

Abstract
Herpesvirus entry functions of the conserved glycoproteins gB and gH–gL have been delineated, but their role in regulating cell–cell fusion is poorly understood. Varicella-zoster virus (VZV) infection provides a valuable model for investigating cell–cell fusion because of the importance of this process for pathogenesis in human skin and sensory ganglia. The present study identifies a canonical immunoreceptor tyrosine-based inhibition motif (ITIM) in the gB cytoplasmic domain (gBcyt) and demonstrates that the gBcyt is a tyrosine kinase substrate. Orbitrap mass spectrometry confirmed that Y881, central to the ITIM, is phosphorylated. To determine whether the gBcyt ITIM regulates gB/gH–gL–induced cell–cell fusion in vitro, tyrosine residues Y881 and Y920 in the gBcyt were substituted with phenylalanine separately or together. Recombinant viruses with these substitutions were generated to establish their effects on syncytia formation in replication in vitro and in the human skin xenograft model of VZV pathogenesis. The Y881F substitution caused significantly increased cell–cell fusion despite reduced cell-surface gB. Importantly, the Y881F or Y881/920F substitutions in VZV caused aggressive syncytia formation, reducing cell–cell spread. These in vitro effects of aggressive syncytia formation translated to severely impaired skin infection in vivo. In contrast, the Y920F substitution did not affect virus replication in vitro or in vivo. These observations suggest that gB modulates cell–cell fusion via an ITIM-mediated Y881 phosphorylation-dependent mechanism, supporting a unique concept that intracellular signaling through this gBcyt motif regulates VZV syncytia formation and is essential for skin pathogenesis.
Keywords: fusogenicity, mutagenesis, polykaryocyte, virulence
The alphaherpesvirus varicella-zoster virus (VZV) is a human pathogen that spreads from mucosal epithelial sites of initial infection to skin via a T cell-associated viremia (1), causing varicella (chicken pox). Viremia and cutaneous infection enable transfer of VZV to sensory nerve ganglia and establishment of latency in neurons (2). Zoster (shingles) is caused by VZV reactivation from latently infected neurons and can lead to the debilitating condition of postherpetic neuralgia. Live attenuated VZV vaccines are effective against varicella and zoster but are not recommended for immunocompromised patients.
Enveloped viruses from several families, including the Herpesviridae, require fusion with cellular membranes for virion entry, and in some cases induce syncytia through cell–cell fusion (2–5). Little is known about the functional role of syncytia during pathogenesis. VZV is a valuable model pathogen for investigating this process because natural infection of the human host involves formation of multinucleated polykaryocytes in skin and fusion of neurons and satellite cells in sensory ganglia (2, 6). In addition, VZV produces syncytia during replication in vitro and triggers fusion between differentiated cells in human skin and dorsal root ganglion xenografts infected in vivo in the severe combined immunodeficiency mouse model of VZV pathogenesis (7, 8).
Herpesvirus fusion is governed by a core complex of virion proteins consisting of the type I membrane glycoprotein B (gB) and the gH–gL heterodimer (9). We have recently shown that this core complex in VZV is sufficient to cause cell–cell fusion in the absence of other viral proteins (10). Functions of gB homologs in herpesviruses, including the other two human alphaherpesviruses, herpes simplex virus (HSV) 1 and 2, have been evaluated in cell–cell fusion assays, primarily as a surrogate for fusion of the virion envelope with the cell membrane during entry (9, 11). These data combined with crystallography studies have identified fusion loops in the gB ectodomain that penetrate the target cell (12, 13). The VZV gB ectodomain has predicted fusion loops essential for replication (14).
The gB cytoplasmic domain (gBcyt) has been implicated in cell–cell fusion regulation by an unknown mechanism. Whereas syncytia formation is typical of VZV, engineered or spontaneous substitutions in the gBcyt in other herpesviruses lead to syncytial herpesvirus phenotypes (15–17). The cytoplasmic domains of gB homologs contain α-helices and endocytosis motifs (18–20). Endocytosis, linked to dileucine and YXXΦ motifs (X, any amino acid; Φ, a large hydrophobic residue), is necessary for gB cell-surface retrieval, trans-Golgi network (TGN) localization, and incorporation into the virion envelope (21). Mutations in the gBcyt can cause elevated cell-surface gB, but this effect does not account for increased fusogenicity of HSV 2 or Epstein–Barr virus gB mutations (17, 22). In VZV, a YXXΦ motif in the gBcyt is implicated in gB endocytosis and endoplasmic reticulum (ER)–Golgi transport (23). VZV isolates with gBcyt mutations have not been identified, but a large C-terminal truncation (Δ896–931), which disrupts predicted α-helices and eliminates dileucine and YXXΦ motifs, had enhanced syncytia formation without increased cell-surface expression (24). These studies suggest that the gBcyt has unexplored regulatory functions important for cell–cell fusion and, therefore, pathogenesis in the human host.
Immunoreceptor tyrosine-based inhibition motifs (ITIMs) located in the cytoplasmic domains of membrane-anchored proteins function to inhibit intracellular signaling cascades in cells of the immune system but also activate some cell signaling pathways and influence cell motility (25–27). ITIMs must be tyrosine-phosphorylated to be functional. In the present study, a canonical ITIM was identified in the VZV gBcyt that was tyrosine-phosphorylated during infection, leading to the hypothesis that cell–cell fusion is regulated through phosphorylation-dependent mechanisms of the gBcyt ITIM. We demonstrate that preserving the native tyrosine, Y881, in this ITIM was necessary for modulating cell–cell fusion when gB is expressed with gH–gL in the absence of other viral proteins. Furthermore, the formation of syncytia in VZV-infected cells in vitro becomes strikingly more extensive unless the gBcyt ITIM has the phosphorylatable Y881 residue, which is critical for viral spread in the microenvironment of human skin in vivo. Thus, the regulation of gB-mediated cell fusion is a previously undescribed ITIM-dependent function required for pathogenesis of this ubiquitous human herpesvirus.
Results
Identification of a Canonical ITIM in the gB Cytoplasmic Domain.
VZV gB, encoded by open reading frame (ORF)31, is predicted to form a trimeric ectodomain with five subdomains, a transmembrane domain, and a 124-amino acid cytoplasmic domain containing three predicted α-helices (Fig. 1 A and B). Analysis of the gBcyt (residues 808–931) with the Web-based server eukaryotic linear motif resource (http://elm.eu.org) identified a canonical ITIM consensus sequence, [ILV]XYXX[LV], in the flanking sequence 879IKYMTL884 around the 881YXXΦ884 motif within the second highly conserved α-helix (Fig. 1B). Tyrosine residue Y881 is central to this canonical ITIM, which is highly conserved among the alphaherpesviruses (Table S1). The flanking sequences of a second YXXΦ motif (920YSRV923) did not form an ITIM.
Fig. 1.

Cytoplasmic domain of VZV gB is a tyrosine kinase substrate. (A) Homology model of the gB ectodomain trimer and predicted transmembrane (TM) domain. The five domains of a single molecule in the gB trimer are as follows: domain I, blue; II, green; III, yellow; IV, orange; V, red; furin cleavage site (RSRR), red spacefill; residues W180 and Y185 in the predicted primary fusion loop, purple spacefill. (B) The gB cytoplasmic domain. The three predicted α-helices (pink cylinders), gB ITIM (879IKYMTL884), two YXXΦ motifs (residues 881–884 and 920–923), and dileucine endocytosis motif (residues 904–905) are shown. (C) Immunoprecipitation of gB from VZV-infected (V) or uninfected (UI) melanoma cells and Western blot for gB (rabbit serum 746-868), gE (mAb 8612), and phosphorylated tyrosine (pY; mAbs 4G10/pY20). Arrowheads indicate the 66- and 124-kDa gB species that are tyrosine-phosphorylated. (D) Coomassie blue staining (Upper) and Western blot (Lower; mAb 4G10) of the same membrane to detect phosphorylated tyrosine on purified GST (negative control) and the GST-tagged cytoplasmic domains of the proteins FcRγ (GST-FcRγ; positive control) and gBcyt (GST-gB) expressed in BL21 or BL21 E. coli expressing the tyrosine kinase Elk-1. Molecular mass standards (kDa) are indicated.
gB Cytoplasmic Domain Is a Tyrosine Kinase Substrate.
gB was demonstrated to be tyrosine-phosphorylated when immunoprecipitated from pOka-infected melanoma cells and probed with monoclonal antibodies against phosphorylated tyrosine residues and a rabbit antibody against the gBcyt epitope 833PEGMDPFAEKPNAT846 (Fig. 1C). The predominant gB species were the immature full-length protein (124 kDa) and a C-terminal cleavage product (66 kDa) resulting from furin cleavage at 491RSRR494 (14), both of which exhibited tyrosine phosphorylation (Fig. 1C). Specificity of the anti-phosphotyrosine and anti-gB antibodies was shown using an anti-VZV gE antibody control. The gBcyt was shown to be a substrate for tyrosine kinases in the absence of other VZV proteins using a GST-tagged construct of the gBcyt (residues 808–931) produced in BL21 Escherichia coli expressing or lacking the exogenous Elk-1 tyrosine kinase. Phosphorylation of the GST negative control was not detected (Fig. 1D). Both the GST-FcRγ positive control and GST-gBcyt were phosphorylated when purified from BL21 E. coli expressing Elk-1 but not when purified from Elk-1 negative BL21 E. coli.
The gBcyt contains two tyrosine residues, Y809 and Y822, near the transmembrane domain in addition to the 881YXXΦ884 within the ITIM and the 920YXXΦ923 motif (Fig. 1B). Y881 was phosphorylated whereas Y809 and Y822 were not, based on Orbitrap mass spectrometry (Fig. S1). Y920 phosphorylation could not be assessed because peptide fragments beginning after gB residue 914 were not recovered. These unique findings demonstrate tyrosine phosphorylation of the gBcyt, in particular at residue Y881, that might influence gB function.
Endocytosis of gB Is Not Impeded by Phenylalanine Substitutions of Tyrosines in gBcyt YXXΦ Motifs.
Herpesvirus replication is dependent on gB endocytosis, and therefore the role of the gBcyt ITIM was analyzed to determine whether phosphorylation of Y881 in the YXXΦ motif within the ITIM or Y920 in the second YXXΦ motif contributed to this essential process. These tyrosines were replaced with phenylalanine to create three mutant gB constructs, Y881F, Y920F, and Y881/920F (Table S2). Phenylalanine rather than alanine substitution was chosen to retain the gBcyt structure, which has been reported to affect HSV-1 gB function (20). These mutants were compared with gB constructs with an L884G mutation that inhibits endocytosis and with two gB ectodomain mutants, W180G and Y185G (Fig. 1A), that disrupt the primary fusion loop (14, 23). Endocytosis was evaluated by the association of wild-type gB or the mutant proteins with early endosomes (EEA1). The anti-gB mAb SG2-2E6 recognizes a gB ectodomain epitope in the endosome lumen, whereas EEA1 is on the cytoplasmic endosome surface, generating a characteristic staining pattern of EEA1 juxtaposed to gB (Fig. 2 and Fig. S2A). As expected, the W180G and W180G fusion-loop mutations did not alter gB endocytosis, as shown by EEA1 and gB localization similar to WT gB. The Y881F, Y920F, and Y881/920F mutants also localized with EEA1 similar to WT gB, indicating intact endocytosis. The Y920F mutant exhibited many filopodia-like protrusions (Fig. S2B), suggesting that Y920 in the second YXXΦ modulates cell-surface morphology. As expected, the L884G mutant was rarely associated with EEA1 despite high levels of gB retained in cytoplasmic structures proximal to the plasma membrane, indicating severe aberrations in gB trafficking (Fig. 2 and Fig. S2A). To determine whether the side chain of residue 881 in the gBcyt ITIM affects endocytosis, aspartic acid (Y881D), glutamic acid (Y881E), and tryptophan (Y881W) mutants were generated. Both Y881D and Y881E demonstrated poor association with EEA1 similar to L884G (Fig. S2A), confirming that the YXXΦ motif was critical for gB trafficking. Importantly, gB localization with EEA1 was unaffected by Y881W, demonstrating that an aromatic side chain at residue 881 in the gBcyt enables endocytosis.
Fig. 2.

Phenylalanine substitution of Y881 in the gBcyt ITIM does not prevent gB endocytosis. Confocal microscopy of melanoma cells at 24 h posttransfection with wild-type or gB mutant expression constructs and stained for gB (red) and early endosome antigen (EEA1; green). These high-magnification images are from boxed areas in Fig. S2A. White arrows highlight representative endocytic vesicles (EEA1) that contain gB. (Scale bar, 10 μm.) Diagram: epitope locations of the mAb SG2-2E6 (gB ectodomain in the endosome lumen, red), EEA1 rabbit polyclonal Ab (cytoplasmic surface, green), and gBcyt with Y881 and Y920 residues (red).
Effects of gBcyt Phenylalanine Mutations on the Intracellular Processing of gB.
The processing of the gBcyt mutants was assessed by comparison with WT gB, the fusion-loop mutants, and the cleavage-defective Δ491RSRR494 mutant (Fig. S3A). The Y881F, Y920F, and Y881/920F mutants produced immature gB (124 kDa) and the cleavage product (66 kDa) at levels similar to WT gB and the W180G and Y185G fusion-loop mutants, whereas the Δ491RSRR494 mutant showed accumulation of an extensively glycosylated form of gB (132 kDa) without a 66-kDa cleavage product. The L884G substitution resulted in accumulation of the 124-kDa form and a cleavage product <66 kDa. Thus, gB intracellular processing was unaffected by the gBcyt tyrosine-to-phenylalanine substitutions, based on the similar molecular masses and relative abundance of gB species produced by these mutants and WT gB.
Mutagenesis of the ITIM Residue Y881 Affects the Fusogenicity of gB/gH–gL.
Expression of gB together with the heterodimer gH–gL in CHO-K1 cells is sufficient to induce cell–cell fusion with target LDL-GFP melanoma cells in our quantitative Cre reporter fusion assay (10), where gB/gH–gL expression induced high frequencies of fusion events (Fig. S3B). In contrast, fusion frequencies were similar to the vector control when gB or gH–gL were expressed alone. The primary fusion-loop mutants W180G and Y185G had cell-surface expression levels similar to WT gB but, as predicted, fusion was abolished (Fig. 3 and Fig. S3C). In contrast, the Y881F substitution increased cell–cell fusion significantly even though gB surface expression was reduced slightly relative to WT gB. The Y881/920F mutation also enhanced fusion significantly, with surface expression comparable to WT gB. In contrast, the Y920F substitution decreased cell–cell fusion despite increased gB surface expression, and L884G prevented fusion (Fig. 3). Unsurprisingly, both Y881D and Y881E abolished gB/gH–gL fusogenicity due to defective intracellular trafficking and poor cell-surface expression similar to L884G. In contrast, the Y881W substitution increased fusogenicity substantially, demonstrating that polarity of the aromatic residue side chain at 881 does not inhibit fusion, whereas the hydroxyl moiety is critical for gBcyt fusion regulation. To determine whether the gBcyt tyrosine mutations might alter gB cell-surface expression by a subtle effect on endocytosis rate, half-lives of cell-surface gB were measured for Y881F, Y920F, and Y881/920F. Surface gB levels were consistent with previous observations (Fig. S3 C and D). Importantly, the half-life of cell-surface gB for Y881F, Y920F, and Y881/920F was similar to WT gB (Fig. S3E). Thus, the gBcyt ITIM has a critical role in cell–cell fusion independent of gB endocytosis.
Fig. 3.
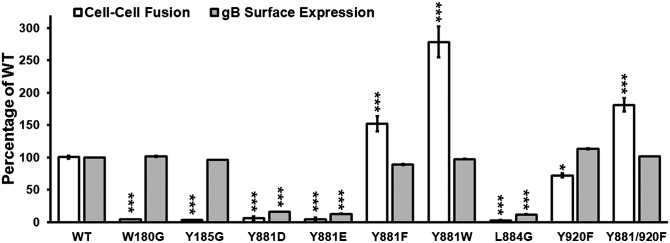
Phenylalanine substitution of Y881 within the gBcyt ITIM enhances gB/gH–gL–induced cell–cell fusion. Quantification of cell–cell fusion and cell-surface expression by flow cytometry of gB mutants compared with WT gB. Statistical differences were evaluated by two-way ANOVA. *P < 0.05, ***P < 0.001. SEM is indicated.
Y881F Substitution in the gBcyt ITIM Enhances VZV Syncytia Formation.
To determine whether the gBcyt modulates cell–cell fusion in the context of viral replication, mutagenesis was performed in the pOka genome using a self-excisable BAC system. Four BAC constructs, carrying the Y881F, L884G, Y920F, or Y881/920F substitutions, were generated in duplicate. Infectious virus was recovered from the gB-Y881F, gB-Y920F, and gB-Y881/920F mutant BACs but, as expected, the gB-L884G substitution was not compatible with VZV replication.
Small foci of infected cells were observed in monolayers at 24 h postinfection (hpi) when syncytia formation was assessed in melanoma cells infected with pOka or the gBcyt mutants by confocal microscopy. The distribution of gB in gB-Y881F, gB-Y920F, or gB-Y881/920F was similar to pOka-infected cells, where gB was localized to plasma membranes and TGN, the site of secondary virion envelopment (Fig. 4 and Fig. S4A). By 48 hpi, single newly infected cells at the leading edge of plaques were identified by gB expression, where typical small VZV-induced syncytia containing multiple nuclei had formed in more central regions of pOka-infected monolayers (Fig. 4). As expected, gB was localized to the plasma membranes and the TGN, which were redistributed to the center of small syncytia. The gB-Y920F mutant had similar pOka characteristics (Fig. S4A). In marked contrast, the Y881F substitution in gB-Y881F and gB-Y881/920F mutants caused dramatic and extensive syncytia formation in infected monolayers at 48 hpi (Fig. 4 and Fig. S4A). These syncytia contained hundreds to thousands of nuclei surrounding the reorganized TGN. The enhanced syncytia formation caused by the Y881F substitution was assessed in composite confocal images, incorporating EEA1 staining for early endosomes along with gB and TGN staining (Fig. S4 B and C). In addition to the TGN, early endosomes were redistributed to central regions of syncytia in monolayers infected with pOka and the three gBcyt mutants (Fig. S4 B and C). The extensive syncytia formation induced by gB-Y881F and gB-Y881/920F created large areas that were predominantly devoid of EEA1 and TGN. In addition, virus penetration into the cell monolayer at the leading edge of gB-Y881 and gB-Y881/920F infection was reduced compared with pOka and gB-Y920F. Thus, tyrosine at residue 881 in the gBcyt ITIM regulates the fusogenic capacity of gB to restrict uncontrolled syncytia formation.
Fig. 4.
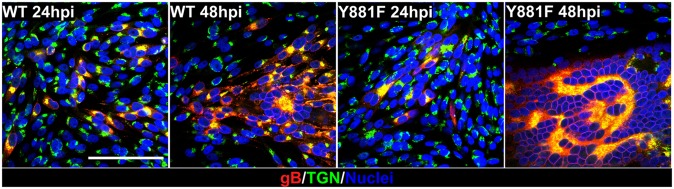
Extensive syncytia formation is triggered by Y881F substitution in the gBcyt ITIM. Confocal microscopy of melanoma cells infected with wild-type pOka and the gBcyt mutant, gB-Y881F, stained for gB (red), trans-Golgi network (green), and nuclei (blue) at 24 and 48 hpi. (Scale bar, 100 μm.)
Increased Syncytia Formation Induced by the gBcyt ITIM Y881F Substitution Impairs VZV Replication and Plaque Size in Vitro.
To determine the effect of gBcyt ITIM-regulated syncytia formation on replication in vitro, replication kinetics, plaque sizes, and plaque morphologies of the three gBcyt mutants and pOka were compared in melanoma cells. As expected, gB-Y920F replication was similar to pOka, which had typical kinetics with a >2 log10 increase in viral titers reaching a plateau between 3 and 5 d postinfection (dpi) (Fig. 5A). In contrast, titers of gB-Y881F and gB-Y881/920F were reduced significantly compared with pOka by 2 dpi and remained lower through 6 dpi for gB-Y881F. This was reflected in the significantly reduced plaque sizes of gB-Y881F and gB-Y881/920F compared with pOka and gB-Y920F (Fig. 5 B and C). Aberrant syncytia formation caused the center of gB-Y881F and gB-Y881/920F plaques to disassociate prematurely, leaving behind a thin outline of infected cells, indicating reduced penetration of the virus into the monolayer at 48 hpi (Fig. 5C), although an earlier rapid viral spread was not excluded. The pOka-like plaque size and morphology of gB-Y920F indicated that the gB-Y881/920F phenotype was due solely to the Y881F mutation (Fig. 5 B and C). Consequently, extensive syncytia formation associated with mutating the ITIM tyrosine, thereby preventing its phosphorylation, in the gB-Y881F and gB-Y881/920F mutants markedly impaired viral spread in vitro.
Fig. 5.

Increased syncytia formation induced by Y881F substitution in the gBcyt ITIM impairs VZV replication and plaque size in vitro. (A) Replication of pOka and the three gBcyt tyrosine mutants as determined by plaque assay. (B) Box and whisker (10–90% percentile) plots of plaques sizes measured (n = 30) in melanoma cells infected with pOka and the three gBcyt mutants. Black dots are outliers in the datasets. (A and B) Statistical differences between pOka and gB-Y881F (red asterisks) or gB-Y881/920F (blue asterisks) were determined by one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001. SEM is indicated. (C) Immunohistochemical staining of plaques in melanoma cells infected with pOka and gB-Y881F, gB-Y920F, and gB-Y881/920F. (Scale bars, 1 mm.)
Y881F Substitution in the gBcyt ITIM Does Not Affect VZV Particle Formation or Egress.
Typical VZV particles were observed in ultrastructure studies of melanoma cells infected with pOka or gB-Y881F at 48 hpi (Fig. S5A). Virus particles were present along cell membranes, and crystalline arrays of nucleocapsids were frequently identified. Although gB contributes to nuclear egress of HSV virions (28), gB-Y881F particles did not accumulate in the perinuclear spaces and nucleocapsids undergoing primary envelopment were readily observed, showing that gBcyt ITIM phosphorylation status did not affect nuclear egress (Fig. S5B). The morphologies of pOka and gB-Y881F virions were similar, but fewer gB-Y881F particles were observed at the periphery of syncytia, which was attributable to TGN sequestration at the center of these very large syncytia (Fig. 4 and Fig. S4).
Enhanced Syncytia Formation Associated with the Y881F ITIM Substitution Severely Impairs VZV Pathogenesis in Skin.
Human skin xenografts were inoculated with the three gBcyt mutant viruses to determine whether dysregulation of syncytia formation affected VZV virulence in vivo. The gB-Y920F mutant, which had a replication phenotype similar to pOka in vitro, was recovered at 10 and 21 dpi with titers similar to those in pOka-infected xenografts (Fig. 6A). Strikingly, infectious virus was not recovered from any xenografts inoculated with the gB-Y881F mutant at 10 or 21 dpi, and only one xenograft inoculated with the Y881/920F mutant was positive at 21 dpi. To further evaluate gB-Y881F and gB-Y881/920F replication, VZV genome copies per nanogram of human DNA in each xenograft were quantified by quantitative PCR. VZV genome copies increased from 10 to 21 dpi in those infected with pOka and gB-Y920F (Fig. 6B). VZV genomes were also detectable in gB-Y881F– and Y881/920F-infected xenografts with a slight increase in frequency from 10 to 21 dpi, but VZV genome copies were significantly reduced compared with pOka.
Fig. 6.

Enhanced syncytia formation triggered by Y881F substitution in the gBcyt ITIM severely impairs VZV infection and spread in human skin xenografts in vivo. (A) Viral titers in skin xenografts at 10 and 21 dpi with pOka, gB-Y881F, gB-Y920F, or gB-Y881/920F. The frequency of virus-positive xenografts/number inoculated is indicated. (B) Viral genome copies/ng human DNA at 10 and 21 dpi. The frequency of VZV DNA-positive xenografts/number inoculated is indicated. (A and B) