

Abstract
Metastasis, the truly lethal aspect of cancer, occurs when metastatic cancer cells in a tumor break through the basement membrane and penetrate the extracellular matrix. We show that MDA-MB-231 metastatic breast cancer cells cooperatively invade a 3D collagen matrix while following a glucose gradient. The invasion front of the cells is a dynamic one, with different cells assuming the lead on a time scale of 70 h. The front cell leadership is dynamic presumably because of metabolic costs associated with a long-range strain field that precedes the invading cell front, which we have imaged using confocal imaging and marker beads imbedded in the collagen matrix. We suggest this could be a quantitative assay for an invasive phenotype tracking a glucose gradient and show that the invading cells act in a cooperative manner by exchanging leaders in the invading front.
Tumor metastasis is obviously of enormous clinical importance. A fundamental physiologic and clinical difference between benign and malignant tumor cells is that the former are usually neither invasive nor fatal. It is currently difficult to predict the probability of metastasis from the morphological or phenotypic properties of tumor cells observed within a primary cancer. Invasive tumor growth, at both primary and secondary sites, requires tumor cells to break through the stromal tissue barrier, evade the immune system, and coordinate signaling among tumor and mesenchymal cells to promote formation of tissue infrastructure, such as angiogenesis, to maintain tumor viability in its newly invaded space (1). We test two hypotheses: (i) The extreme thermodynamic costs of the oxidative Warburg cycle used by cancer cells forces them to follow glucose gradients; and (ii) minimization of the thermodynamic costs can be achieved by collective invasion strategies.
Invading cells are subject to significant potential fitness costs that may be minimized by collective behavior. Metastatic tumor cells that leave the primary tumor have to break through the stromal tissue barrier, evade the immune system, and coordinate with other local cells during angiogenesis to finally set up a viable remote tumor. It has been estimated that less than 1% of the primary tumor cells are able to finish the metastasis cycle (2), and yet they contribute to more than 90% of the cancer-related deaths (3). Although the molecular details are still unclear, it is significant that the malignant transition is accompanied by a change in the metabolism pathway from the mitochondrial oxidation of pyruvate in mitochondria (an aerobic process) to the far less energy-efficient and ancient anaerobic pathway of glycolysis followed by lactic acid fermentation in the cytosol (4), known as the Warburg cycle (5).
Most, if not all, metastatic cancer cells use the Warburg cycle, which is a far less efficient metabolism in terms of ATP production (6), consuming glucose (glycolysis) rather than using the usual oxidative Krebs phosphorylation cycle, although, thermodynamically, the process is efficient in the storage of chemical energy. Why would a metastatic cell activate an anaerobic metabolic pathway and bypass the mitochondria? One answer seems to be that the interior of a cancer tumor is very acidic and hypoxic (7–9). In this highly stressful anaerobic microenvironment, fitness is conferred by the Warburg cycle despite its low thermodynamic efficiency. However, with time, the increasing stress of the interior of a tumor will lead even cancer cells adapted to high-stress environments to seek new microenvironments, giving rise to the process of metastasis (10, 11). Fig. 1 presents a schematic of how we view the collective metastatic process of invasion (12, 13).
Fig. 1.

Schematic of the process of invasion of metastatic cells into an elastic medium in the presence of a glucose gradient. Metastatic breast cancer cells MDA-MB-231, running the Warburg cycle, invade through the collagen matrix in a collective manner to minimize metabolic expenditure.
Unfortunately, it has proved difficult to quantitatively characterize in vivo the metastatic process outlined in Fig. 1, because it requires tracking the movements of single cells in a 3D structure over times of the order of a week, so our actual understanding of the metastatic process is poor (14), despite the lethal nature of metastasis. We can hope that basic thermodynamic and physical laws governing the process can be discovered by carefully designed in vitro synthetic, 3D microenvironments in which the invasion of cancer cells can be quantitatively characterized, and the evolutionary advantages behind this behavior understood (15–17). Whereas there have been recent measurements of 3D invasion of metastatic cells using confocal imaging (18) in this work, we focus on the chemotactic driving force of this invasion.
Results
The primary phenotype of metastasis is the ability to invade the extracellular matrix (ECM) surrounding a tumor (19–21). It makes a certain amount of qualitative physical sense to assume that the more rigid the ECM, the greater the thermodynamic difficulty a cancer cell would have in penetrating it, and presumably metastatic cells would show greater ability in penetrating a rigid ECM than would a nonmetastatic but tumorigenic cancer cell (22). We choose to work with two different breast cancer cell lines: MDA-MB-231 cells that have the metastatic phenotype (23) and tumorigenic but nonmetastatic MCF-7 cells. The MDA-MB-231 cells were transfected with a gene for red fluorescent protein (RFP), whereas the MCF-7 cells were transfected with a gene for green fluorescent protein (GFP). This differential labeling allowed us to perform 3D imaging of the cells as they penetrated the collagen, and so we can characterize the spatiotemporal collective dynamics of the metastatic invasion.
It is, of course, more complex to work with 3D elastic media rather than 2D media to assay metastasis and track cells in 3D rather than 2D; however, we believe that quasi-2D continuum elastic sheets are not a good indicator of metastatic potential for a simple physical reason of stiffness or the restoring force for a given strain. The elastic restoring force F of an elastic medium of Young’s modulus Y with strain 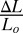 is:
is: 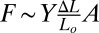 , where Lo is the unstrained length of the material, and A is the cross-sectional area of the material. In a thin slab, defined as a relatively small area A, the restoring force is relatively small and penetration facile. This can be shown by the basic ability of tumorigenic cells such as the MCF-7 cell line to penetrate a ECM matrix slab in a modified “wound-healing” experiment. Typically, in such an experiment, a wound is created by dragging a sharp tip through a confluent surface of cells (24). However, here we use a simple microfabrication technique to create an ECM barrier for cancer cells to penetrate. The healing mechanism in conventional wound healing research can be divided into two parts (25): (i) the contraction of a multicellular actomyosin belt generated along the wound, resulting in the closure of the wound and mediated at the molecular level by Rho1 and its regulators Rho kinase and myosin light chain kinase (26–29); and (ii) the lamellipodium-mediated migration regulated by Rac1 GTPase and others (30). In wound healing for normal cells, the border cells maintain their overall shape and cell structure (31).
, where Lo is the unstrained length of the material, and A is the cross-sectional area of the material. In a thin slab, defined as a relatively small area A, the restoring force is relatively small and penetration facile. This can be shown by the basic ability of tumorigenic cells such as the MCF-7 cell line to penetrate a ECM matrix slab in a modified “wound-healing” experiment. Typically, in such an experiment, a wound is created by dragging a sharp tip through a confluent surface of cells (24). However, here we use a simple microfabrication technique to create an ECM barrier for cancer cells to penetrate. The healing mechanism in conventional wound healing research can be divided into two parts (25): (i) the contraction of a multicellular actomyosin belt generated along the wound, resulting in the closure of the wound and mediated at the molecular level by Rho1 and its regulators Rho kinase and myosin light chain kinase (26–29); and (ii) the lamellipodium-mediated migration regulated by Rac1 GTPase and others (30). In wound healing for normal cells, the border cells maintain their overall shape and cell structure (31).
For the 2D experiments to test MCF-7 cells for intrinsic motility in collagen, we used a microfabricated soft elastic microstencil that is simply removed from a confluent surrounding lawn of cells (32). In particular, a polydimethylsiloxane (PDMS) stamp is attached on the surface at the beginning of the experiment, followed by the culturing of the MCF-7 cell line along the boundary of the stamp. When the desired 100% confluence is reached, the stamp is removed from the surface, generating a rectangular gap with a clean border. Immediately upon removal of the stamp, a solution of the desired concentration of collagen is poured over the stenciled area, where it sets in a matter of several minutes at 37 °C. The Young’s modulus of collagen is sensitive to the temperature at which the collagen is set, at a 37 °C setting temperature the Young’s modulus E should scale in pascals (i.e., newtons per square meter) as 3.5 c2.1, where c is the concentration of collagen in milligrams per milliliter (33).
After several hours, the cells begin to penetrate the stencil slab. For these experiments, we used MCF-7 tumorigenic cells. Fig. 2 shows the basic process of ECM penetration by the tumorigenic cell line MCF-7 as a function of the collagen concentration. Although the MCF-7 cells respond positively to collagen and are able to penetrate it, they can only penetrate at relatively low collagen concentrations. Furthermore, the MCF-7 cells do not present a uniform penetration front at 1.2 mg/mL but, rather, act like a random gas of cells moving into the collagen because of the lack of clearly defined invasion front. We have presented in 2D, mean propagation rates 〈vt〉 as a function of time and vs. the collagen concentration. Because the MCF-7 cells do not present a defined invasion front, we calculated the mean speed in MATLAB (MathWorks) to mask cell-free region first, and then tracked cells using Image J [National Institutes of Health (NIH)].
Fig. 2.

Invasion of MCF-7 cells vs. time in 2D. (Scale bar: 200 μm.) (A) MCF-7 for no collagen. (B) MCF-7 penetration for 1.2 mg/mL. (C) MCF-7 penetration for 2.3 mg/mL. (D) Average speed vs. time in hours for three different collagen concentrations.
The mean speed 〈vt〉 is a complex function of time after removal of the PDMS block and is highly sensitive to the concentration of the collagen. Somewhat nonintuitively, the MCF-7 cells do not invade a simple open space quickly. (The invasion speed for no collagen is one-eighth the maximum speed for collagen of 1.8 mg/mL, and it is not a simple function of time.) This kind of cell–cell–independent movement is to be expected from cancer cells because one of the basic phenotypes of solid tumor cancer cells is a loss of contact inhibition (34), but we believe it is compromised by the 2D nature of the experiment, because the dynamics that we show next are greatly different in 3D.
However, the critical point here, which is important in the discussion below, is that MCF-7 cancer cells, although they are not metastatic, are certainly highly mobile and capable of moving through collagen in a 2D slab topology.
It has been shown that there are some fundamental differences between the 2D collective cell migration on flat rigid substrate and the 3D migration in gels (35, 36), both in intrinsic rigidity changes attributable to the dimensions of a 2D slab vs. a 3D solid as discussed above and how the cells communicate with each other. Having shown that MCF-7 cancer cells, although highly motile, do not form a uniform invading front in a 2D system, we move to a more realistic 3D microstructure (37). We designed a mesoscopic ecology, as shown in Fig. 3, to mimic one aspect of the metabolic forces driving cells to metastasis, and we compared metastatic vs. nonmetastatic cells as they moved up this glucose gradient. Briefly, the cancer cells are seeded on the surface of a collagen gel through which the cells must move to obtain the glucose nutrients they need if they are locked on the Warburg cycle. When the cells penetrate the collagen to follow the glucose gradient, a physical strain field is generated that involves the consumption of energy. This device allows us to move to three dimensions and still maintain an initial front of cells and the presence of a glucose gradient driving the metastatic process.
Fig. 3.

Experimental setup used to study the collective invasion of cancer cells in a 3D microenvironment. The cells are cultured on the top of a collagen gel inside a 2-mm hole drilled in a 1-mm-thick glass slide. The gel contains 2-μm-diameter green fluorescent beads to observe the deformation of the gel. A stable glucose gradient is established through the gel with low-glucose medium on the top of the device and high-glucose medium on the bottom. The metastatic cells are invading from the top to the bottom of the gel. (A) Schematic 2D view of the device. (B) Three-dimensional view of the collagen gel with the green beads inside and the cancer cells on top.
To measure the strain forces acting on the collagen during the metastatic process, we distributed randomly 2-μm-diameter green fluorescent beads inside the matrix. The displacement of these beads as a function of the movement of the cells through the 3D space allows us to compute the elastic strain energy stored in the matrix and from that the effective “drag” that the penetrating cells feel (38–40). Several aspects of the metastatic phenotype can be probed in such a 3D structure: penetration of the collagen, movement along a glucose gradient, and possible cooperative behavior of the metastatic cells. Time-dependent imaging of the movement of the cells in this 3D structure was done by confocal imaging as discussed in Materials and Methods.
To test chemotaxis of metastatic cells toward glucose, we created a glucose gradient through the collagen by using glucose-free medium (RPMI 1640; Life Technologies) with added 10% (vol/vol) FBS serum (Invitrogen) at the top, where the cells were initially seeded, and glucose-rich medium (2 mg/mL glucose plus RPMI 1640 plus FBS) at the bottom. The external medium was changed every 3 d to maintain a stable glucose gradient throughout the experiment. The cells were continuously cultured for more than 4 wk at 37 °C and 5% CO2 and imaged on an inverted confocal microscope every 12 h. Because the cells are seeded on the top of the gel, the cells are presented with a free surface to invade. In that sense, this experiment is closely related to wound-healing assays (see above), where collective migration is triggered on a 2D flat substrate by only presenting some free surface to the epithelium (41).
The 2D experiments showed that a collagen concentration of 2.3 mg/mL (corresponding to an E = 15 Pa) is sufficient to stop the invasion of tumorigenic cells; however, this is a collagen elasticity that is much softer than normal tissue (42, 43), which has a E of ∼70 Pa. There is some debate about the value of the Young’s modulus of normal human breast tissue, indeed, if one can even characterize it accurately from macroscopic measurements as such because of nonlinearities (44) and differences between in vivo and ex vivo measurements (45). We choose for our 3D experiments a collagen concentration of 4.7 mg/mL, which is twice the concentration and 4× the elasticity that can stop MCF-7 cells in a 2D configuration.
Fig. 4 shows the invasion of metastatic RFP-tagged MDA-MB-231 cells into the 70-Pa collagen gel over a 5-d period. The images are constructed by a maximum projection of 20 equally spaced (15 μm apart) slices along the y direction. The front position at time 0, when the cells started the invasion process of the collagen, is shown in blue to serve as a reference surface. The cells grew on the surface of the collagen until confluence was reached (defined as time 0). Tumorigenic but nonmetastatic cells (the MCF-7 cell line) did not penetrate the collagen matrix after confluence was reached but, instead, remained on top (Fig. 4 B1 and B2) even in the presence of a glucose gradient as shown in Fig. 4 D1 and D2. The 3D images also show that, unlike the 2D experiments, the cells form a collective front upon invasion: they travel as a well-defined wave.
Fig. 4.

Invasion dynamics of human breast cancer cells into collagen gel. RFP-tagged MDA-MB-231 cells were seeded on the top surface of the gel. (A1–A4) The cross-section (xy plane) of the invasion front of every 48 h. The red channel is the fluorescent image of the cells, whereas the green channel is the image of the tracing beads. (B1–B4) The side view (xz plane) of the whole gel at the same time of A1–A4. (C1–C2) When the collagen gel was exposed to glucose rich medium on both sides, there was no invasion by the MDA-MB-231 cells. Here, time 0 is defined as the fifth day after seeding the cells. (D1–D2) Under the same condition, in the presence of glucose gradient, no invasion was observed for MCF-7 cells, cancerous nonmetastatic cells. Here, again, time 0 is defined as the fifth day.
Furthermore, the metastatic cells MDA-MB-231 will only invade the collagen if they are chasing a glucose gradient: if there is no gradient, they will not enter the collagen, as is shown in Fig. 4 C1 and C2. Thus, it is clear that only the metastatic cell line will penetrate a collagen matrix that is roughly the stiffness of normal tissue and only in the presence of a glucose gradient, thus showing the active glucose chemotaxis of metastatic cells (Fig. 4 B3 and B4). These experiments show that the glucose gradient is necessary: even in the presence of high uniform glucose concentrations, the metastatic cells will not penetrate the collagen.
To further study the dynamics of the invasion front, we chose to tag only a subset of the MDA-MB-231 cells (∼ 50%), so we could identify them in the cell-invasion front and track them individually. As we explain in Materials and Methods, only the leading front of the invading cells can be identified. Tracking of individual cells in the metastatic invasion revealed a curious phenomenon: unlike the fingering phenomenon seen in epithelial wound-healing experiments (46) (and not observed in the 2D experiment above using cancer cells), in the case of metastatic invasions, no fingering instabilities occur, and yet the front is still dynamic: the leading cells in the invasion front relinquish their leadership position over a 70-h period, roughly. The trajectory of a leading cell persisted for about 70 h (Fig. 5A), followed by a new leading cell at a different place in the invasion front, indicating a dynamic rearrangement in the cell colony: new cells continuously took over the leading positions, so the invasion front was refreshed every ∼70 h. Fig. 5 shows how this process occurred; the previous leader subsequently fell back into the middle of the pack.
