

Abstract
Most P-type ATPases pump specific cations or heavy metals across a membrane to form ion gradients. However, the type IV P-type ATPases evolved the ability to transport specific phospholipid substrates rather than cations and function to establish plasma membrane asymmetry in eukaryotic cells. The mechanism for how a P-type ATPase, or any other transporter, can recognize and flip a phospholipid substrate is unclear. Here, through a combination of genetic screening and directed mutagenesis with the type IV P-type ATPases Dnf1 and Drs2 from budding yeast, we identify more than a dozen residues that determine headgroup specificity for phospholipid transport. These residues cluster at two interfacial regions flanking transmembrane segments 1–4 and lie outside of the canonical substrate binding site operating in cation pumps. Our data imply the presence of two substrate-selecting gates acting sequentially on opposite sides of the membrane: an entry gate, where phospholipid is initially selected from the extracellular leaflet, and an exit gate at the cytosolic leaflet. The entry and exit gates act cooperatively but imperfectly, with neither being able to restrict phosphatidylserine selection completely when the opposing gate is tuned to permit it. This work describes a unique transport mechanism for a P-type ATPase and provides insight into how integral membrane proteins can recognize and transport phospholipid substrate across a lipid bilayer.
Keywords: flippase, P4-ATPase, ATP8A1, ATP8A2
All living cells are encapsulated by a plasma membrane that separates the inside of the cell from the external environment. In eukaryotic cells, the plasma membrane is a conglomerate of protein and lipid, with its lipid component composed of glycerophospholipids, sphingolipids, and sterols. The amphipathic nature of these lipids causes spontaneous formation of a bilayer structure with a hydrophobic core that prevents passage of charged or bulky polar molecules, ions, and many toxins into and out of the cell. This barrier function of the bilayer allows the formation of concentration gradients across the membrane, including the component phospholipid molecules themselves. Phosphatidylserine (PS), phosphatidylethanolamine (PE), and phosphatidylinositol are enriched in the cytosolic face of the plasma membrane, whereas the glycosphingolipids are exposed on the exofacial side. The polar headgroups of the phospholipids prevent spontaneous flip-flop to maintain this asymmetrical structure. However, establishing membrane asymmetry requires an energetic input with ATP-dependent transporters to facilitate movement of phospholipids up the concentration gradient. The uneven distribution of phospholipids between the two membrane faces generates unique properties on each face, and controlled disruption is critical in physiological processes, such as blood coagulation, apoptosis, cytokinesis, and host–viral interactions (1).
Establishing membrane asymmetry requires vectorial transport of the component lipids; the proteins capable of catalyzing this transport are ATP-binding cassette (ABC) transporters and type IV P-type ATPases (P4-ATPases). ABC transporters “flop” lipids from the cytosolic to exofacial leaflet opposing the P4-ATPases, which “flip” phospholipids from the exofacial to cytosolic side of the membrane. The glycerophospholipid specificity of the P4-ATPases restricts PE and PS to the cytoplasmic leaflet, establishing and maintaining the characteristic plasma membrane asymmetry. The first ABC transporter identified was a P-glycoprotein capable of transporting a remarkable range of substrates out of the cell (2). However, most ABC transporters have a much narrower range of substrate specificities, with some capable of lipid transport. ABCA1, ABCA2, ABCB4, and ABCG1 are all capable of transporting both phosphatidylcholine (PC) and cholesterol, ABCG5/G8 is specific for sterols, and MsbA transports the bulky lipid A components across the Escherichia coli membrane (3). X-ray crystal structures have been solved for several members of the ABC transporter family, highlighting some of the extreme conformational changes within their catalytic cycle (4, 5). Even with the extensive biochemical and structural understanding of many ABC transporters, a lack of understanding of how these proteins recognize and transport lipid substrates remains an overriding theme.
P-type ATPases also have an amazing, albeit more compact, range of substrate specificities. Many transport only a specific set of ions or heavy metals (P1, P2, and P3), whereas others transport a subset of phospholipids (P4). These proteins have a transmembrane (TM) domain typically composed of 10 TM segments, along with cytosolic actuator, phosphorylation, and nucleotide-binding domains. X-ray crystal structures with bound substrate have been solved for ion-transporting P-type ATPases (P1–P3), and they use a highly conserved ion binding pocket within the center of the TM domain (6–9). Accessibility of this canonical ion binding site is classically described within the E1 and E2 states of the Post–Albers cycle (10). Access to this binding site alternates between the cytosolic side (E1), temporary occlusion (E1P/E2P), and the exofacial side of the membrane (E2) (11). Ion-transporting P-type ATPases use a collection of charged or polar residues in conserved positions within TM4, TM5, TM6, TM8, and TM9 to coordinate the ions (12). In the P4-ATPases, the corresponding residues are bulky or hydrophobic amino acids that render this site useless for coordinating ions. The canonical binding site is also spatially restrictive, accommodating only ions of the appropriate charge and size. It is difficult to imagine how this canonical binding site may have evolved the capacity to bind a bulky amphipathic phospholipid, giving rise to the P4-ATPase “giant substrate problem” of how phospholipid is transported (13, 14).
Two opposing models have been proposed for how P4-ATPases could transport substrate, with the first model using the canonical substrate binding site and the second model using a noncanonical pathway formed at the protein/lipid interface. Support for the first model was based on an observation that mutation of a conserved lysine residue in the P4-ATPase Atp8a2, which is predicted to be near the canonical binding site within TM5, reduced the apparent affinity of this pump for substrate (15). In support of the noncanonical transport pathway, we mapped residues involved in substrate selection by the yeast P4-ATPases Dnf1, a PC and PE flippase, and Drs2, a PS flippase, to TM3–4 (13). Within TM4, we identified a single amino acid involved in substrate selection that is four residues from a proline conserved in all P-type ATPases. The TM4 “proline + 4” position controls PS selection by these P4-ATPases; the presence of a Tyr restricts PS flip, whereas a Phe allows PS flip. We also identified a second residue within the exofacial loop between TM3 and TM4 that specifically affected the recognition of PC by Dnf1. The orientation of Tyr618 in the Dnf1 homology model and the analogous residues in each available P-type ATPase X-ray crystal structure suggest this residue faces away from the canonical ion binding pocket and permits a distinctive transport pathway (6–9). Among the integral membrane proteins capable of transporting phospholipid, P4-ATPases are the only set of proteins with any residues involved in substrate selection identified. However, with only two to three residues implicated in substrate recognition, distinguishing between the noncanonical and canonical transport pathway models is premature (16).
