

Abstract
DNA methylation is a common feature of eukaryotic genomes and is especially common in noncoding regions of plants. Protein coding regions of plants are often methylated also, but the extent, function, and evolutionary consequences of gene body methylation remain unclear. Here we investigate gene body methylation using an explicit comparative evolutionary approach. We generated bisulfite sequencing data from two tissues of Brachypodium distachyon and compared genic methylation patterns to those of rice (Oryza sativa ssp. japonica). Gene body methylation was strongly conserved between orthologs of the two species and affected a biased subset of long, slowly evolving genes. Because gene body methylation is conserved over evolutionary time, it shapes important features of plant genome evolution, such as the bimodality of G+C content among grass genes. Our results superficially contradict previous observations of high cytosine methylation polymorphism within Arabidopsis thaliana genes, but reanalyses of these data are consistent with conservation of methylation within gene regions. Overall, our results indicate that the methylation level is a long-term property of individual genes and therefore of evolutionary consequence.
Keywords: epigenetics, methylome, Poaceae, molecular evolution
Cytosine methylation is a heritable modification of DNA that is associated with additional epigenetic markers, including histone modification (1) and nucleosome positioning (2). Together these epigenetic modifications regulate transcription, providing a flexible mechanism to adjust expression during development and stress (3, 4). In plants, DNA methylation is especially pervasive in intergenic regions, where it acts to limit transcription and proliferation of transposable elements (TEs) (5). Cytosines within TEs are typically methylated in three sequence contexts: CG, CHG, and CHH, where H = A, C, or T.
Cytosines are also methylated within protein-coding regions (i.e., between start and stop codons), but typically gene body methylation (gbM) is limited to the CG context (6–8). The molecular mechanisms that produce gbM are not yet fully characterized, but studies suggest it is under different mechanistic and regulatory controls than TE methylation (9–11). As a result, TE and gene body methylation demonstrate different evolutionary distributions. Although TE methylation has been acquired independently in several evolutionary lineages, gbM is a basal evolutionary feature of eukaryotes (12, 13). Nonetheless, gbM may be evolutionary labile in plants, based on two observations. First, it is wholly absent from a fern and a moss (12, 13), suggesting that there is variation in the presence and extent of gbM. Second, it is “highly polymorphic” (14) between accessions of Arabidopsis thaliana (8, 15). These methylation polymorphisms can accrue rapidly. For example, 60% of 2,485 differentially methylated regions among A. thaliana mutation accumulation (MA) lines are located within genes (16, 17).
The uncertain evolutionary dynamics of gbM are matched by uncertainty in function. Because gbM tends to be associated with genes of intermediate expression (18, 19), one hypothesis is that gbM is a functionless byproduct of transcription (20, 21). Alternative hypotheses include the ideas that gbM increases the accuracy of splicing (22–24) or prevents aberrant transcription within genes (19, 25). If gbM is indeed functional, one expects its distribution to be nonrandom among genes. This expectation holds in A. thaliana, where body-methylated genes are expressed at intermediate levels (18, 19), but are longer, evolve more slowly, and are more apt to exhibit phenotypic effects when knocked out (26). These observations are consistent with a gbM function related to transcription efficiency or accuracy, but this conclusion is at odds with the lability and polymorphism of gbM in plants.
If gbM does indeed play a crucial functional role, we postulate that it should be constrained, and thus highly correlated, between orthologs across species. However, there have been no detailed comparisons of gbM patterns among orthologous genes, largely because existing methylome data are too taxonomically distant. To make such a comparison, we generated bisulfite sequencing (BS-seq) data for Brachypodium distachyon from two tissues (leaves and immature floral buds) to compare gbM patterns both between B. distachyon tissues and between B distachyon and rice (Oryza sativa ssp. japonica) (12). These two species represent separate subfamilies of the economically important grass family (Poaceae). They last shared a common ancestor 40–53 million years ago (27) but are closely enough related to permit molecular evolutionary comparisons.
With BS-seq data from B. distachyon, rice, and A. thaliana, we address questions central to understanding the evolutionary dynamics of gene body methylation. Do methylated genes in the grasses exhibit biases similar to those of A. thaliana? Is gbM conserved between orthologs of highly diverged grass species? If so, what might be the long-term evolutionary consequences of gbM for these genes, and how might observations of long-term gbM conservation be synthesized with previous observations of high gbM polymorphism? Finally, what do the answers to these questions imply about the evolutionary forces that act on gbM?