Fig. 5.

MDA-MB-231 cells in a glucose gradient. (A) z position of leading cells changing as a function of time. (B) Trajectories in 3D view of the leading cells as a function of time; for this view, the cells are moving upwards. (C) Emergence location of the leaders in the xy plane. (D) Three-dimensional image of the invading cell front as a function of time. The leaders are marked using the same color code as in A. (Scale bar: 150 μm.)
There is a strain field preceding the cells as they move through the collagen, which we can measure via the displacement of the tracer beads in the collagen matrix. As we describe in Materials and Methods, the fluorescent beads that are randomly dispersed in the matrix can be tracked using confocal imaging and the relative movement of the beads (strain) tracked vs. time. Fig. 6 shows that the cell front produces a long-range strain field as it moves through the collagen, as measured by the displacement of beads within the matrix at sites remote from the cells. This strain field precedes the arrival of the current leader cell, with displacements on the order of 2 μm. It is also interesting to note that the strain field displaces the marker beads toward the invading front, not away from it, in effect pulling the matrix toward the front.
Fig. 6.

Displacement of fluorescent marker beads as the metastatic cells invade from the top down in our confocal image. Two particular beads are high-lighted with the yellow disk. Note the invasion of the cells at C; they appear as red blobs. In D, the front has reformed itself in terms of leaders. The net strain field displacement is shown by the white arrows; note that the cells pull the matrix toward the front. (E–H) Displacement of the highlighted marker beads in A–C versus time.
Discussion
The coupled strain field that we observed, as shown in Fig. 6, means that there is elastic strain energy Es stored in the ECM as the cells penetrate the matrix, giving rise to a stress (force) acting on the cells that the malignant cells must contend with (47). The amount of energy stored because of this strain varies because the square of the deformation for a linear medium, and the total stored energy, would need to be evaluated by integrating over all of space, which is a difficult task. In addition to this strain energy, there is also a surface energy term Eγ attributable to the interaction between the surface and the matrix. We do not know enough about these terms to be able to do a quantitive calculation, but we can make some semiquantitative observations. First, it is the leading cells that bear the brunt of energy costs of the matrix formation, because the strain field propagates ahead of the leading cell. Second, because it is long-ranged, this strain field will couple the cells together, and the following cells can ride the formation created by the leading cells. Furthermore, the more rigid the matrix, the greater the energy cost of the deformation; based on our 2D work, nonmetastatic cells cannot afford this kind of energy cost, and metastatic cells will only do so if they are following a gradient to greater metabolite resources, which, for them, is more glucose. It is clear from Fig. 6 that the strain field is very anisotropic and complex, and we do not attempt such a detailed calculation in this paper. However, we can attempt a rough order-of-magnitude estimate. The force F acting on each cell is roughly equal to AcEɛ, where Ac is the cross-sectional area of the cell, and 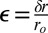 is the fractional strain. Typical values for ɛ taken from Fig. 6 are about 0.1, yielding for a cell of area Ac = 100 μm2 a force of about 100 pN.
is the fractional strain. Typical values for ɛ taken from Fig. 6 are about 0.1, yielding for a cell of area Ac = 100 μm2 a force of about 100 pN.
The dynamics of the leaders in the advancing front are coupled with the strain field because it represents energy expenditure by the cells in the invasion process. In the 2D wound-healing experiment, the cancer cells appeared to expand outward into the soft matrix, basically as a free gas of cells, which is consistent with the idea that cancer cells do not obey contact inhibition (34, 48, 49). However, for the 3D metastatic invading front, it would appear that there are correlations between the invading cells on the front because it remains a fairly tightly focused front and does not disperse. Our experiments show aspects of the metastatic invasion of collagen in three dimensions that may, in the future, be of some use in identifying the onset of metastasis in a cell population. The three primary phenotypes that we have identified in this paper are: (i) metastatic breast cancer cells will penetrate a 3D matrix if there is a glucose gradient present; (ii) there is a coupling strain field between the cells; and (iii) the metastatic cells moving through the matrix appear to exchange leading roles as they penetrate through the matrix.
Materials and Methods
Cell Preparation.
The RFP-tagged MDA-MA-231 cells and GFP-tagged MCF-7 cells were provided by Robert Gillies (H. Lee Moffitt Cancer Center, Tampa, FL). The cells were originally prepared through transfection using FuGene 6 transfection reagent (11815091001; Roche) and pcDNA3.1(+)/zeo containing GFP and RFP coding regions. The RFP was cut from the pDsRed2-N1 vector (632406; Clontech), and the GFP sequence was from pCMV-EGFP (6085; Clontech). They were first seeded in six-well plates at a cell density of 4 × 104 cells per well and grown until cells reached 80% confluence. For transfection, 2 μg of plasmid DNA was first mixed in 100 μL of OPTI-MEM I reduced-serum medium (31985070; Invitrogen). Eight microliters of FuGene 6 transfection reagent were then added to the diluted plasmid DNA and mixed by vortexing for 2 s. After that, the mixture was added to cells. The GFP- or RFP-expressing clones were selected by Zeocin (R350-05; Invitrogen). The expression of GFP or RFP was confirmed by using a Nikon TE-2000 fluorescence microscope before use.
Cell Culture.
The cells were cultured in RPMI 1640 (11875-093; Invitrogen) supplemented with 10% FBS (900108; Gemini) and 1% antibiotic solution [penicillin (10,000 units/mL) and streptomycin (10 mg/mL)]. Cells were seeded and maintained at 37 °C, 5% CO2, and 80% humidity throughout the experiments. Before loading into the microchips, MDA-MB-231 cells and MCF-7 cells were grown in flasks in a monolayer to ∼80% confluence and trypsinized with 0.25% trypsin-EDTA (Mediatech) solution for collection. Cell suspensions were then centrifuged at 400 × g, 4 °C for 5 min. Cells were pelleted, resuspended again in RPMI media with no glucose (RPMI 1640[−]) (11879-020; Invitrogen) and later loaded into the microdevice.
Preparation of the 3D Microdevice.
A 2-mm-diameter hole was drilled into a 1-mm-thick glass slide. Then a 2-mm-thick PDMS layer with a 4-mm hole was plasma treated and bonded to the glass slide to create a reservoir on the top. RPMI 1640[−] and collagen were cooled to 4 °C for 10 min. A total of 200 μL of RPMI 1640[−] was then taken to mix, respectively, with 3 μL of 1 M NaOH solution (Sigma), 1 μL of nonfunctionalized microspheres (2 μm in diameter; Bangs Labs), and 200 μL of collagen (9.8 mg/mL; 354249; BD). After that, it was vortexed at 1,500 rpm for 30 s. Finally, 20 μL of the solution was used to fill the hole from the chip top as soon as possible. The device was placed in the incubator at 37 °C for another 30 min, therefore creating an in vitro ECM with 4.7mg/mL collagen. When the device was ready, previously prepared cancer cells with RPMI 1640[−] were seeded on the top of the gel and fill the upper reservoir. Simultaneously, the bottom of the chip was filled with RPMI-1640 containing glucose to create a glucose gradient.
Time-Dependent Imaging.
The 3D imaging was performed on an inverted confocal microscope (SP5; Leica) with a 10× air objective lens. Typically, a volume of 1 mm × 1 mm × 1 mm was scanned with spatial resolution of 2 μm × 2 μm × 2 μm. We have chosen the tracing beads (Bangs Labs) to be spectrally distinct from the RFP-expressing cells; thus, the gel configuration can be captured simultaneously with the cancer cell morphology. Each volumetric scan took 6 min, after which the device was put into the incubator until the next scan took place. Time delay between two scans was kept at 12 h, an optimal condition we found to maximize the sampling rate without noticeable photodamaging to the cells. Within each scan, fluorescent intensity of the tracing beads was used as a calibration standard to compensate the signal loss attributable to diffraction at different focal depth. However, when imaging deep behind the invasion front, scattering by the cells significantly impaired the signal-to-noise ratio, making it inaccessible for the low NA objective lens. As a result, data are only limited close to and outside of the invasion front.