Here, we present an extensive screen for residues in TM1–6 that define phospholipid specificity in Dnf1 and Drs2. We have identified mutations in TM1–4 that cause a gain of PS flip by Dnf1 or a loss of PS flip by Drs2. Other mutations reduce PC recognition by Dnf1 without perturbing PE flip. These residues form two clusters (or gates) in TM1–4, one on the exofacial membrane face, where substrate is initially selected, and the second near the cytosolic membrane face, where substrate is released. The positioning of residues involved in phospholipid selection within TM1–2 and TM3–4 are incompatible with the canonical substrate binding site. These results support a noncanonical phospholipid transport pathway along a groove between TM1, TM3, and TM4 and imply a two-gate mechanism for the phospholipid selection by P4-ATPases.
Results
TM1–2 Residues Defining Substrate Specificity.
We sought to address the mechanism through which P4-ATPases select and transport phospholipid by identifying additional residues in TM1–6 responsible for determining phospholipid specificity. We previously generated a series of chimeras by exchanging segments of Drs2, a PS flippase, into Dnf1, which preferentially flips PC and PE (mutations exchanging residues between Dnf1 and Drs2 are indicated by brackets; for example, Dnf1[Drs2]). To assay flippase activity, the Dnf1[Drs2] chimeras were expressed in a dnf1,2Δ strain, and relative uptake of 7-nitro-2-1,3-benzoxadiazol-4-yl phospholipid (NBD-PL) across the plasma membrane was recorded by flow cytometry. A swap of TM1–2 from Drs2 to Dnf1 resulted in a Dnf1 chimera (Dnf1[TM1–2]) with low NBD-PC and NBD-PE uptake activity, as well as a minor increase in NBD-PS activity relative to WT Dnf1 (13) (Fig. 1A). This Dnf1[TM1,2] chimera localizes normally to the plasma membrane; thus, the low overall activity is not caused by misfolding and retention in the endoplasmic reticulum (ER) (13). However, the low activity made interpretation of the substrate specificity difficult; therefore, smaller substitutions were generated. Exchanging TM1 resulted in a chimera (Dnf1[TM1]) that had intermediate NBD-PC and NBD-PE activity compared with WT Dnf1 and Dnf1[TM1–2] but displayed an increase in PS uptake activity (Fig. 1A). The activity of the Dnf1[TM2] was also very low compared with that of WT Dnf1, but it had no PS uptake (Fig. 1A). These data implied TM1–2 harbored residues important for substrate selection.
Fig. 1.
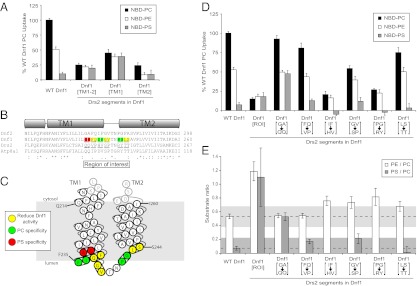
TM1–2 contributes to phospholipid selection. (A) NBD-PL uptake by WT Dnf1 and Dnf1[Drs2] chimeras. TM1 and TM2 contain residues involved in phospholipid selection. (B) Primary sequence alignment of TM1–2 from Dnf1, Dnf2, Drs2, and Atp8a1. Predicted TM helices are shown above the sequence alignment. Underlined residues are pairs exchanged from Drs2 into Dnf1. The colors indicate positions where mutations caused a change in specificity (red for changes altering PS recognition and green for changes altering PC recognition) or a ≥20% reduction in activity (yellow) based on D and E. (C) Topology diagram of Dnf1 TM1–2 indicates residues implicated in substrate preference based on E. (D) NBD-PL uptake by Dnf1[Drs2] ROI chimeras identifies Drs2 residues involved in PS selection (QQ) and Dnf1 residues involved in PC selection (IF and PG). (E) PE/PC and PS/PC uptake ratios provide a measure of substrate preference of Dnf1 and chimeras, independent of the overall activity or the number of transporters at the plasma membrane. The gray zones indicate a confidence interval, and values outside this interval define a change of specificity. For all experiments, values are the mean (±SEM). Ratios less than 0 are not shown.
An alignment of TM1–2 sequences from P4-ATPases preferring PS (Drs2 and Atp8a1) (17–19) and those preferring PC (Dnf1 and Dnf2) (20) identified a region of interest (ROI) near the exofacial leaflet where conservation strongly correlated with substrate preference (Fig. 1 B and C). Replacing the ROI in Dnf1 with the corresponding sequence in Drs2 yielded a chimera (Dnf1[ROI], Fig. 1D) with low activity and substrate specificity similar to Dnf1[TM1–2] and Dnf1[TM1] (Fig. 1A). Exchanging pairs of residues in the ROI illuminated key residues for substrate selection (Fig. 1D). Substitution of G230Q and A231Q allowed Dnf1[GA→QQ] to translocate PS as efficiently as PE, without disturbing PC or PE uptake. The IF→HV and PG→RY substitutions reduced uptake of PC and PE and altered the specificity because these substrates were transported almost equally well. The FQ→VP and LS→TT substitutions modestly reduced activity without significantly altering substrate specificity. A measure of substrate specificity independent of total activity or transporter numbers at the plasma membrane (13) was determined by graphing the ratio of PE to PC uptake and the ratio of PS to PC uptake for each chimera. We defined a change in substrate specificity as a change in the PE/PC ratio or a PS/PC ratio of 15% or greater without error bars (±SEM) overlapping with this zone of confidence (gray areas, Fig. 1E). Each value falling outside of the gray zone was determined to be statistically different from WT Dnf1 by the Student’s t test (P < 0.05). Residues color-coded red and green in Fig. 1 B and C indicate the substitutions at these positions cause a significant change in specificity, and residues are color-coded yellow if activity is significantly reduced without a change in specificity. These data indicate that residues at the base of TM1 and TM2, far removed from the canonical binding site, are critical determinants of substrate specificity.
TM3–4 Residues Defining Substrate Specificity.
