problem becomes even more challenging in the case of mammalian genomes, which contain a high fraction of repetitive elements (17). In general, the procedures of de novo assembly from the short reads that are generated by NGS can be described as follows (reviewed in ref. 18). First, the reads are grouped into contigs based on their overlaps. The most widely used approaches are based on either (i) the overlap graph, in which nodes represent the reads, and edges connect overlapping reads, or (ii) the de Bruijn graph (19), in which nodes are fixed-length sequences (k-mers), and edges indicate the predecessor and successor relationships of the k-mers. These contigs are further assembled into scaffolds by using paired-end reads, i.e., pairs of sequence reads from the two ends of inserts contained in a library of cloned genomic fragments. Many scaffolding tools have been developed, such as SSPACE (20) as well as scaffolding modules in de novo assemblers such as SGA (21). It is noteworthy that the paired-end reads can also be used in the contig production step (e.g., in Velvet) to correctly extend contigs through repetitive regions. The larger paired-end libraries or longer sequence reads are more beneficial for correctly assembling repetitive regions. The final output of the assembly algorithms are sequence scaffolds, and the further assembly of the scaffolds to generate chromosome sequences is usually done by integrating with genetic or physical maps (10, 22, 23). The lack of genetic or physical maps for most of the newly sequenced species makes the correct ordering of scaffolds along chromosomes an extremely pressing challenge. As a result, most genomes generated by large-scale projects such as G10K and i5K will lack chromosome assemblies and will be unsuitable to study chromosome evolution (10).
There are now representative genome assemblies of most of the major phylogenetic clades of vertebrates. These assemblies present a powerful resource to address the problem of chromosome assembly of newly sequenced species that do not have a physical map. Herein, we describe a method, called reference-assisted chromosome assembly (RACA), which can be used to further assemble de novo assembled sequence scaffolds into longer chromosomal fragments. This method will be valuable for large-scale genome assembly projects and will greatly facilitate better understanding of the mechanisms and consequences of chromosome rearrangements during evolution.
Results
Algorithm Overview.
The reconstruction of chromosome fragments using RACA uses an alignment of the target, reference, and outgroup genomes as input (Fig. 1A) produced with a pairwise alignment program such as LASTZ (24). The RACA algorithm merges colinear alignments into syntenic fragments (SFs), and keeps the SFs of length greater than a given threshold (Fig. 1B). For each pair of SFs, the adjacency score that represents how likely the two SFs are adjacent in the target genome is calculated by combining (i) the posterior probability of the adjacency in the target genome given its adjacencies in a reference and outgroup genomes together with the phylogenetic relationship among input genomes (Methods and SI Appendix), and (ii) the amount of paired-end reads that support the adjacency, which may not be effectively used to join or split sequence contigs produced by other assembly algorithms (Fig. 1C).
Fig. 1.

Overview of the RACA algorithm. (A) RACA takes a reference, a de novo sequenced target (as scaffolds), and one or more outgroup genomes as input data. (B) Syntenic fragments (SFs) delimited by vertical dashed lines are constructed by first aligning reference and target genome sequences and next merging colinear alignments. The outgroup is not always aligned to SFs (e.g., sf2) and may contain rearrangements compared with one SF (e.g., sf10). Pluses and minuses represent the orientations of the target and outgroup on the reference, and three groups of SFs represent three reference chromosomes. (C) For each pair of SFs, the adjacency scores (edge weights) that combine (i) the posterior probability [PostProb(i,j)] of the adjacency and (ii) the coverage of paired-end reads [Link(i,j)] are calculated. Only a portion of the edge weight matrix is shown on the Left, and this matrix can represent all four adjacency cases: (i, j), (−i, j), (i, −j), and (−i, −j), where i and j are the indexes of two SFs sfi and sfj, respectively. (D) The SF graph is built by connecting SFs whose edge weight in C is higher than a certain threshold (0.1 was used in the case of Tibetan antelope). Head (closed circle) and tail (open circle) vertices from the same SF are always connected with a maximum weight (dashed edge). (E) Constructed chains of SFs that are extracted by the RACA algorithm.
Once the computation of the adjacency scores is completed, the SF graph is constructed, which consists of head and tail vertices that represent the head and tail of each SF, and their undirected edges (Fig. 1D). In the SF graph, different edges have different weights (the adjacency scores), and the head and tail vertices from the same SF are always connected with a maximum weight. Here, the distinction between the head and tail of a SF is essential because each SF can be connected to either the head or tail of another SF. The RACA is a greedy algorithm in that it constructs the chains of SFs by merging two adjacent SFs with the highest edge weight first at each step (Fig. 1E). By using the order and orientation of SFs that are inferred from the chains of SFs, RACA finally concatenates the scaffolds of the de novo target assembly that the SFs belong to (see Methods for the details of the RACA algorithm).
Evaluation of the RACA Algorithm Using Simulated Genome Assemblies.
To evaluate RACA we simulated genome sequences using Evolver (25). From an ancestral genome that consists of 69 mbp of human genome sequence (chromosomes HSA21 and HSA22), Evolver simulated the evolution of the input genome sequences according to a given phylogeny (Fig. 2A) by applying all possible small-scale as well as large-scale mutations. The results of the simulation produced 12 derived genomes, R and D0–D10, (SI Appendix). In this evaluation, the paired-end read data were not used and the main focus was to evaluate the adjacency reconstructions by using the posterior probabilities of SF adjacencies (Discussion). For each target dataset D0–D9, the dataset R was used as a reference genome, and more divergent datasets than the chosen target from R were used as outgroup genomes (SI Appendix, Table S1 for statistics of the simulated datasets). The dataset D10 was used only as an outgroup genome. To produce a comparable number of breakpoints that would be found in real genome data we used 5 kbp as a minimum SF size (SI Appendix).
Fig. 2.

Estimation of the accuracy of RACA using simulated genome assemblies. (A) Phylogenetic tree used to generate simulated datasets. Average substitution rates of the generated datasets in comparison with R are D0: 0.059, D1: 0.064, D2: 0.0724, D3: 0.081, D4: 0.090, D5: 0.098, D6: 0.106, D7: 0.114, D8: 0.121, D9: 0.129, D10: 0.136. (B) For all sequence fragments that were generated from the datasets D0–D9, RACA predicted the order and orientation of the sequence fragments by using the dataset R as a reference and more divergent datasets as an outgroup, which were then compared with the true order and orientation. Two evaluation measures were used: (i) recall (x axis), which is the fraction of the true order and orientation of sequence fragments that was found in the predicted sequence fragments, and (ii) precision (y axis), which is the fraction of the predicted order and orientation of sequence fragments that agree with the true order and orientation. For each dataset, the average across five different fragments was displayed, and error bars (horizontal for recall and vertical for precision) represent ±1 SD from the average.
The simulated datasets D0–D9 were partitioned into multiple sequence fragments based on the down-scaled length distribution of scaffolds (SI Appendix) that was estimated from a real de novo assembly, and randomly chosen fragments were combined pairwise to simulate chimeric scaffolds (SI Appendix). The fragmentation was repeated five times to produce five different sets of sequence fragments for each dataset. We predicted the order and orientation of the sequence fragments and compared them with the true order and orientation that were known from the fragmentation step, in terms of (i) recall, which is the fraction of the true order and orientation of sequence fragments that was found in the predicted sequence fragments, and (ii) precision, which is the fraction of the predicted order and orientation of sequence fragments that agree with the true order and orientation (Fig. 2B). We ran RACA using different simulated outgroup datasets to measure their effect on accuracy. However, the observed difference was marginal (less than 0.5%), and therefore the results with the one closest outgroup dataset were reported here. The recall and precision of our method with the datasets D0 and D1 were about 98% and 94%, respectively. Even with the dataset D5, which roughly corresponds to the divergence between human and rhesus (SI Appendix, Table S2), RACA produced almost 80% recall and precision. In this evaluation, 96–99% of total sequences in each dataset were aligned and used for the reconstruction (SI Appendix, Table S1). There was significant negative correlation between the accuracy and the number of breakpoints in the datasets (P value <1e-08 for both recall and precision; Pearson’s correlation test).
Evaluation of the RACA Algorithm Using Real Genome Assemblies.
To examine the potential for improving real genome assemblies produced by various genome assemblers, we applied RACA to seven different assemblies of human chromosome 14 produced from paired-end reads used as part of the Genome Assembly Gold-Standard Evaluations (GAGE) competition (26): ALLPATHS-LG, Bambus2, CABOG, MSR-CA, SGA, SOAPdenovo, and Velvet. Using different resolutions of SF sizes (100, 50, 10, and 1 kbp) and two independent reference species (orangutan and mouse) with cattle as an outgroup, we compared the original and RACA assemblies in terms of total number of scaffolds, the N50 statistic, the number of adjacency errors, and coverage (Methods).
When using orangutan as the reference genome and cattle as an outgroup (Fig. 3; SI Appendix, Table S3), RACA further assembled many of the original scaffolds, resulting in substantial improvement in N50 and dramatically reducing the number of adjacency errors in almost all cases. For example, in the case of the SGA assembly, RACA increased the N50 >900-fold while reducing the adjacency errors >170-fold (orangutan used as a reference; 10 kbp resolution of SFs; SI Appendix, Table S3). Increase in the N50 of predicted chromosome fragments (PCFs) produced by RACA compared with the ALLPATHS-LG assembly was not as pronounced compared with the other assemblers because of the high N50 of the original assembly of human chromosome 14 data used in GAGE. These results for HSA14 were attributed to the sequencing strategy that was specifically designed for ALLPATHS-LG (26) to maximize scaffold size. Despite this inherent advantage for ALLPATHS-LG, at most resolution thresholds, RACA performed equally or better to ALLPATHS-LG. The genome coverage of the original GAGE and RACA assemblies was very similar. When using mouse as a reference, which is ten times more divergent from human than the orangutan reference based on the neutral substitution rate, we observed similar patterns as with the orangutan reference (Fig. 3; SI Appendix, Table S4; Discussion). However, the orangutan reference was more effective in increasing N50 of sequence scaffolds and decreasing adjacency errors.
Fig. 3.

Evaluating RACA improvement of the GAGE assemblies. RACA improved the original assemblies created by seven genome assemblers in the GAGE datasets. The final RACA assemblies were compared with the original assemblies in terms of N50 and the number of adjacency errors. Heat maps show the log ratio of RACA N50 to the N50 of the original assembly (Upper horizontal block), and the log ratio of RACA adjacency errors to the errors of the original assembly (Lower horizontal block), with orangutan genome as a reference (vertical block on the Left) as well as mouse genome as a reference (vertical block on the Right). Four different resolutions of SF size were used: 100, 50, 10, and 1 kbp; gray blocks represent the results where there were no N50 data due to low coverage at certain resolutions. For the complete dataset, see SI Appendix, Tables S3 and S4.
The novelty of the RACA algorithm is that it uses both paired-end read mapping and comparative genome information to create chromosome-scale assemblies. An outgroup species, which provides more information in the context of evolution, can be used to resolve ambiguity in determining the location of evolutionary breakpoints in chromosomes. Therefore, we also evaluated the effect of the outgroup species on adjacency errors. When we used GAGE HSA14 data as the target, orangutan as the reference, and cattle as the outgroup species, reduced adjacency errors were found in most cases (SI Appendix, Table S5). In addition, as the SF resolution was increased from 100 kbp to 1 kbp, the outgroup information was helpful in reducing adjacency errors. This is because with higher resolution, there are more ambiguous cases due to the alignment uncertainty. When mouse was used as the reference, we observed an even clearer pattern of the benefit of outgroup information (SI Appendix, Table S6). Finally, when we evaluated RACA without using paired-end mapping information, we observed similar performance with only a minor increase of errors, indicating that the comparative genome information was critical for producing robust results with RACA (SI Appendix, Fig. S1 and Tables S7 and S8).
Reconstruction of Tibetan Antelope PCFs.
We applied RACA to reconstruct the PCFs of the recently sequenced and assembled Tibetan antelope (Pantholops hodgsonii; 2N = 60) genome (SI Appendix). We used a cattle genome assembly (University of Maryland, UMD3.0) as a reference and a human genome assembly (National Center for Biotechnology Information, NCBI36/hg18) as an outgroup. Selecting a minimum SF size of 150 kbp, 1,434 Tibetan antelope scaffolds of 15,996 (96% coverage) were used and 1,597 SFs were identified. These SFs cover 95% of the assembled Tibetan antelope genome sequence, all 29 cattle autosomes and the X chromosome (SI Appendix, Table S9). Our method predicted 1,537 SF adjacencies, of which 73 were recovered only by RACA and not from direct mapping of scaffolds to the cattle genome. The RACA algorithm reconstructed 60 Tibetan antelope PCFs, of which 16 were homologous to complete cattle chromosomes (2, 3, 6, 8, 9, 11, 12, 15, 18–20, 23–25, 28, and 29) (Table 1 and SI Appendix, Fig. S2). The N50 of PCFs is 87 mbp (Table 1), with the size of the longest PCF being 193 mbp and consisting of 111 Tibetan antelope scaffolds. If we were more conservative in reconstructing SF adjacencies by requiring them to have paired-end read support (1,046 adjacencies), we were able to reconstruct 512 PCFs (N50 = 9.5 mbp) (SI Appendix, Table S10 and S11). We visualized the reconstructed Tibetan antelope PCFs with mapped cattle, Tibetan antelope, and human genome assemblies (see mapping results in Datasets S1, S2, S3, S4, and S5) using the Evolution Highway comparative chromosome browser (5) (http://eh-demo.ncsa.uiuc.edu/TA).
Table 1.
Statistics of Tibetan antelope predicted chromosome fragments
| Category | Value |
| No. PCFs | 60 |
| No. PCFs that are homologous to complete cattle chromosomes* | 16 |
| No. PCFs without outgroup (human) matches† | 1 |
| Total length of PCFs | 2.601 gbp |
| Maximum length of PCFs | 193 mbp |
| Minimum length of PCFs | 251 kbp |
| PCF N50 | 87 mbp |
| Maximum no. Tibetan antelope scaffolds in PCFs | 111 |
| Minimim no. Tibetan antelope scaffolds in PCFs | 1 |
| No. cattle EBRs | 64 |
| No. other EBRs | 411 |
| No. Tibetan antelope scaffolds that have more than one SF | 130 (9%)‡ |
| No. Tibetan antelope scaffolds predicted as chimeric | 84 (6%)§ |
EBRs, evolutionary breakpoint regions; PCFs, predicted chromosome fragments; SFs, syntenic fragments.
*These correspond to entire cattle chromosomes 2, 3, 6, 8, 9, 11, 12, 15, 18, 19, 20, 23, 24, 25, 28, and 29.
†There was no mapped human genome fragment to these PCFs.
‡Percentage of the total number of aligned Tibetan antelope scaffolds.
§Among 84 scaffolds, 6 were mapped to three different PCFs, 69 were mapped to two different PCFs, and the remaining 9 were mapped to the same PCF at different and nonadjacent locations.
The accuracy of this reconstruction can be estimated by first computing the number of breakpoints between cattle and Tibetan antelope and then interpolating the accuracy (Fig. 2B) on the basis of the number of breakpoints in the simulated datasets. However, the accurate computation of the number of evolutionary chromosome breakpoints between cattle and Tibetan antelope is not possible because the assembly of Tibetan antelope is highly fragmented. Therefore, as an alternative measure, we calculated the ratio of the number of SFs to the number of Tibetan antelope scaffolds and compared it to the same ratio derived from the sequence fragments of the simulated datasets (SI Appendix, Table S12). This measure can be used to directly compare two sets of sequence fragments in terms of the difficulty of chromosome reconstruction because a higher ratio means that more scaffolds have multiple SFs, which could make the chromosome reconstruction more challenging. We found significant negative correlation between the accuracy (Fig. 2B) and the above ratio of the datasets (P value <1e-10 for both recall and precision; Pearson’s correlation test), which confirms the efficiency of the measure. The ratio of the number of SFs to the number of scaffolds in the Tibetan antelope assembly was 1.1137, which is larger than the ratio of the dataset D0 (recall and precision ∼99%) but smaller than the same ratio of dataset D1 (recall and precision ∼95%) (Fig. 2B; SI Appendix, Table S12). Therefore, the reconstruction of the Tibetan antelope PCFs may have recall and precision higher than 95%.
We performed PCR to validate the predicted adjacencies and misassemblies. Of 14 primer pairs used to test predicted adjacencies between Tibetan antelope scaffolds, 11 pairs (11/14 = 79%) produced single products, of which 4 (36.4%) were very similar to the expected gap size in the cattle genome as determined by the alignment of the Tibetan antelope scaffolds to the cattle genome sequence (Dataset S6); the rest of the PCR products were larger than expected, indicating insertions in the Tibetan antelope genome relative to the cattle genome at these coordinates. In the same way, we confirmed misassemblies in two Tibetan antelope scaffolds (63 and 358) that were marked as chimeric (misjoins) by RACA (SI Appendix).
Analysis of Evolutionary Breakpoint Regions in Cattle and Tibetan Antelope Chromosomes.
We mapped the cattle and human genomes to the reconstructed PCFs and found 64 cattle-specific and 411 other evolutionary breakpoint regions (EBRs) (Table 1; see an example in SI Appendix, Fig. S2). Of 64 cattle-specific EBRs, there were two interchromosomal EBRs that join two pairs of cattle chromosomes (7, 10 and 21, 27, respectively) in Tibetan antelope PCFs (SI Appendix, Fig. S2). We note that the actual number of EBRs could be larger than what is reported here due to the fragmented nature of the Tibetan antelope assembly. We next analyzed how many Tibetan antelope scaffolds span known EBRs that were discovered in the cattle genome (27) (SI Appendix). Of 73 known cattle-specific EBRs, at least 46 were spanned by the Tibetan antelope scaffolds, indicating that these are ancestral bovid- or ruminant-specific (in cattle and Tibetan antelope, but not in pig) EBRs (SI Appendix, Table S13). We also found that 35% (6/17) of known cetartiodactyl-specific EBRs were spanned by the Tibetan antelope scaffolds (SI Appendix, Table S13).