Results and Discussion
B. distachyon Methylome.
We generated B. distachyon BS-seq data from three biological replicates and two tissues (leaf and immature flower buds), resulting in ≥15 times coverage for each replicate of each tissue (SI Appendix, Table S1). Based on comparisons to unmethylated chloroplast DNA, the data had a low, 1.05% average error rate of conversion error across replicates. We used these error rates to infer whether a particular cytosine site was methylated, based on the binomial test of Lister et al. (7) (Materials and Methods). This approach provides a reasonable assignment of methylation status relative to the proportion of nonconverted reads at each site (SI Appendix, Fig. S1).
Based on this approach, we found that cytosine methylation differences were low among our replicate samples, with 2.44% differences on average, for entire chromosomes. We also found that the B. distachyon methylome is typical of higher eukaryotes in at least three respects (6, 7, 18, 19, 28): (i) levels of cytosine methylation are highest in the CG (56.5%) and CHG (35.3%) contexts relative to the CHH context (1.8%); (ii) the total level of methylation is highest near centromeres (Fig. 1; SI Appendix, Fig. S2); and (iii) CG and CHG methylation levels are correlated negatively with gene density (r = −0.692 for CG; r = −0.788 for CHG; P < 10−5) and positively with TE density (r = 0.511 for CG; r = 0.539 for CHG; P < 10−5; Fig. 1; SI Appendix, Table S2).
Fig. 1.
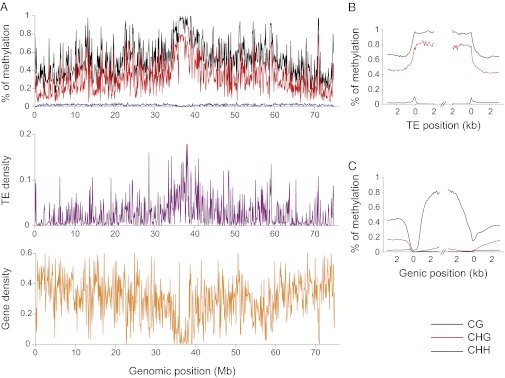
DNA methylation in B. distachyon. (A) The pattern of CG (black), CHG (red), and CHH (blue) methylation (Top), gene density (Middle), and TE density (Bottom) across chromosome 1 (see SI Appendix, Fig. S2 for other chromosomes). (B) The pattern of DNA methylation within and around TEs. (C) The pattern of DNA methylation within and around genes. For this figure, we used only genes for which 5′ and 3′ untranslated regions were annotated. In B and C, the colors represent cytosine sequence contexts as in A), and the x axes represent length along TEs and genes, respectively. For TEs, the zeros represent element boundaries; for genes, the zero on the left corresponds to the transcription-start sites and that on the right to the 3′ end of transcripts.
However, both the pattern and level of CHH methylation differs between species. B. distachyon CHH levels are 5- to 10-fold lower than those of rice (12, 13). Unlike other plant species (29), B. distachyon CHH levels are lowest near centromeres (Fig. 1; SI Appendix, Fig. S2) and positively correlated with gene density (SI Appendix, Table S2), despite the lack of CHH methylation within gene bodies (Fig. 1). This unexpected difference in pattern between species cannot be ascribed to the loss of homologs to known CHH-methylating genes, such as DRD1, DDM1, or HOG1, in B. distachyon (SI Appendix, Table S3). It does suggest, however, that the pathways that mediate CHH methylation differ in targeting, regulation, or mechanism between species. The unexpected pattern of CHH methylation illustrates that there is much to discover about the differences between, and significance of, methylation patterns among plant species (30).
gbM patterns in B. distachyon are also broadly similar to those of other plant species. For example, methylation in CHH and CHG contexts are low within genes, and CG levels are low near transcription start sites but peak within the protein coding region (12, 13) (Fig. 1). These patterns are conserved between tissues. Only 0.77% of genic cytosines differ in methylation status between leaf and flower buds, compared with an average of 0.74% among replicates within tissues; thus, over entire chromosomes, there is little evidence of gbM differentiation between tissues (SI Appendix, Table S4), just as there is little divergence in total levels of methylation between tissues.
Gene Body Methylation Is Conserved Between Orthologs.
To compare gbM between species, we mapped existing BS-seq data (12) to the rice genome and identified 7,826 colinear orthologs with sufficient methylation data for comparison (Materials and Methods). Levels of CG methylation were highly positively correlated across the 7,826 ortholog pairs (r = 0.755, P < 10−5; permutation test; Fig. 2). This correlation indicates that the gbM characteristics of orthologs are typically conserved between B. distachyon and rice.
Fig. 2.

Comparisons between rice and Brachypodium ortholog pairs. (A) Distribution of CG methylation level of 7,826 ortholog pairs. (B) The correlation between the differentiation of CG dinucleotide sites between rice and B. distachyon orthologs (y axis) and the level of CG methylation in B. distachyon genes (x axis).
Although our study focuses on comparisons between rice and B. distachyon, we also extended the contrast to maize (Zea mays ssp. mays) orthologs to assess conservation of gbM across a broader evolutionary expanse. Comparisons to maize were complicated by the fact that maize has two subgenomes (31) and also by the fact that fewer genes had sufficient BS-seq coverage for comparison (32) (SI Appendix). Despite these limitations, we identified ∼900 orthologs for comparison with rice and B. distachyon. There were again significant (P < 10−5) and strongly positive correlations across orthologs between species (maize vs. rice, r = 0.510; maize vs. B. distachyon, r = 0.541; SI Appendix, Fig. S3). These correlations suggest that the gbM characteristics of orthologs are typically conserved throughout the grass family.
For rice and B. distachyon, we also tested the null hypothesis that a gene had a methylation level equal to the genomic average. The purpose of this test was to identify genes with high CG methylation but without correspondingly high CHG and CHH methylation levels, because the latter could be indicative of either mis-annotation of repetitive DNA or genes that have heterochromatic properties. After removing annotated genes with high CHG and CHH methylation, the probability distribution of CG methylation (PCG) was strikingly bimodal for both rice and B. distachyon, indicating that the distribution of CG methylation is both nonrandom and autocorrelated (6, 7) (SI Appendix, Fig. S4). After defining body-methylated (BM) and undermethylated (UM) genes as PCG < 0.05 and PCG >0.95 (26), respectively, we identified 3,712 BM and 18,787 UM genes in O. sativa and 3,564 BM and 15,739 UM genes in B. distachyon. In addition, both species contained genes intermediate (IM) between the two well-defined categories, with 2,505 IM genes in rice and 1,781 in B. distachyon. The three classifications (BM, IM, and UM) were conserved between species; 76% of the 7,826 ortholog pairs retained their classification, a proportion far higher than random (P < 10−5; permutation tests). To sum, both correlative (Fig. 2) and probabilistic approaches suggest that gbM is a conserved property of grass orthologs.
Implications of gbM Conservation.
These comparisons paint an overarching picture of conservation of gbM levels among orthologs, even after ∼100 My or more of evolutionary divergence. This conservation may be driven either by functional requirements to methylate particular genes or by sequence characteristics such as shared CG sites between orthologs. Although the latter may contribute, we believe the former is predominant for two reasons. First, only 27% of CG sites were conserved between B. distachyon and rice orthologs; in fact, the proportion of shared CG sites between orthologs was negatively correlated with methylation levels (r = −0.593, P < 10−5; permutation test). In other words, highly methylated orthologs share fewer CG dinucleotides, on average, than lowly methylated genes (Fig. 2).
Second, gbM affects a nonrandom subset of genes in both rice and B. distachyon. As a group, BM genes are biased for longer lengths, lower evolutionary rates (KA), and lower CG [O/E] ratios (Fig. 3). The CG [O/E] ratio is a measure of the observed number of CG dinucleotides relative to that expected given the overall G+C content of a gene; it has been used as a proxy for methylation, with low values consistent with heavy methylation (33). In this context, it is interesting to note that the subset of nonconserved genes (i.e., genes that are not conserved as either BM or UM between species) exhibit intermediate values for all three characteristics: length, KA, and CG [O/E] (Fig. 3). We note, however, that BM and UM genes are not biased with respect to their location near methylated TEs (SI Appendix). For example, in the B. distachyon genome, BM genes are 23.0 kb from the nearest annotated and methylated TE, whereas UM genes are 22.2 kb from the nearest such TE (P > 0.10; permutation test).
Fig. 3.

Evolutionary analysis of BM (red) vs. UM (blue) genes. Gene sets are defined by their category in B. distychon and rice, respectively. Box plots show that gene designated BM in both species are longer (A) and have lower CG [O/E] ratios (B) in each species relative to UM genes. BM genes also diverge more slowly as measured by nonsynonymous divergence (KA) (C). Letters above box plots denote significance groups at P < 0.001.