The gray-scale, multichannel raw images were further processed with MATLAB (MathWorks) and ImageJ (NIH) to render 3D views of the invading cells within the collagen gel. The tracing beads at the bottom of the device (high-glucose side) were used to colocalize all of the volumetric scans of the same device with submicron accuracy.
Acknowledgments
We thank Jerry Lee, Thea Tlsty, Salvatore Torquato, Yang Jiao, Clare Yu, and Elliot Botvinick for helpful discussions. This work was supported by the National Science Foundation and the National Cancer Institute.
Footnotes
The authors declare no conflict of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Hunter KW, Crawford NP, Alsarraj J. Mechanisms of metastasis. Breast Cancer Res. 2008;10(Suppl 1):S2. doi: 10.1186/bcr1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sleeman J, Steeg PS. Cancer metastasis as a therapeutic target. Eur J Cancer. 2010;46(7):1177–1180. doi: 10.1016/j.ejca.2010.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warburg O, Posener K, Negelein E. On the metabolism of carcinoma cells. Biochem Z. 1924;152:309–344. [Google Scholar]
- 6.Vazquez A, Liu JX, Zhou Y, Oltvai ZN. Catabolic efficiency of aerobic glycolysis: The Warburg effect revisited. BMC Syst Biol. 2010;4:58. doi: 10.1186/1752-0509-4-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 8.Khain E, et al. Collective behavior of brain tumor cells: The role of hypoxia. Phys Rev E Stat Nonlin Soft Matter Phys. 2011;83(3 Pt 1):031920. doi: 10.1103/PhysRevE.83.031920. [DOI] [PubMed] [Google Scholar]
- 9.Pennacchietti S, et al. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3(4):347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 10.Scheel C, Onder T, Karnoub A, Weinberg RA. Adaptation versus selection: The origins of metastatic behavior. Cancer Res. 2007;67(24):11476–11479. doi: 10.1158/0008-5472.CAN-07-1653. [DOI] [PubMed] [Google Scholar]
- 11.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8(1):56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 12.Rørth P. Collective cell migration. Annu Rev Cell Dev Biol. 2009;25:407–429. doi: 10.1146/annurev.cellbio.042308.113231. [DOI] [PubMed] [Google Scholar]
- 13.Bidard FC, Pierga JY, Vincent-Salomon A, Poupon MF. A “class action” against the microenvironment: Do cancer cells cooperate in metastasis? Cancer Metastasis Rev. 2008;27(1):5–10. doi: 10.1007/s10555-007-9103-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langley RR, Fidler IJ. The seed and soil hypothesis revisited—the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer. 2011;128(11):2527–2535. doi: 10.1002/ijc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science. 1977;197(4306):893–895. doi: 10.1126/science.887927. [DOI] [PubMed] [Google Scholar]
- 16.Bernards R, Weinberg RA. A progression puzzle. Nature. 2002;418(6900):823–823. doi: 10.1038/418823a. [DOI] [PubMed] [Google Scholar]
- 17.Anderson ARA, Weaver AM, Cummings PT, Quaranta V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell. 2006;127(5):905–915. doi: 10.1016/j.cell.2006.09.042. [DOI] [PubMed] [Google Scholar]
- 18.Sakai K, et al. Invasion of carcinoma cells into reconstituted type I collagen gels: Visual real-time analysis by time-lapse microscopy. Biosci Trends. 2011;5(1):10–16. doi: 10.5582/bst.2011.v5.1.10. [DOI] [PubMed] [Google Scholar]
- 19.Friedl P, Wolf K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat Rev Cancer. 2003;3(5):362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 20.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 21.Friedl P, et al. Migration of coordinated cell clusters in mesenchymal and epithelial cancer explants in vitro. Cancer Res. 1995;55(20):4557–4560. [PubMed] [Google Scholar]
- 22.Peyton SR, Putnam AJ. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J Cell Physiol. 2005;204(1):198–209. doi: 10.1002/jcp.20274. [DOI] [PubMed] [Google Scholar]
- 23.Abdelkarim M, et al. Invading basement membrane matrix is sufficient for MDA-MB-231 breast cancer cells to develop a stable in vivo metastatic phenotype. PLoS ONE. 2011;6(8):e23334. doi: 10.1371/journal.pone.0023334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fenteany G, Janmey PA, Stossel TP. Signaling pathways and cell mechanics involved in wound closure by epithelial cell sheets. Curr Biol. 2000;10(14):831–838. doi: 10.1016/s0960-9822(00)00579-0. [DOI] [PubMed] [Google Scholar]
- 25.Jacinto A, Martinez-Arias A, Martin P. Mechanisms of epithelial fusion and repair. Nat Cell Biol. 2001;3(5):E117–E123. doi: 10.1038/35074643. [DOI] [PubMed] [Google Scholar]
- 26.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 27.Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582(14):2093–2101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 28.Omelchenko T, Vasiliev JM, Gelfand IM, Feder HH, Bonder EM. Rho-dependent formation of epithelial “leader” cells during wound healing. Proc Natl Acad Sci USA. 2003;100(19):10788–10793. doi: 10.1073/pnas.1834401100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Contact guidance mediated three-dimensional cell migration is regulated by Rho/ROCK-dependent matrix reorganization. Biophys J. 2008;95(11):5374–5384. doi: 10.1529/biophysj.108.133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81(1):53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 31.Lauffenburger DA, Horwitz AF. Cell migration: A physically integrated molecular process. Cell. 1996;84(3):359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 32.Petitjean L, et al. Velocity fields in a collectively migrating epithelium. Biophys J. 2010;98(9):1790–1800. doi: 10.1016/j.bpj.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang YL, Leone LM, Kaufman LJ. Elastic moduli of collagen gels can be predicted from two-dimensional confocal microscopy. Biophys J. 2009;97(7):2051–2060. doi: 10.1016/j.bpj.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu LY, et al. Probing the invasiveness of prostate cancer cells in a 3D microfabricated landscape. Proc Natl Acad Sci USA. 2011;108(17):6853–6856. doi: 10.1073/pnas.1102808108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lämmermann T, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 2008;453(7191):51–55. doi: 10.1038/nature06887. [DOI] [PubMed] [Google Scholar]
- 36.Kenny PA, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1(1):84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol. 2006;7(3):211–224. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 38.Trepat X, et al. Physical forces during collective cell migration. Nat Phys. 2009;5:426–430. [Google Scholar]
- 39.Gov NS. Traction forces during collective cell motion. HFSP J. 2009;3(4):223–227. doi: 10.2976/1.3185785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butler JP, Tolić-Nørrelykke IM, Fabry B, Fredberg JJ. Traction fields, moments, and strain energy that cells exert on their surroundings. Am J Physiol Cell Physiol. 2002;282(3):C595–C605. doi: 10.1152/ajpcell.00270.2001. [DOI] [PubMed] [Google Scholar]
- 41.Poujade M, et al. Collective migration of an epithelial monolayer in response to a model wound. Proc Natl Acad Sci USA. 2007;104(41):15988–15993. doi: 10.1073/pnas.0705062104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levental KR, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ng MR, Brugge JS. A stiff blow from the stroma: Collagen crosslinking drives tumor progression. Cancer Cell. 2009;16(6):455–457. doi: 10.1016/j.ccr.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 44.Wang ZG, Liu Y, Wang G, Sun LZ. Nonlinear elasto-mammography for characterization of breast tissue properties. Int J Biomed Imaging. 2011;2011:540820. doi: 10.1155/2011/540820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samani A, Bishop J, Luginbuhl C, Plewes DB. Measuring the elastic modulus of ex vivo small tissue samples. Phys Med Biol. 2003;48(14):2183–2198. doi: 10.1088/0031-9155/48/14/310. [DOI] [PubMed] [Google Scholar]
- 46.Gov NS. Collective cell migration patterns: Follow the leader. Proc Natl Acad Sci USA. 2007;104(41):15970–15971. doi: 10.1073/pnas.0708037104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paszek MJ, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8(3):241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 48.Carmona-Fontaine C, et al. Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature. 2008;456(7224):957–961. doi: 10.1038/nature07441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abercrombie M. Contact inhibition and malignancy. Nature. 1979;281(5729):259–262. doi: 10.1038/281259a0. [DOI] [PubMed] [Google Scholar]