Discussion
Chromosome-anchored assemblies of mammalian genomes have facilitated the discovery of important features of chromosome evolution (10). A critical limiting factor in gaining even greater insight into the biology of genome evolution is that the genome assemblies produced from NGS are highly fragmented. Recent improvements in NGS strategies, such as sequencing of paired ends from large insert libraries, and the development of new assembly algorithms have produced scaffold assemblies with larger N50, which in turn increases the likelihood that such assemblies will span EBRs. However, a reliable method for reconstructing chromosome-scale fragments from NGS assemblies has remained a critical limitation of the current technology. The RACA algorithm can further assemble sequence scaffolds into chromosome-scale assemblies. Our method uses comparative evolutionary inference together with support from paired-end reads in de novo-generated scaffolds to reconstruct chromosomal architecture with high accuracy. The framework is generic enough to accommodate other available information, such as known EBRs and partial genetic mapping data.
Evaluation of RACA using 10 synthetic genomes with different simulated divergence times from the reference genome demonstrated that our approach generates chromosome blocks with 65–98% accuracy of adjacency given that the reference and target genomes can be aligned with ∼95% coverage of both genomes within SFs, and when the ratio of SFs to the total number of scaffolds in the de novo assembled genome is between 1.79 and 1.09, respectively. Therefore, the accuracy of RACA can be adjusted by either selecting a reference genome(s) that is phylogenetically closer to the de novo assembled species or by increasing the scaffold size in the de novo assembly. We evaluated how well our method reconstructs the original genomes from the sequence scaffolds by considering only the posterior probabilities of SF adjacencies. The potential drawback of this evaluation is that the paired-end reads were not simulated and their effect on prediction was not measured. However, we did merge 6% of simulated scaffolds randomly to analyze effects resulting from errors in the assembly. An important feature of our framework is that it provides a reliability score of all SF adjacencies in the de novo assembly as a natural consequence of their posterior probabilities (based on the reference and outgroup genomes) and the number of paired-end reads that support each adjacency. In addition, our approach uses the number of paired-end reads to distinguish between true and chimeric adjacencies in the scaffolds that contain multiple SFs. Therefore, the accuracy of our algorithm is likely to be higher than the evaluation results that considered only the posterior probabilities.
In comparison with the GAGE datasets, our results show that the original N50 has an impact on the final RACA result. If the N50 of an assembly was very small then the genome coverage obtained using RACA was also limited when low RACA resolution (e.g., 100 kbp SFs) was used. This is because scaffolds smaller than the user-defined SF resolution are not included in the analysis. However, as long as the original N50 was reasonable (e.g., greater than 80 kbp), RACA always significantly improved N50 scaffold size and the number of adjacency errors was reduced (unless the original N50 was already very high, such as with ALLPATHS-LG). However, it is noteworthy that RACA still has advantage over ALLPATHS-LG, although the HSA14 data used in GAGE may not reflect that because ALLPATHS-LG is specifically tuned to the Broad Institute genome sequencing pipeline, which was the source of the HSA14 data used for the GAGE comparison (26). For example, in genomes with larger repetitive regions, we expect such regions will have an effect on ALLPATHS-LG performance if the insert sizes of the paired-end libraries are significantly lower than the size of large repetitive regions in the target genome. By comparison, RACA has the ability to use comparative information from other species in the context of evolution to further predict scaffold orders so that the chromosome organization of the assembled sequence scaffolds of the target species can be resolved. In this evaluation, we can improve the results from various assemblers when we used both the orangutan as a reference and the more divergent mouse as a reference. This demonstrates that if we have a good outgroup, RACA can still greatly improve the original assembly even though the reference has a relatively long evolutionary distance (e.g., 80 MYA divergence time) from the target genome.
The utility and effectiveness of RACA was demonstrated by the reconstruction of Tibetan antelope chromosomes from the scaffold assembly produced by SOAPdenovo (Table 1). Sixteen Tibetan antelope PCFs were homologous to full-length cattle autosomes, whereas the remaining 27 PCFs covered the remaining 13 cattle autosomes, and 17 PCFs were aligned to the cattle X chromosome BTAX. Fragmentation of the Tibetan antelope X chromosome into many PCFs is likely due to problems in the assembly of BTAX (28), resulting in many unsupported adjacencies. The vast majority (79%) of adjacencies in the Tibetan antelope genome predicted by RACA and tested by PCR were confirmed by a single PCR product, providing strong experimental support for the method. The few unconfirmed adjacencies were supported by paired-end read data and so the multiple bands obtained by PCR likely represent true adjacencies. Furthermore, on the basis of alignments of scaffolds to multiple chromosomes, or different sites in the same chromosome, we predicted 6% of scaffolds to be chimeric, of which adjacencies in two were also not confirmed by PCR. These results show that RACA can also be used to flag problematic parts of an assembly, and to properly align components of chimeric scaffolds to their proper location on the target genome chromosomes. We also demonstrated that RACA is useful in resolving lineage- and clade-specific chromosomal rearrangements. At least half of what were previously believed to be cattle-specific EBRs (27) were found to have occurred before the divergence of cattle and Tibetan antelope from a common ancestor of bovids ∼26 MYA. With the availability of RACA results for other artiodactyls (11), it will now be possible to address questions about the rates of chromosomal rearrangements and other features of chromosome evolution within this clade, which exhibits many exquisite adaptations.
In the present study, we demonstrated that when the reference and target genomes have evolutionary distance (in terms of number of EBRs) similar to cattle and Tibetan antelope, we can reach high accuracy for both precision and recall of the PCFs assembled by RACA. Also, in our analysis based on the GAGE dataset, we used mouse as a reference (in addition to orangutan), which has about 80 MYA divergence with human. Our results show that RACA can significantly improve the results obtained with different de novo assemblers (both N50 and adjacency errors) when they are used in tandem. One important aspect to consider when picking a reference genome is the alignability of the target and reference sequences. As our results indicate, the more closely related the target and reference are, the better the results with RACA will be.
A pressing challenge for genomes assembled using NGS technologies is that the assemblies are highly fragmented due to limited length of sequence reads. As we have shown, RACA permits the prediction of chromosome organization of SFs on a genome-wide basis in a de novo sequenced species without the aid of a genetic or physical map. Simulation studies and comparison with the GAGE data support our conclusion that RACA will be extremely useful for large-scale sequencing projects now underway (11, 12). Proper identification, classification, and characterization of EBRs within and between the major taxa will allow detailed studies of genome evolution and a better understanding of the unique adaptations that have occurred in different lineages (10).
Methods
RACA.
SF graph.
Given a set of SFs B, the SF graph is an undirected graph G = (V, E), where V = {bh, bt|b ∈ B} is the set of vertices that represents the head bh and tail bt of a SF b, and E is the set of undirected edges. The idea is analogous to the breakpoint graph that was used in genome rearrangement analysis (29, 30). The edge between two SFs can be created by connecting either the head or tail vertices of each SF, which represents both the order and orientation of SFs. Each edge has a weight in the range of 0–1 that indicates how confident the connection is. The head and tail vertices from the same SF are always connected with a maximum weight, and other vertices are connected only when their weights are greater than 0 (see SI Appendix, Fig. S3A for an example).
Edge weight.
Each edge has a weight that represents the confidence of an adjacency between two SFs. We defined the edge weight by considering two sources of information: (i) the posterior probability of an adjacency in terms of the evolution of a SF configuration and (ii) the support from the paired-end read mapping. For each pair of two SFs, bi and bj, there exist four different types of edges to connect them: (i) edge between 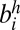 and
and 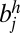 , represented as (−i, j) or (−j, i); (ii) edge between
, represented as (−i, j) or (−j, i); (ii) edge between 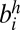 and
and  , represented as (−i, −j) or (j, i); (iii) edge between
, represented as (−i, −j) or (j, i); (iii) edge between 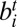 and
and 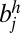 , represented as (i, j) or (−j, i); and (iv) edge between
, represented as (i, j) or (−j, i); and (iv) edge between 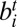 and
and 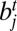 , represented as (i, −j) or (j, −i). In other words, the i and j are the indexes of SFs, and the positive and negative signs indicate the relative orientations of SFs. For example, the two solid edges in SI Appendix, Fig. S3A can be represented as (1, −2) or (2, −1), and (−2, 3) or (−3, 2). The weight w(i, j) of an edge (i, j) is defined as:
, represented as (i, −j) or (j, −i). In other words, the i and j are the indexes of SFs, and the positive and negative signs indicate the relative orientations of SFs. For example, the two solid edges in SI Appendix, Fig. S3A can be represented as (1, −2) or (2, −1), and (−2, 3) or (−3, 2). The weight w(i, j) of an edge (i, j) is defined as:
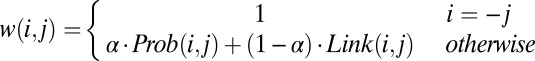 |
The head and tail vertices from the same SF are always connected with a weight 1 (Eq. 1). An edge weight w(i, j) between two different SFs (Eq. 2) is computed by merging (i) the posterior probability of the adjacency between two SFs bi and bj [Prob(i, j); SI Appendix] and (ii) the support from paired-end reads that link them [Link(i, j); SI Appendix]. The parameter α controls the relative contribution of the two scores (SI Appendix).
SF ordering algorithm.
We defined the SF ordering problem as: given a SF graph G, find a set of connected components with the degree of each vertex at most two and without a cycle that maximizes the sum of edge weights. The degree and cycle constraints ensure that each connected component is actually a chain of SFs (without cycle), and each SF is adjacent with only one SF. The SF ordering problem is analogous to the minimum path cover problem and it is known as NP-hard (30). Therefore, we developed a greedy algorithm as an approximate solution to solve the SF ordering problem, which constructs the chains of SFs by merging two adjacent SFs with the highest edge weight first at each step (SI Appendix).
Evaluation of RACA Using the GAGE Datasets.
The paired-end reads and assembly results for human chromosome 14 produced by seven genome assemblers, ALLPATHS-LG, Bambus2, CABOG, MSR-CA, SGA, SOAPdenovo, and Velvet, were downloaded from the GAGE website (http://gage.cbcb.umd.edu). To construct the paired-end read mapping information, we aligned the paired-end reads against assembly sequences by using the BWA (v 0.5.9) program (31) with default parameters except for “-t 10 -q 15”. Paired-end reads that were not mapped uniquely were discarded. RACA was tested on two different datasets: one with an orangutan (ponAbe2 assembly) reference and the other with a mouse (mm9 assembly) reference, both with the cattle (umd3 assembly) genome as an outgroup. SFs were constructed at four different resolutions, 100, 50, 10, and 1 kbp. Then RACA was applied to each dataset to further assemble the original assemblies. The performance of RACA was mainly examined by measuring the fraction of N50 increase and the number of adjacency errors that were computed by using the GAGE evaluation pipeline. We defined two evaluation measures: misjoin and unjoin errors. The misjoin errors occur when two adjacent contigs in the predicted assembly are not actually adjacent in the human genome assembly. The unjoin errors occur when two contigs are actually adjacent in the human genome assembly, but they are in separate scaffolds in the predicted assembly. We considered the sum of the misjoin and unjoin errors as the final adjacency errors (see SI Appendix for details).
Analysis of Known Evolutionary Breakpoint Regions.
We obtained the coordinates (bosTau4 assembly) of 135 cow- and cetartiodactyl-specific EBRs from Elsik et al. (27). A lineage-specific EBR is defined as an interval between the two boundaries of a lineage-specific breakpoint inferred from cross-species sequence comparison. After removing 12 EBRs that could result from assembly errors, we converted the coordinates (5, 32) to the UMD 3.0 assembly by using the liftOver program in the UCSC Genome Browser (33). This produced 90 coordinates that were successfully mapped to the UMD 3.0 assembly. A Tibetan antelope scaffold was determined to span a known EBR only when the distances from its two boundaries to the known EBR were greater than 100 kbp. This threshold was set to minimize the possibility of artifacts in sequence alignment.
Supplementary Material
Acknowledgments
We thank G. Robinson for useful comments on the manuscript. This work was supported by a National Science Foundation CAREER Award 1054309 (to J.M.); National Institutes of Health/National Human Genome Research Institute Grant 1R21HG006464 (to J.M.); US Department of Agriculture Grants 538 AG2009-34480-19875 and 538 AG 58-1265-0-031 (to H.A.L.); National Research Foundation of Korea Grant 2012R1A1A1015186 (to J.K.); Polish Grid Infrastructure Project Grant POIG.02.03.00-00-007/08-00 (to D.M.L.); National Basic Research Program of China Grant No. 2012CB518200 (to R.-L.G.), Program of International S&T Cooperation of China Grant No. 0S2012GR0195 (to R.-L.G.), and National Natural Science Foundation of China Grant No. 30393133 (to R.-L.G.). J.K. was an Institute for Genomic Biology fellow at the University of Illinois.
Footnotes
The authors declare no conflict of interest.
Data deposition: The data reported in this paper have been deposited in the Reference-Assisted Chromosome Assembly (RACA) website, http://bioen-compbio.bioen.illinois.edu/RACA/.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1220349110/-/DCSupplemental.
References
- 1.Hardison RC. Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet. 2000;16(9):369–372. doi: 10.1016/s0168-9525(00)02081-3. [DOI] [PubMed] [Google Scholar]
- 2.Bejerano G, et al. Ultraconserved elements in the human genome. Science. 2004;304(5675):1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- 3.Pollard KS, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443(7108):167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- 4.Bourque G, Zdobnov EM, Bork P, Pevzner PA, Tesler G. Comparative architectures of mammalian and chicken genomes reveal highly variable rates of genomic rearrangements across different lineages. Genome Res. 2005;15(1):98–110. doi: 10.1101/gr.3002305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy WJ, et al. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science. 2005;309(5734):613–617. doi: 10.1126/science.1111387. [DOI] [PubMed] [Google Scholar]
- 6.Ma J, et al. Reconstructing contiguous regions of an ancestral genome. Genome Res. 2006;16(12):1557–1565. doi: 10.1101/gr.5383506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma J, et al. The infinite sites model of genome evolution. Proc Natl Acad Sci USA. 2008;105(38):14254–14261. doi: 10.1073/pnas.0805217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’haene B, et al. Disease-causing 7.4 kb cis-regulatory deletion disrupting conserved non-coding sequences and their interaction with the FOXL2 promotor: Implications for mutation screening. PLoS Genet. 2009;5(6):e1000522. doi: 10.1371/journal.pgen.1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goode DL, et al. Evolutionary constraint facilitates interpretation of genetic variation in resequenced human genomes. Genome Res. 2010;20(3):301–310. doi: 10.1101/gr.102210.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewin HA, Larkin DM, Pontius J, O’Brien SJ. Every genome sequence needs a good map. Genome Res. 2009;19(11):1925–1928. doi: 10.1101/gr.094557.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genome 10K Community of Scientists Genome 10K: A proposal to obtain whole-genome sequence for 10,000 vertebrate species. J Hered. 2009;100(6):659–674. doi: 10.1093/jhered/esp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson GE, et al. Creating a buzz about insect genomes. Science. 2011;331(6023):1386. doi: 10.1126/science.331.6023.1386. [DOI] [PubMed] [Google Scholar]
- 13.Simpson JT, et al. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009;19(6):1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gnerre S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA. 2011;108(4):1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li R, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20(2):265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jurka J, et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1-4):462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 18.Miller JR, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics. 2010;95(6):315–327. doi: 10.1016/j.ygeno.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pevzner PA, Tang H, Waterman MS. An Eulerian path approach to DNA fragment assembly. Proc Natl Acad Sci USA. 2001;98(17):9748–9753. doi: 10.1073/pnas.171285098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27(4):578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 21.Simpson JT, Durbin R. Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 2012;22(3):549–556. doi: 10.1101/gr.126953.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trask BJ. Human cytogenetics: 46 chromosomes, 46 years and counting. Nat Rev Genet. 2002;3(10):769–778. doi: 10.1038/nrg905. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz DC, et al. Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science. 1993;262(5130):110–114. doi: 10.1126/science.8211116. [DOI] [PubMed] [Google Scholar]
- 24.Harris RS. 2007. Improved pairwise alignment of genomic DNA. PhD thesis (Pennsylvania State Univ, University Park, PA)
- 25.Edgar RC, Asimenos G, Batzoglou S, Sidow A. 2010. Evolver: A whole-genome sequence evolution simulator. Available at www.drive5.com/evolver. Accessed December 15, 2011.
- 26.Salzberg SL, et al. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012;22(3):557–567. doi: 10.1101/gr.131383.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elsik CG, et al. Bovine Genome Sequencing and Analysis Consortium The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science. 2009;324(5926):522–528. doi: 10.1126/science.1169588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimin AV, et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009;10(4):R42. doi: 10.1186/gb-2009-10-4-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alekseyev MA, Pevzner PA. Breakpoint graphs and ancestral genome reconstructions. Genome Res. 2009;19(5):943–957. doi: 10.1101/gr.082784.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma J, et al. DUPCAR: Reconstructing contiguous ancestral regions with duplications. J Comput Biol. 2008;15(8):1007–1027. doi: 10.1089/cmb.2008.0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larkin DM, et al. Breakpoint regions and homologous synteny blocks in chromosomes have different evolutionary histories. Genome Res. 2009;19(5):770–777. doi: 10.1101/gr.086546.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.