BM genes also represent a biased set of functions relative to UM genes. Based on GO analyses, BM genes are enriched for eight categories, including critical functions like nucleic acid, nucleotide, and protein binding (Table 1). Analyses from A. thaliana (26) and invertebrate genomes also suggest that methylated genes are enriched for critical functions (34). This bias toward methylation of critical, long, and conserved genes is consistent with hypothesized functional roles for body methylation. For example, long genes are more likely than short genes to have either aberrant transcription sites or complex structures that are prone to mis-splicing.
Table 1.
GO categories enriched for BM vs. UM genes
| Function | Proportion in BM genes | Proportion in UM genes | P value* | Corrected P value† |
| Nucleic acid binding | 0.2750 | 0.1854 | <10−12 | <10−11 |
| Nucleotide binding | 0.1042 | 0.0517 | <10−11 | <10−9 |
| Protein binding | 0.2021 | 0.1509 | <10−5 | <10−3 |
| Nucleobase-containing compound metabolic process | 0.0174 | 0.0079 | <0.01 | NS |
| Cytoplasm | 0.0451 | 0.0306 | <0.01 | NS |
| Kinase activity | 0.0069 | 0.0029 | <0.05 | NS |
NS, not significant.
*P values by Fisher’s exact test.
†P values after Bonferroni correction.
Given that gbM status is evolutionarily conserved between orthologs, it has the potential to influence the evolutionary properties of genes. One such property is the bimodality of G+C content among grass (and monocot) genes (35), which is most apparent in the third codon position (36). The cause of this pattern has been much debated, and three hypotheses are commonly invoked: selection on codon use, neutral mutational heterogeneity, and biased gene conversion (37). Although all of these may contribute to bimodality—particularly to the broader isochore structure of grass genomes (35)—BM and UM genes also demonstrate marked bimodality. G+C content across coding regions and at fourfold degenerate sites are much reduced in BM relative to UM genes for both species (Fig. 4), consistent with cytosine deamination leading to C->T transitions (33). Thus, gbM may cause the G+C bimodality of grass genes. This explanation makes sense only because it is now clear that gbM is evolutionarily conserved, such that mutational heterogeneities between BM vs. UM genes can become apparent over time. Also note that gbM effects may contribute to the fact that G+C content within grass genomes are correlated between introns and codons but not with flanking sequences (38).
Fig. 4.

Frequency distributions of G+C content in rice and B. distachyon genes for the entire coding region and fourfold degenerate sites (GC4). Red and blue lines represent BM and UM genes, respectively, which differ for every comparison (P < 10−5 by permutation test).
Resolving the Paradox of Polymorphism Data.
Our grass comparison, which reveals gbM conservation for orthologs, superficially contradict those from A. thaliana, in which genes are highly polymorphic for cytosine methylation (14–17). On reanalysis, however, Arabidopsis may not differ from rice and B. distachyon. Two observations support this statement. The first is based on analysis of A. lyrata orthologs to the BM genes of A. thaliana Col-0 (7, 26). These 3,492 A. lyrata orthologs also represent a biased gene set with respect to high length, low evolutionary rate (KA), and low CG [O/E] ratio (SI Appendix, Fig. S5), consistent with the maintenance of gbM over ∼26 My of divergence between the two Arabidopsis sister species (39).
The second observation is based on reanalysis of BS-seq data from eight A. thaliana MA lines (16). We defined BM, IM, and UM genes for each of the eight lines and then classified genes by the number of lines in which they were designated in different classifications (i.e., from zero to eight; Table 2). Categories were well conserved across the eight lines; 82% of genes held consistent categories (P < 10−5 based on permutation). Less than 0.3% of genes varied between the BM and UM categories in one or more of the eight lines (Table 2).
Table 2.
Methylation classification and statistics for 21,792 A. thaliana genes among 8 MA lines
| BM | IM | UM | No. of genes | Gene length | CG [O/E] | KA |
| 8 | 0 | 0 | 2,633 | 3,779.3 (NC,**)† | 0.572 (NC,**) | 0.0230 (NC,**) |
| 1–7* | 1–7 | 0 | 1,571 | 2,721.0 (**,**) | 0.591 (**,**) | 0.0235 (**,**) |
| 0 | 8 | 0 | 1,268 | 2,408.1 (**,**) | 0.588 (**,**) | 0.0262 (**,**) |
| 0 | 1–7 | 1–7 | 2,111 | 2,131.2 (**,**) | 0.627 (**,**) | 0.0267 (**,**) |
| 0 | 0 | 8 | 14,137 | 1,664.2 (**,NC) | 0.784 (**,NC) | 0.0312 (**,NC) |
| 1–7 | 1–7 | 1–7 | 72 | 1,867.1 (**,NS) | 0.705 (**,*) | 0.0336 (**,NS) |
BM, body methylated; IM, intermediate methylated; UM, undermethylated.