Dnf1 recognizes NBD-PC and NBD-PE with short acyl chains (C6) and an NBD group in the sn2 position (20), lyso-PC, lacking the sn2 acyl chain (21), as well as the toxic lyso-PC analog edelfosine (22). Toxicity of edelfosine to yeast requires uptake by Dnf1 or Dnf2. To identify additional residues in Dnf1 involved in recognizing the phosphocholine headgroup, we randomly mutagenized Dnf1 and selected for functional variants with reduced ability to take up edelfosine. Initially, mutagenesis was targeted to the TM3–4 region because prior chimera studies indicated residues important for PC selection were present in this region (13). The mutagenized dnf1 alleles were transformed into a dnf1,2,3Δdrs2Δ pRS416-DRS2 strain and screened for mutants resistant to edelfosine (Fig. S1 A and B). Next, functional alleles were selected by counterscreening on 5-fluoroorotic acid (5-FOA; which selects for loss of URA3 encoded on pRS416-DRS2). Of the mutants resistant to both edelfosine and 5-FOA, we selected one-third of the clones with the strongest resistance to edelfosine for sequencing and recovered 25 point mutations within TM3–4 (Fig. S1C, a summary is provided in Fig. S1D).
The dnf1 alleles with point mutations were transferred into a dnf1,2Δ strain, and the cells were assayed for NBD-PL uptake activity. For the point mutations present in TM3, most caused a general reduction in activity (F551L, I555L, V558E, and F565L; Fig. 2C) without altering substrate specificity (Fig. 2D). Importantly, these residues should line one face of the TM3 helix. Dnf1 I545T caused a greater reduction in PC uptake activity than in PE activity (Fig. 2C), which was apparent in the PE/PC uptake ratio (Fig. 2D). Therefore, this position has a strong effect on PC selection by Dnf1. The second position of interest is Asn550, which was recovered from the screen as alleles encoding four different amino acid substitutions. Unexpectedly, in each case, the substitutions for Asn550 allowed a substantial increase in NBD-PS transport by Dnf1 with minor changes in NBD-PC or NBD-PE transport (Fig. 2C). The Asn550 residue is predicted to be in close proximity to Tyr618 near the cytoplasmic face of the membrane, and mutation of Tyr618 also increases PS activity of Dnf1 (13). However, in the case of Tyr618, acquisition of NBD-PS transport seems to require a specific substitution to Phe (Y618F), whereas multiple substitutions at Asn550 (Ile, Lys, Ser, or Tyr) allow NBD-PS transport.
Fig. 2.

TM3 mutations conferring edelfosine resistance alter Dnf1 substrate specificity. (A) Primary sequence alignment of TM1–2 from Dnf1 and Drs2. TM sequences are indicated below the sequence alignment. (B) Topology diagram of Dnf1 TM3 indicates residues implicated in substrate preference based on D. (C) NBD-PL uptake by edelfosine-resistant Dnf1 mutants. Substitutions at I545 decrease PC recognition by Dnf1, and substitutions at Asn550 increase PS uptake by Dnf1. (D) PC/PE and PS/PC uptake ratios describe two residues involved in substrate specificity (Ile545 and Asn550). For all experiments, values are the mean (±SEM). Ratios less than 0 are not shown.
For the point mutations recovered in the luminal loop between TM3 and TM4 (LL3–4), we identified two residues important for activity (Y575 and Phe583) and at least two residues having an impact on specificity. In addition to Phe587 we previously reported, we repeatedly recovered mutations in a second position (Ile590), some of which altered the specificity of Dnf1 (Fig. 3 C and D). K578E was the only mutation in this region that enhanced NBD-PS transport, although the change in specificity indicated by the PS/PC ratio did not extend above our zone of confidence. Within TM4, we have previously reported the proline + 4 position as critical in phospholipid selection by Dnf1 (Tyr618) and Drs2 (Phe511). In this screen, we recovered a single Dnf1 mutation in this position (Y618C), which caused an overall reduction in NBD-PL uptake activity but no significant change in specificity. Two mutations in TM4 caused a change in phospholipid specificity. Dnf1 V621A acquired minor NBD-PS activity, and Dnf1 E622V showed substantial increases in both the PE/PC and PS/PC ratios as a result of a reduced PC activity and enhanced PS activity (Fig. 3 C and D). Val621A and Glu622 are approximately a full turn of the helix above Tyr618, which, along with N550 and/or I545 in TM3, may represent a cluster of vicinal residues controlling exit of substrate to the cytosolic leaflet.
Fig. 3.
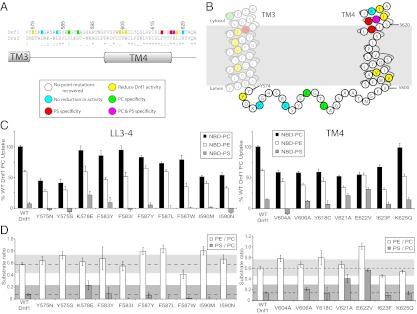
LL3–4 and TM4 mutations conferring edelfosine resistance alter Dnf1 substrate specificity. (A) Primary sequence alignment of LL3–4, TM4 from Dnf1 and Drs2 with the position of TM helices. (B) Topology diagram of Dnf1 TM3–4 indicates residues implicated in substrate preference based on D. (C) NBD-PL uptake by edelfosine-resistant Dnf1 mutants with mutations in LL3–4 (Left) and TM4 (Right). (D) PC/PE and PS/PC uptake ratios implicate four residues involved in substrate specificity (Phe587, Ile590, Val621, and Glu622). For all experiments, values are the mean (±SEM). Ratios less than 0 are not shown.
To assess the saturation of the directed evolution screen on TM3–4 and to identify any periodicity for residues having an impact on function, we also performed an alanine scan across a portion of TM3 and LL3–4 (Fig. S2A). Within the scanned region (24 residues), three mutations (N571A, Y575A, and K578A) prevented complementation of a dnf1,2,3Δdrs2Δ strain and two weakly complemented (Y574A and S581A). These inactivating mutations were found at a periodicity that might suggest a critical α-helix face at the beginning of LL3–4. Several of the alanine mutations conferred variable degrees of resistance to edelfosine (Fig. S2B). As expected, each allele that did not complement also conferred edelfosine resistance, consistent with a loss of function. Several other alanine scan mutants showed weak resistance to edelfosine and would not have been selected through the screen. However, one mutation (G588A) caused strong resistance to both edelfosine and 5-FOA. The G588A and several other mutants with weaker resistance to edelfosine were assayed for NBD-PL uptake activity (Fig. S2D). Most displayed a general reduction in activity (G568A, N571A, Y574A, K578A, and F587A), but Y575A, E586A, and G588A actually altered the phospholipid specificity of Dnf1 by reducing PC uptake to a greater extent than PE uptake (Fig. S2 D and E). These LL3–4 residues, along with the substrate-defining exofacial residues in TM1–2, represent a selective gate for substrate entry.