*By way of example, this row tallies genes that were classified UM in zero of the eight lines, BM in from one to seven of the eight lines, and IM from one to seven of the eight lines. Thus, a single gene in this row was classified as both BM and IM among the eight MA lines.
†The two symbols in parentheses represent P values, respectively, of differences vs. the statistics from a configuration of {8,0,0} and of differences vs. the statistics from configuration vs. {0,0,8}: **P < 10−5; *P < 10−2; NC, no comparison; NS, nonsignificant.
These categories follow a now-familiar trend: genes conserved as BM across all eight lines are longer, have lower CG [O/E] ratios, and evolve more slowly than other genes. Moreover, the remaining genes follow a gradation in these three statistics: IM genes are intermediate between BM and UM genes in length, CG [O/E], and KA (Table 2). Interestingly, as a group, the 72 genes that include at least one accession with a BM allele and another accession with a UM allele most closely approximate genes that are consistently UM (Table 2). In other words, the 72 genes with alleles that vary between UM and BM do not have the structural and evolutionary signatures of other BM genes.
Of course, the conservation of gbM status among accessions could simply be a function of the recent, ∼30-generation divergence among the eight A. thaliana MA lines. However, 22.4% of all CG sites are polymorphic for methylation among the 2,633 genes classified as BM in all eight lines. Thus, BM genes are highly polymorphic for individual cytosine methylation, but not to the extent that it affects the classification of genes that are significantly highly methylated.
Evolutionary Questions Raised by gbM Conservation.
Our comparative analyses between B. distachyon and rice reveal at least four patterns that impact our understanding of the evolution, prevalence, and consequences of gbM. First, patterns of CHH methylation differ substantially between B. distachyon and the other plant species for which methylome data are available. We know neither the cause nor the taxonomic extent of this atypical pattern. Second, gbM does not differ substantially among B. distachyon leaves and floral buds. We do not know whether this is a general trend, because surprisingly few papers have compared gbM among plant tissues, particularly with robust biological replication to measure error. Those few studies that have examined different tissues detect few differences except for highly specialized tissues like the endosperm (40). One exception is Populus trichocarpa, which has high gbM differentiation among tissues (30). Third, gbM affects a biased subset of genes typified by longer length, higher numbers of exons, and slower evolutionary rates, on average (Fig. 3; SI Appendix, Fig. S5). Many BM genes have transcriptional levels similar to UM genes (26); hence, it seems unlikely that gbM is just a byproduct of transcription (20, 21). It seems more likely that gbM plays a functional role that has yet to be fully elucidated. Finally, gbM levels are well conserved between orthologs (Fig. 2), indicating that methylation is of evolutionary consequence.
These series of observations raise two interesting evolutionary issues. The first is the apparent paradox between high gbM polymorphism vs. long-term conservation. Assuming gbM is functional, its extent and distribution must be shaped by natural selection. We conjecture that three features characterize this selection. First, it is primarily a property of regions rather than individual methylated sites. Second, regions are subjected to site-to-site stochasticity in the methylation process and also a threshold effect caused by natural selection. Under this model (Fig. 5), individual cytosine polymorphisms may vary without functional consequence as long as some minimal (or maximal) level of gbM is maintained throughout a genic region. In theory, a threshold effect resolves the paradox of high polymorphism but strong conservation. Finally, selection for gbM applies to a subset of genes, probably because gbM is metabolically costly and thus maintained only for those genes for which transcriptional disruption confers an even greater cost.
Fig. 5.

A schematic of the fitness of alleles within a gene for which gbM is favored by selection; i.e., a BM gene. Alleles A, B, and C are shown with circles denoting methylated cytosines within the coding region. Given that selection is on a region, alleles A and B have similar fitness effects despite detectable methylation polymorphism, because their overall methylation exceeds some threshold (vertical dashed line on the fitness graph). In contrast, allele C is undermethylated and has lower fitness.
If our threshold model is correct, it implies that many, and perhaps most, of the gbM polymorphisms characterized in A. thaliana lack functional consequences. This idea is consistent with our observation that methylation polymorphisms among MA lines rarely affects the gbM status of the entire gene (Table 2), but it also requires further testing through functional analyses. We also note that some cytosine methylation polymorphisms are correlated with functional effects, such as responses to stress (4, 41). However, thus far, these polymorphisms have been shown to lie primarily within promoter and intergenic regions rather than within gene bodies (4, 8). The ratio of consequential vs. nonconsequential cytosine methylation polymorphisms within genes remains an open question.
The second evolutionary issue is that of the maintenance of gbM over evolutionary time, because deamination removes CG dinucleotides sites via spontaneous C->T mutation. Because of this mutation pressure, it is not surprising that highly methylated orthologs share fewer CG sites in common than less methylated genes (Fig. 2). However, how is gbM maintained against this mutation pressure? A key factor is the C->T mutation rate relative to the rate of mutation to cytosines. Recent studies suggest these mutation rates differ by less than fivefold (42), a difference that may be low enough to maintain equilibrium CG [O/E] values similar to those observed in methylated grass and Arabidopsis genes (SI Appendix, Fig. S6). Another possibility is that weak positive or negative selection maintains CG sites when the number of sites becomes too low to maintain a threshold gbM level (Fig. 5). These possibilities require further theoretical modeling, coupled with additional evolutionary analysis of DNA methylation data from throughout the plant kingdom.
Materials and Methods
Generating BS-seq Data in Brachypodium distachyon.
Three B. distachyon plants of the reference Bd21 line (16) were grown under identical greenhouse conditions, including 20-h days to induce rapid flowering. Spikes and leaves were harvested at the beginning of anthesis. BS-seq libraries were generated for each plant and each tissue, for six total libraries, following ref. 16. Additional details are provided in the SI Appendix.
Analyzing BS-seq Data and Identification of Body-Methylated Genes.
We mapped published and original BS-seq data from B. distachyon, A. thaliana (7, 16), and O. sativa (12) using previously published methods (26). Briefly, low-quality reads and bases (q < 20) were filtered, and reads were mapped with BRAT software (43) to reference genomes, allowing mismatches only at potentially methylated sites. Uniquely mapping reads were used for analysis, and clonal biases were removed probabilistically (26). Reference genomes and gene annotations were retrieved from TAIR for A. thaliana [TAIR9 (44)], RAP-DB for O. sativa ssp. japonica [build 5 (45)], JGI for A. lyrata [Filtered Model 6 (46)], and Brachypodium.org for B. distachyon (version 1.0). Although all six B. distachyon replicates (three for leaf, three for flower bud) were mapped to the reference, we used a single leaf replicate as the basis for most inferences (replicate 1 in SI Appendix, Table S1).
BS-seq conversion error rates were estimated by mapping reads to the unmethylated chloroplast DNA (7). Error rates for the B. distachyon data ranged from 0.89% to 1.33% among the six replicates (SI Appendix, Table S1) and from 0.6% to 2.8% for the A. thaliana MA lines. The error rate for rice was 0.11%. We used the error rate to test support for methylation of each nuclear cytosine residue with more than one read after collapsing reads with clonal bias, following ref. 7. The test was based on binomial probabilities, and cytosines with P < 0.01 were considered methylated.
A probabilistic approach was then used to identify body-methylated genes, also following published methods (26). Briefly, we separately assessed cytosine methylation levels for each of the three sequence contexts, CG, CHG, and CHH, where H is A, T, or C, using P values that denote the departure from genomic averages. Within bona fide genes, body methylation is enhanced at only CG sites (6, 7), so we discarded genes that were significantly enriched for CHG and/or CHH methylation. We then classified the remaining genes into three categories: BM (PCG < 0.05), IM (0.05≤ PCG ≤0.95), and UM (PCG > 0.95). We only considered genes with sufficient CG information (ncg ≥ 20) and genes for which ≥40% and 60% of cytosine residues were covered by at least two reads for rice and B. distachyon, respectively (SI Appendix, Fig. S7). Additional details about the analysis and interpretation of BS-seq data, including those from maize, are provided in SI Appendix.
Identifying Orthologs and Calculating Evolutionary Rates.
We calculated substitution rates between A. thaliana–A. lyrata ortholog pairs and between O. sativa–B. distachyon ortholog pairs. The list of 18,330 orthologs for the A. thaliana–A. lyrata pair was taken from ref. 47, with the BM genes in A. thaliana being previously defined (26). For O. sativa–B. distachyon, we inferred orthologous relationships based on both homology and on collinearity following ref. 47 with slight modifications (SI Appendix ).
We ultimately detected 9,531 orthologs between rice and B. distachyon. This set was further pared to 7,826 ortholog pairs based on two criteria: sufficient levels of methylation data and exclusion of genes with PCHG < 0.05 and/or PCHH < 0.05. The set of 7,826 orthologs was also used as a reference by which to identify maize orthologs (SI Appendix). For all suitable orthologs, we calculated KA and KS using the Nei and Gojobori method after alignment with ClustalW (48), limiting our analyses to ortholog alignments that included ≥100 bp of synonymous change sites. We also calculated CG [O/E] (33) from this set of orthologs.
Gene Ontology Analyses.
The plant-specific GO_slim library (goslim_plant.obo) was retrieved from the Gene Ontology (GO) web site (www.geneontology.org) in July 2012. The GO terms for all rice genes were retrieved from the RAP-DB database (http://rapdb.dna.affrc.go.jp). We slimmed these RAP-DB GO annotations using map2slim software, downloaded from http://search.cpan.org/~cmungall/go-perl/scripts/map2slim, and used the third level of slimmed GO categories.
Reanalysis of A. thaliana Mutation Accumulation Lines.
The data from ref. 16 contain BS-seq information from eight lines of A. thaliana. Using the methods described above, we filtered and mapped reads to the Col-0 reference and then calculated PCG values in each of the eight MA lines for genes with ncg ≥ 20 and for which ≥60% of cytosine residues were covered by at least two BS-seq reads. We discarded genes with PCHH or PCHG < 0.05 and classified genes as BM, IM, or UM. Sequence statistics (length, KA, and CG [O/E]) were calculated as above.
Supplementary Material
Acknowledgments
We thank R. Gaut for technical assistance and D. Garvin for tissue samples. J. Hollister, C. Muñoz-Díez, T. Sasaki, J. Fawcett, and H. Sakai provided helpful comments. K. Dawe and J. Gent provided unpublished maize BS-seq data. S.T. is a postdoctoral fellow for research abroad of the Japan Society for the Promotion of Science.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequence reported in this paper has been deposited in the Short Read Archive database (accession nos. SRX208151–SRX208156).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1215380110/-/DCSupplemental.
References
- 1.Bernatavichute YV, Zhang X, Cokus S, Pellegrini M, Jacobsen SE. Genome-wide association of histone H3 lysine nine methylation with CHG DNA methylation in Arabidopsis thaliana. PLoS ONE. 2008;3(9):e3156. doi: 10.1371/journal.pone.0003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chodavarapu RK, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466(7304):388–392. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11(3):204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dowen RH, et al. Widespread dynamic DNA methylation in response to biotic stress. Proc Natl Acad Sci USA. 2012;109(32):E2183–E2191. doi: 10.1073/pnas.1209329109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lippman Z, et al. Role of transposable elements in heterochromatin and epigenetic control. Nature. 2004;430(6998):471–476. doi: 10.1038/nature02651. [DOI] [PubMed] [Google Scholar]
- 6.Cokus SJ, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452(7184):215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lister R, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133(3):523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greaves IK, et al. Trans chromosomal methylation in Arabidopsis hybrids. Proc Natl Acad Sci USA. 2012;109(9):3570–3575. doi: 10.1073/pnas.1201043109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miura A, et al. An Arabidopsis jmjC domain protein protects transcribed genes from DNA methylation at CHG sites. EMBO J. 2009;28(8):1078–1086. doi: 10.1038/emboj.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inagaki S, et al. Autocatalytic differentiation of epigenetic modifications within the Arabidopsis genome. EMBO J. 2010;29(20):3496–3506. doi: 10.1038/emboj.2010.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coleman-Derr D, Zilberman D. Deposition of histone variant H2A.Z within gene bodies regulates responsive genes. PLoS Genet. 2012;8(10):e1002988. doi: 10.1371/journal.pgen.1002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328(5980):916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- 13.Feng S, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci USA. 2010;107(19):8689–8694. doi: 10.1073/pnas.1002720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaughn MW, et al. Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol. 2007;5(7):e174. doi: 10.1371/journal.pbio.0050174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X, Shiu S-H, Cal A, Borevitz JO. Global analysis of genetic, epigenetic and transcriptional polymorphisms in Arabidopsis thaliana using whole genome tiling arrays. PLoS Genet. 2008;4(3):e1000032. doi: 10.1371/journal.pgen.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitz RJ, et al. Transgenerational epigenetic instability is a source of novel methylation variants. Science. 2011;334(6054):369–373. doi: 10.1126/science.1212959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Becker C, et al. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature. 2011;480(7376):245–249. doi: 10.1038/nature10555. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell. 2006;126(6):1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39(1):61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 20.Roudier F, Teixeira FK, Colot V. Chromatin indexing in Arabidopsis: an epigenomic tale of tails and more. Trends Genet. 2009;25(11):511–517. doi: 10.1016/j.tig.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 21.Teixeira FK, Colot V. Gene body DNA methylation in plants: A means to an end or an end to a means? EMBO J. 2009;28(8):997–998. doi: 10.1038/emboj.2009.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol. 2004;11(11):1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- 23.Luco RF, et al. Regulation of alternative splicing by histone modifications. Science. 2010;327(5968):996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shukla S, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479(7371):74–79. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maunakea AK, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takuno S, Gaut BS. Body-methylated genes in Arabidopsis thaliana are functionally important and evolve slowly. Mol Biol Evol. 2012;29(1):219–227. doi: 10.1093/molbev/msr188. [DOI] [PubMed] [Google Scholar]
- 27.International Brachypodium Initiative Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463(7282):763–768. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- 28.Shen H, et al. Genome-wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell. 2012;24(3):875–892. doi: 10.1105/tpc.111.094870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng S, Jacobsen SE. Epigenetic modifications in plants: An evolutionary perspective. Curr Opin Plant Biol. 2011;14(2):179–186. doi: 10.1016/j.pbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vining KJ, et al. Dynamic DNA cytosine methylation in the Populus trichocarpa genome: Tissue-level variation and relationship to gene expression. BMC Genomics. 2012;13:27. doi: 10.1186/1471-2164-13-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schnable JC, Springer NM, Freeling M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc Natl Acad Sci USA. 2011;108(10):4069–4074. doi: 10.1073/pnas.1101368108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghent JI, et al. CHH islands: De novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013 doi: 10.1101/gr.146985.112. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bird AP. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980;8(7):1499–1504. doi: 10.1093/nar/8.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nanty L, et al. Comparative methylomics reveals gene-body H3K36me3 in Drosophila predicts DNA methylation and CpG landscapes in other invertebrates. Genome Res. 2011;21(11):1841–1850. doi: 10.1101/gr.121640.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serres-Giardi L, Belkhir K, David J, Glémin S. Patterns and evolution of nucleotide landscapes in seed plants. Plant Cell. 2012;24(4):1379–1397. doi: 10.1105/tpc.111.093674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carels N, Bernardi G. Two classes of genes in plants. Genetics. 2000;154(4):1819–1825. doi: 10.1093/genetics/154.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- 38.Tatarinova TV, Alexandrov NN, Bouck JB, Feldmann KA. GC3 biology in corn, rice, sorghum and other grasses. BMC Genomics. 2010;11:308. doi: 10.1186/1471-2164-11-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beilstein MA, Nagalingum NS, Clements MD, Manchester SR, Mathews S. Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc Natl Acad Sci USA. 2010;107(43):18724–18728. doi: 10.1073/pnas.0909766107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zemach A, et al. Local DNA hypomethylation activates genes in rice endosperm. Proc Natl Acad Sci USA. 2010;107(43):18729–18734. doi: 10.1073/pnas.1009695107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mirouze M, Paszkowski J. Epigenetic contribution to stress adaptation in plants. Curr Opin Plant Biol. 2011;14(3):267–274. doi: 10.1016/j.pbi.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 42.Gaut B, Yang L, Takuno S, Eguiarte LE. The patterns and causes of variation in plant nucleotide substitution rates. Annu Rev Ecol Evol Syst. 2011;42:245–266. [Google Scholar]
- 43.Harris EY, Ponts N, Levchuk A, Roch KL, Lonardi S. BRAT: Bisulfite-treated reads analysis tool. Bioinformatics. 2010;26(4):572–573. doi: 10.1093/bioinformatics/btp706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408(6814):796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka T, et al. Rice Annotation Project The Rice Annotation Project Database (RAP-DB): 2008 update. Nucleic Acids Res. 2008;36(Database issue):D1028–D1033. doi: 10.1093/nar/gkm978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu TT, et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet. 2011;43(5):476–481. doi: 10.1038/ng.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fawcett JA, Rouzé P, Van de Peer Y. Higher intron loss rate in Arabidopsis thaliana than A. lyrata is consistent with stronger selection for a smaller genome. Mol Biol Evol. 2012;29(2):849–859. doi: 10.1093/molbev/msr254. [DOI] [PubMed] [Google Scholar]
- 48.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.