TM5–6 Residues Modulate Dnf1 Activity.
TM5 and TM6 form a portion of the canonical transport pathway used by the ion-specific P-type ATPases. However, Dnf1[TM5–6] chimeras previously characterized were retained within the ER and likely misfolded (13). To address the potential role(s) of TM5–6 in transport specificity further, we applied the same directed evolution edelfosine resistance screen with mutagenesis targeted to TM5–6 (Fig. 4 A and B). Most clones with mutations in TM5–6 reduced activity but did not alter the specificity (n > 20) of Dnf1 whether alleles with point mutations (Fig. 4 C–H) or multiple mutations (Fig. S3 C and D) were assayed. However, one TM5–6 mutant, I1235F, displayed an increase in overall activity and an increase in the PS/PC ratio indicating change of specificity (Fig. 4 E and F).
Fig. 4.

Substrate preference of TM5–6 edelfosine-resistant Dnf1 mutants. (A) Primary sequence alignment of TM5–6 from Dnf1 and Drs2 with the position of TM helices. (B) Topology diagram of Dnf1 TM1–2 indicates residues implicated in substrate preference based on D and F. (C) NBD-PL uptake by Dnf1 TM5 point mutants recovered from edelfosine resistance screen targeted to TM5–6. These mutations reduced activity but did not change specificity. (D) PE/PC and PS/PC uptake ratios provide the substrate preference based on C. (E) NBD-PL uptake by Dnf1 TM6 point mutants recovered from the edelfosine resistance screen targeted to TM5–6. (F) PE/PC and PS/PC uptake ratios depict the substrate preference based on E. A single mutation (I1235F) affected the specificity of Dnf1 by enhancing PS uptake. (G) Dnf1[GA→>QQ]; Dnf1 N550K; Dnf1 N550S; and Dnf1[GA→>QQ], N550S, [Y618F] enhance PS uptake while maintaining specificity for the phospholipid headgroup and glycerol backbone. However, the Dnf1 I1235F mutation perturbs recognition of the glycerol backbone, as indicated by increased uptake of the nonsubstrate SM. For all experiments, values are the mean (±SEM). Ratios less than 0 are not shown.
We assayed Dnf1[GA→QQ], several Dnf1 Asn550* alleles, and Dnf1 I1235F to determine if the gain-of-function activity was specific for PS or also included the nonsubstrate NBD lipids phosphatidic acid (PA) and sphingomyelin (SM). For Dnf1[GA→QQ], Dnf1 N550K, and Dnf1 N550S, we observed no significant uptake activity for NBD-PA or NBD-SM, demonstrating the specific acquisition of NBD-PS as a substrate (Fig. 5A). Dnf1 I1235F had no activity for NBD-PA but did display a significant increase in NBD-SM uptake activity relative to WT Dnf1 (Fig. 4G). We also combined the three TM1–4 PS gain-of-function mutations into a single allele (Dnf1[GA→QQ], N550S, [Y618F]) and observed enhanced overall activity, a substantial increase in NBD-PS activity, and minor increases in NBD-PA or NBD-SM activity. Thus, Dnf1 I1235F showed a partial loss of specificity for glycerophospholipid that was not simply a consequence of increased overall activity.
Fig. 5.

Specificity determinants on both sides of the membrane cooperate to transport phospholipid. (A) Combination of the [GA→QQ] substitution (exofacial) with either N550S or [Y618F] (cytosolic) results in additive increases in PS uptake activity relative to either substitution alone. Combining the two cytosolic residues (exit gate) N550S and [Y618F] results in a minor increase in PS uptake, suggesting they alter the same site. For all experiments, values are the mean (±SEM). (B) Expression levels of Flag-WT Dnf1, Dnf1[GA→QQ], Dnf1 N550S, and Dnf1[Y618F] are similar. The closed arrow indicates Flag-Dnf1 protein, and the open arrow indicates a background band in Saccharomyces cerevisiae whole-cell extracts. (C) GFP-tagged WT Dnf1, Dnf1[GA→QQ], Dnf1 N550S, and Dnf1[Y618F] display similar localization patterns, polarizing to the budding daughter cell. (Scale bar: 5 μm.)
Previously with Atp8a2, a TM5 lysine (Lys873) was targeted and mutated, altering the apparent affinity for substrate. This lysine was suggested to be critical for substrate interaction and was proposed to be in the canonical binding pocket (15). This lysine residue is highly conserved among P4-ATPases; thus, we generated substitutions at this position in the context of Dnf1 (Lys1194). In each case, we observed a reduction in activity with no change in specificity (Fig. S3).
Cytosolic and Exofacial Gates Cooperate to Restrict PS Flip by Dnf1.
We identified clusters of residues on both the exofacial side (entry gate) and cytosolic side (exit gate) of the membrane that contribute to phospholipid selection. Therefore, we sought to define the relationship between these gates to understand better the mechanism for phospholipid transport. Dnf1 variants harboring combinations of GA→QQ, N550S, and Y618F mutations were assayed for NBD-PL uptake activity to determine if tuning each position to allow NBD-PS flip would increase PS uptake. As shown in Figs. 4G and 5A, the triple mutant displays greatly enhanced PS uptake (25-fold greater than WT Dnf1 and at least twofold more than each single mutant). The greatest increase in PS uptake occurred when we combined the exofacial substitution (GA→QQ) with a cytosolic substitution (N550S or Y618F). Interestingly, when we combined the N550S with the Y618F mutation, there was no significant increase in the PS uptake relative to Y618F, indicating that the mutations in the cytosolic gate were not additive. These data imply that the exofacial entry gate and cytosolic exit gate act sequentially, but each imperfectly, to select substrate for transport.
Next, we tested the expression of each of the strong NBD-PS–selective Dnf1 variants. In each case, the expression level was similar to WT Dnf1 and unlikely to account for the differences in substrate specificity or activity (Fig. 5B). The localization of GFP-labeled versions of Dnf1, Dnf1[GA→QQ], Dnf1 N550S, and Dnf1[Y618F] were similar and, again, unlikely to account for the difference in substrate specificity at the plasma membrane (Fig. 5C). Examination of NBD-PC and NBD-PS by TLC after incubation with cells confirmed no significant degradation (Fig. S4), as previously reported (23). In addition, these NBD-PS–selective variants of Dnf1 maintained the Lem3 β-subunit requirement (Fig. S5).
Drs2 Uses a Similar Two-Gate Mechanism for PS Selection and Transport.
The presence of an Asn550 in TM3 of Dnf1 seems to prevent NBD-PS transport, because several different mutations in this position cause enhanced NBD-PS uptake. Surprisingly, this Asn is conserved among yeast and mammalian P4-ATPases, including Drs2 (Asn445). We wondered what consequence the N445S mutation might have on Drs2 activity, by itself and in combination with F511Y (the proline + 4 mutation that abrogates PS recognition). Each single mutant and the double mutant complemented the cold-sensitive growth defect of a drs2Δ strain (Fig. 6A). Deletion of DRS2 also causes disruption of the plasma membrane asymmetry for PS and PE, which can be monitored by sensitivity to pore-forming toxins that specifically interact with PS [Papuamide B (PapB)] or PE (duramycin) exposed on the outer leaflet (24, 25). The single and double mutants were also tested for PapB sensitivity to measure the influence of the mutations on PS asymmetry. As previously reported, Drs2[F511Y] displayed a PapB sensitivity intermediate between the empty vector (drs2∆) control and cells containing WT Drs2 (Fig. 6B) (13). The Drs2 N445S single mutant provided WT resistance to PapB, and the double mutant (Drs2 N445S, [F511Y]) was slightly more sensitive to PapB than F511Y. Thus, N445S could not restore PS recognition to the F511Y exit gate mutant. To determine the influence of these mutations on PE asymmetry, we monitored resistance to duramycin, a toxin that specifically permeabilizes cells exposing PE. As previously reported, Drs2[F511Y] displayed no PE asymmetry loss relative to WT Drs2. The same was true for Drs2 N445S and N445S, [F511Y] (Fig. S6A).
Fig. 6.

Control of Drs2 PS recognition by both cytosolic and exofacial residues. (A) DRS2 alleles harboring cytosolic cluster (exit gate) mutations complement the cold sensitivity of a drs2Δ strain. (B) Influence of exit gate mutations on PS asymmetry measured by sensitivity to PapB. Relative to WT Drs2, Drs2[F511Y] displays a defect in PS asymmetry. Drs2 N445S maintains WT resistance to PapB, whereas Drs2 N445S, [F511Y] slightly exacerbates the Drs2[F511Y] defect. (C) DRS2 alleles harboring combination entry and exit gate mutations complement the cold sensitivity of a drs2Δ strain. (D) Relative to WT Drs2, Drs2[QQ→GA] displays a major loss of PS asymmetry, which can be restored by mutation of a cytosolic cluster residue (Drs2[QQ→GA], N445S) to permit increased PS recognition. (E) Influence of cytosolic cluster mutations on PE asymmetry measured by sensitivity to duramycin. Relative to WT Drs2 and drs2∆, each mutant maintains normal PE asymmetry. Therefore, Drs2[QQ→GA] exhibits a specific defect in PS transport. (F) Combining the QQ→GA substitution with either D473K or F511Y does not disrupt PS asymmetry further or restore PS asymmetry. However, the exit gate mutation N445S can restore PS asymmetry to Drs2[QQ→GA] and Drs2[QQ→GA], [D473K]. Compare Drs2[QQ→GA], N445S with Drs2[QQ→GA] and Drs2[QQ→GA], [D473K], N445S with Drs2[QQ→GA], [D473K]. For all experiments, values are the mean (±SEM).
The TM1 GA→QQ substitution in Dnf1 conferred gain-of-function NBD-PS recognition; thus, we tested if the reciprocal Drs2[QQ→GA] mutation would cause a loss of PS recognition. This entry gate mutant was functional in vivo because it complemented the drs2∆ cold-sensitive growth defect (Fig. 6C), but Drs2[QQ→GA] cells displayed a substantial loss of PS asymmetry (Fig. 6D). Remarkably, the N445S mutation completely suppressed QQ→GA such that the Drs2[QQ→GA], N445S double mutant displayed WT resistance to PapB (Fig. 6D). Importantly, each of these Drs2 mutants was capable of maintaining PE asymmetry similar to WT Drs2 (Fig. 6E), indicating that the QQ→GA entry gate substitution specifically perturbs endogenous PS recognition by Drs2.
An LL3–4 entry gate mutation in Dnf1 (K578E) provided a marginal increase in NBD-PS uptake, and the analogous Drs2 residue also carries a negative charge (Asp473). Therefore, we tested the Drs2[D473K] substitution and consistently found partial loss of PS asymmetry for this mutant. Again, N445S completely suppressed the loss of PS asymmetry caused by [D473K] (Fig. S6B). We then combined the two entry gate mutations (Drs2[QQ→GA] [D473K]) and found they were not additive; the double mutant displayed the same PapB sensitivity as the QQ→GA mutant. Even in this context ([QQ→GA] [D473K]), N445S could restore PS asymmetry to the triple mutant (Fig. 6F). We would not expect the F511Y exit gate mutation to have this suppressive capacity because it causes loss of asymmetry; F511Y neither suppressed nor exacerbated the QQ→GA influence on PS asymmetry (Fig. 6F). In sum, these data indicate the same relationship between entry gate and exit gate mutations in Drs2 and Dnf1. In each case, the exit gate Asn mutations in TM3 can allow PS transport even when entry gate residues are present that normally restrict PS transport.
Discussion
A number of different proteins are thought to mediate the translocation of lipid across a membrane bilayer. However, very little is known about how these transporters recognize and flip their specific substrates. This issue is particularly interesting for P-type ATPases, for which the mechanism of ion recognition and transport is known in exquisite detail for P2-ATPases; however, whether P4-ATPases use this canonical pathway or a noncanonical mechanism for phospholipid transport is a subject of debate.
Using an unbiased screening procedure, we identified two clusters of residues involved in phospholipid selection by P4-ATPases (Fig. 7A). One cluster is located on the exofacial side of the membrane, where phospholipid is initially selected, and we propose that this cluster forms an “entry gate” (Fig. 7). The residues composing the entry gate are present on TM1, LL1–2, TM2, and LL3–4, none of which correspond to residues in the canonical ion-binding site of the P2-ATPases. Within the entry gate, we have identified residues specifically involved in the PC preference of Dnf1 (Ile234, Phe235, Pro240, Gly241, Tyr575, Glu586, Phe587, Gly588, and Ile590) and suppression of PS recognition (Gly230, Ala231, and K578) (Fig. 7A). Swapping the Gln237 and Gln238 residues from TM1 in Drs2 for the analogous Gly230 and Ala231 residues in Dnf1 (or Dnf1 Lys578 for Drs2 Asp473) conferred PS transport to Dnf1 and abrogated PS transport by Drs2 (Fig. 6D and Fig. S6B). These results support the noncanonical transport pathway for Dnf1 and Drs2.
Fig. 7.

Residues affecting the phospholipid specificity of P4-ATPases cluster into two gates. (A) Topological diagram of Dnf1 TM1–2 and TM3–4 indicating positions of residues where mutations lead to increased PS recognition (red), reduced PC recognition (green), or reduction of overall activity (yellow). These residues cluster around a phospholipid entry gate (exofacial membrane face) and an exit gate (cytosolic membrane face). (B) Dnf1 modeled on the E1 conformational state of sarco/endoplasmic reticulum Ca2+-ATPase-1 (SERCA1) (11) indicates the relative positioning of residues involved in substrate specificity. The residues cluster into two gates, where phospholipid is selected on each side of the membrane. Light blue boxes indicate the entry and exit gates, and black lines denote the membrane boundaries. A, actuator domain; N, nucleotide-binding domain; P, phosphorylation domain. (C) Dnf1 modeled on the E2-P conformational state of SERCA1 (11) highlights the conformational changes in TM1–4 during the pumping cycle driven by transfer of phosphate from ATP to the P domain. (D and E) Proposed mechanism for phospholipid transport by P4-ATPases. Phospholipid is initially selected at the entry gate, perhaps when the pump is in the E1 conformation. On transfer of phosphate from ATP to the P domain to induce the E2 conformation, TM1–2 shift up dramatically in the membrane, feasibly providing the physical force to flip the phospholipid headgroup across the lipid bilayer and feed the substrate into the exit gate. Substrate would be released from the exit gate to the cytosolic leaflet in the E2 → E1 transition.
The second cluster of residues is located in TM3 and TM4 near the cytosolic side of the membrane, and we propose that this cluster forms an “exit gate” for the phospholipid (Fig. 7). Within the exit gate, we identified residues responsible for PC preference (Ile545 and Glu622) and suppression of PS transport by Dnf1 (Tyr618, Asn550, Val621, and Glu622). Val621 and Glu622 are predicted to lie one turn of the helix above Tyr618 in TM4, all of which are in the vicinity of Asn550 in TM3. A specific Tyr→Phe mutation seems to be required at position 618 to confer NBD-PS activity to Dnf1; however, surprisingly, several different substitutions at position 550 allow PS transport. A number of mutations were recovered in TM3 between the two gates, and it is probably significant that these mutations only reduce Dnf1 activity and line one face of the helix (Fig. 7A), bookended by Asn571 (which is essential) and Asn550 (a specificity determinant).
The recovery of many mutations that reduce PC transport without significantly altering PE transport is not surprising because the edelfosine screen was designed to identify residues involved in PC selection. An interesting conundrum is why the Asn550, V621A, and K578E mutations allowing PS recognition came through the edelfosine resistance screen. It is possible that strains expressing these mutants could be resistant to edelfosine because the gain-of-function for PS transport created competition in vivo between endogenous PS and edelfosine for Dnf1. Regardless of the basis for edelfosine resistance, this screen illuminated additional residues involved in allowing PS recognition in Dnf1 and Drs2.
Whereas each of these TM3 and TM4 exit gate resides outside of the canonical binding site, we did recover a single mutation in TM6, I1235F, which enhances PS transport and corresponds to a canonical site residue (Asp800 in sarco/endoplasmic reticulum Ca2+-ATPase). There are several reasons why we think the I1235F mutation is altering the function of the exit gate in a noncanonical pathway rather than perturbing binding of phospholipid substrate in the canonical site. The first is that I1235F increased the overall activity of Dnf1 for all substrates and caused partial loss of specificity for glycerophospholipid, unlike the other point mutations in TM1–4, which enhanced PS uptake with no significant increase in SM uptake, including Dnf1[GA→QQ], N550S, [Y618F], which has overall higher uptake activity but no increase in NBD-SM uptake. I1235 in TM6 is modeled very close to the TM4 exit gate residues, and the enhanced SM uptake caused by I1235F is similar to that previously observed with Dnf1[YIS→FVT], a triple mutation in the TM4 exit gate region (13). It is possible that altering the packing of the TM helices in this region perturbs recognition of the glycerol backbone of the substrate.
The second reason why we believe our data support the noncanonical pathway has to do with the frequency of specificity-altering mutations recovered in the edelfosine screen. Within TM3–4, we assayed 26 point mutants (Figs. 2 and 3) derived from random mutagenesis. Of these 26 mutants assayed, 10 exhibited a change of specificity (∼40% of the mutants). Within TM5–6, we assayed 25 mutants in total [18 point mutations (Fig. 4 and Fig. S3A) and 7 multiple mutations (Fig. S3C)]. Of these mutants assayed, only I1235F displayed a change in specificity (4%). This sample size should have identified at least 9 to 10 clones with altered specificity if the rate was similar to that of TM3–4 mutants (∼40%). Our data indicate that TM5–6 residues are important for activity, because many of the mutations reduce activity without altering patterns of substrate recognition (Fig. 4 and Fig. S3). In general, it seems most likely that the TM5–6 mutations that reduce activity are due to a defect in transducing the mechanics of phospholipid transport between the ATP-consuming cytosolic domains and substrate-selecting gates in TM1–4.
Finally, we note that a crystal structure of the Ca2+-ATPase in the E2 conformation contains a PE molecule specifically bound in a cleft between TM1–4, in the region we describe as the exit gate (Protein Data Bank ID code 2AGV) (26) (Fig. S7). For the Ca2+-ATPase, PE likely enters and exits this binding site from the cytosolic leaflet as the pump transitions from E1→E2→E1 conformations. We suggest that this phospholipid binding site is conserved in the P4-ATPases as part of the exit gate but that the flippases evolved the ability to load this site with phospholipid selected from the exofacial leaflet using entry gate residues and to expel the substrate to the cytosolic leaflet in the E2→E1 transition.
The two gates appear to act in tandem sequentially to select substrate for transport. For mutations that enhance PS transport in Dnf1, we find that combining two exit gate mutations does not have an additive effect on transport. However, combining entry gate and exit gate mutations does have an additive effect. We interpret this observation to mean that phospholipid is imperfectly selected at each gate. Thus, an entry gate mutation in Dnf1 can allow PS transport even when the exit gate is tuned for PC, and vice versa, but maximal PS transport requires residues at each gate that select for PS. The mechanistic coupling of the two gates seems to be conserved for Drs2. For example, the entry gate QQ→GA mutation causes a specific loss of PS asymmetry that can be restored by the N445S exit gate mutation. For Dnf1, N550S also allows PS transport when GA is present at the base of TM1.
Using the canonical ion-binding site for phospholipid transport seems unlikely, given the positioning of residues within the entry and exit gates along TM1–4. Based on the general location and cooperation of the substrate specificity determinants we have identified, we propose a two-gate mechanism for phospholipid flip. In our preferred model, phospholipid is initially selected at the entry gate in the E1-P state (Fig. 7 B and D). As the pump transitions from E1-P→E2-P, TM1–2 shifts up into the membrane, carrying the phospholipid near the exofacial side of the membrane and the exit gate (Fig. 7 C and E). The phospholipid is again selected at the exit gate before (or concurrently with) dephosphorylation (E2-P→E2) and release of substrate to the cytosolic leaflet (E2→E1). Substrate is required for dephosphorylation of the P4-ATPase (15, 17, 27), probably because the phospholipid must bind at the exit gate to induce movement of the actuator domain to dephosphorylate the pump.
These studies provide important unique mechanistic insight into how integral membrane proteins recognize and select phospholipid substrate within a membrane environment. It remains to be seen whether a similar two-gate selection mechanism is used by other lipid transporters (e.g., the ABC transporter family) or if this represents a unique mechanism for the P4-ATPases.
Materials and Methods
Strains and Culture.
E. coli strain DH5α was used for molecular cloning. Each plasmid used in this study is listed in Table S1. Strains used in this study are listed in Table S2. Yeast was grown in standard rich medium [yeast peptone dextrose (YPD)] or synthetic minimal glucose medium (SD). Yeast was transformed using the lithium acetate method (28). To test complementation, 50,000 cells were spotted with 10-fold serial dilutions onto SD or SD + 5-FOA. Plates were grown at 20 °C or 30 °C for 3 to 4 d before imaging.
Edelfosine Resistance Screen.
To generate random mutations, we used procedures described previously (13). We cotransformed ZHY704 with mutagenized PCR products and pRS313-DNF1 gapped with restriction enzymes MfeI and NruI [or for TM5–6, we used pRS313-Dnf1(w/SfoI) site-gapped with SfoI] to allow for gap repair through homologous recombination. The transformations were plated onto synthetic dropout plates and grown for 3 d; colonies were transferred into 96-well plates and spotted onto synthetic dropout plates containing 20 μg/mL edelfosine or 5-FOA. We sequenced dnf1* in strains that grew strongly on both edelfosine and 5-FOA.
Viability Assay.
To test for PapB and duramycin sensitivity, midlog phase yeast was seeded in 96-well plates at 0.1 OD600/mL ± drug. Plates were incubated at 30 °C for 20 h, and the OD600/mL was measured with a Multimode Plate Reader Synergy HT (Bio-Tek). Relative growth was based on mock-treated cells in the assay.
Fluorescence Microscopy.
Images were collected with an Axioplan microscope (Carl Zeiss) using a Hamamatsu CCD camera and MetaMorph software (Molecular Devices). To observe GFP-tagged proteins, cells were grown to midlog phase, pelleted, and resuspended in 10 mM Tris⋅HCl (pH 7.4) and 2% glucose (wt/vol). An aliquot of cells was spread onto slides and visualized using a GFP filter set.
Cell Extracts and Western Blotting.
Yeast expressing N-terminal 3× FLAG-tagged Dnf1 was grown in YPD to midlog phase and harvested in SDS/urea sample buffer [40 mM Tris⋅HCl (pH 6.8), 8 M urea, 0.1 mM EDTA, 1% 2-mercaptoethanol (vol/vol), 5% SDS (wt/vol), and 0.25% bromophenol blue (wt/vol)]. Cell lysate from 0.2 OD(600) was separated by SDS/PAGE, transferred to PVDF membrane, and blotted with anti-FLAG M2 antibody (Sigma–Aldrich).
NBD-Lipid Uptake.
Lipid uptake was performed essentially as described previously (13). Briefly, overnight cultures were subcultured to 0.15 OD(600)/mL and allowed to grow to early-midlog phase. Five hundred microliters of cells was harvested, resuspended in ice-cold SD media containing 2 μg/mL NBD-lipid (∼2.5 μM), and incubated on ice for 30 min. Cells were washed twice with SA media [SD media + 2% sorbitol (wt/vol), + 20 mM NaN3] + 4% BSA (wt/vol), once with SA media, and resuspended in SA media before analysis by flow cytometry.
Flow Cytometry.
Flow cytometry was performed with a BD LSRII-3 laser (BD Biosciences) using BD FACSDiva v6.1.3 (BD Biosciences). NBD-lipid uptake was measured with the FITC filter set (530/30 bandpass with a 525 long pass). Immediately before analysis, propidium iodide was added at a final concentration of 5 μM. At least 10,000 events were analyzed using forward/side scatter to identify single cells and propidium iodide fluorescence to exclude dead cells.
Data Analysis.
Within each NBD-PL uptake experiment, at least three independently isolated transformants harboring the same construct were assayed, as previously described (13). The mean of these values from at least three separate experiments was reported (±SEM). Uptake of NBD-PL for a dnf1,2Δ strain with an empty vector was subtracted, and NBD-PC uptake by WT Dnf1 was used to normalize the data. Each value is reported relative to WT Dnf1 PC uptake at 30 min. Substrate preference ratios were determined by dividing the NBD-PE or NBD-PS uptake value by the NBD-PC uptake value for each independent replicate (n ≥ 9). The mean of each substrate preference ratio is reported (±SEM). All Dnf1 alleles showing a substrate preference ratio falling outside of the 15% confidence interval with no overlap of the error bars are significantly different (P < 0.05) from WT Dnf1 determined by the Student’s t test. For drug-sensitivity assays, at least two independently isolated transformants were assayed in two separate experiments and reported as the mean ± SEM.
Supplementary Material
Acknowledgments
These studies were supported by National Institutes of Health Grant GM62367 (to T.R.G.) and Cellular and Molecular Microbiology Training Program T32AI007611-11 (to R.D.B.). The Vanderbilt Medical Center Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (Grant P30 CA68485) and the Vanderbilt Digestive Disease Research Center (Grant DK058404).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Author Summary on page 1577 (volume 110, number 5).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1216948110/-/DCSupplemental.
References
- 1.Sebastian TT, Baldridge RD, Xu P, Graham TR. Phospholipid flippases: Building asymmetric membranes and transport vesicles. Biochim Biophys Acta. 2012;1821(8):1068–1077. doi: 10.1016/j.bbalip.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455(1):152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 3.Dean M, Annilo T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu Rev Genomics Hum Genet. 2005;6:123–142. doi: 10.1146/annurev.genom.6.080604.162122. [DOI] [PubMed] [Google Scholar]
- 4.Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: A framework for ABC transporter architecture and mechanism. Science. 2002;296(5570):1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 5.Ward A, Reyes CL, Yu J, Roth CB, Chang G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci USA. 2007;104(48):19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gourdon P, et al. Crystal structure of a copper-transporting PIB-type ATPase. Nature. 2011;475(7354):59–64. doi: 10.1038/nature10191. [DOI] [PubMed] [Google Scholar]
- 7.Morth JP, et al. Crystal structure of the sodium-potassium pump. Nature. 2007;450(7172):1043–1049. doi: 10.1038/nature06419. [DOI] [PubMed] [Google Scholar]
- 8.Pedersen BP, Buch-Pedersen MJ, Morth JP, Palmgren MG, Nissen P. Crystal structure of the plasma membrane proton pump. Nature. 2007;450(7172):1111–1114. doi: 10.1038/nature06417. [DOI] [PubMed] [Google Scholar]
- 9.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature. 2000;405(6787):647–655. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 10.Post RL, Hegyvary C, Kume S. Activation by adenosine triphosphate in the phosphorylation kinetics of sodium and potassium ion transport adenosine triphosphatase. J Biol Chem. 1972;247(20):6530–6540. [PubMed] [Google Scholar]
- 11.Toyoshima C, Nomura H, Tsuda T. Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature. 2004;432(7015):361–368. doi: 10.1038/nature02981. [DOI] [PubMed] [Google Scholar]
- 12.Morth JP, et al. A structural overview of the plasma membrane Na+,K+-ATPase and H+-ATPase ion pumps. Nat Rev Mol Cell Biol. 2011;12(1):60–70. doi: 10.1038/nrm3031. [DOI] [PubMed] [Google Scholar]
- 13.Baldridge RD, Graham TR. Identification of residues defining phospholipid flippase substrate specificity of type IV P-type ATPases. Proc Natl Acad Sci USA. 2012;109(6):E290–E298. doi: 10.1073/pnas.1115725109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puts CF, Holthuis JC. Mechanism and significance of P4 ATPase-catalyzed lipid transport: Lessons from a Na+/K+-pump. Biochim Biophys Acta. 2009;1791(7):603–611. doi: 10.1016/j.bbalip.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Coleman JA, Vestergaard AL, Molday RS, Vilsen B, Peter Andersen J. Critical role of a transmembrane lysine in aminophospholipid transport by mammalian photoreceptor P4-ATPase ATP8A2. Proc Natl Acad Sci USA. 2012;109(5):1449–1454. doi: 10.1073/pnas.1108862109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thøgersen L, Nissen P. Flexible P-type ATPases interacting with the membrane. Curr Opin Struct Biol. 2012;22(4):491–499. doi: 10.1016/j.sbi.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Ding J, et al. Identification and functional expression of four isoforms of ATPase II, the putative aminophospholipid translocase. Effect of isoform variation on the ATPase activity and phospholipid specificity. J Biol Chem. 2000;275(30):23378–23386. doi: 10.1074/jbc.M910319199. [DOI] [PubMed] [Google Scholar]
- 18.Natarajan P, Wang J, Hua Z, Graham TR. Drs2p-coupled aminophospholipid translocase activity in yeast Golgi membranes and relationship to in vivo function. Proc Natl Acad Sci USA. 2004;101(29):10614–10619. doi: 10.1073/pnas.0404146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou X, Graham TR. Reconstitution of phospholipid translocase activity with purified Drs2p, a type-IV P-type ATPase from budding yeast. Proc Natl Acad Sci USA. 2009;106(39):16586–16591. doi: 10.1073/pnas.0904293106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pomorski T, et al. Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol Biol Cell. 2003;14(3):1240–1254. doi: 10.1091/mbc.E02-08-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riekhof WR, et al. Lysophosphatidylcholine metabolism in Saccharomyces cerevisiae: The role of P-type ATPases in transport and a broad specificity acyltransferase in acylation. J Biol Chem. 2007;282(51):36853–36861. doi: 10.1074/jbc.M706718200. [DOI] [PubMed] [Google Scholar]
- 22.Hanson PK, Malone L, Birchmore JL, Nichols JW. Lem3p is essential for the uptake and potency of alkylphosphocholine drugs, edelfosine and miltefosine. J Biol Chem. 2003;278(38):36041–36050. doi: 10.1074/jbc.M305263200. [DOI] [PubMed] [Google Scholar]
- 23.Kean LS, Fuller RS, Nichols JW. Retrograde lipid traffic in yeast: Identification of two distinct pathways for internalization of fluorescent-labeled phosphatidylcholine from the plasma membrane. J Cell Biol. 1993;123(6 Pt 1):1403–1419. doi: 10.1083/jcb.123.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Navarro J, et al. Interaction of duramycin with artificial and natural membranes. Biochemistry. 1985;24(17):4645–4650. doi: 10.1021/bi00338a025. [DOI] [PubMed] [Google Scholar]
- 25.Parsons AB, et al. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell. 2006;126(3):611–625. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 26.Obara K, et al. Structural role of countertransport revealed in Ca(2+) pump crystal structure in the absence of Ca(2+) Proc Natl Acad Sci USA. 2005;102(41):14489–14496. doi: 10.1073/pnas.0506222102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paterson JK, et al. Lipid specific activation of the murine P4-ATPase Atp8a1 (ATPase II) Biochemistry. 2006;45(16):5367–5376. doi: 10.1021/bi052359b. [DOI] [PubMed] [Google Scholar]
- 28.Gietz RD, Woods RA. Yeast transformation by the LiAc/SS Carrier DNA/PEG method. Methods Mol Biol. 2006;313:107–120. doi: 10.1385/1-59259-958-3:107. [DOI] [PubMed] [Google Scholar]
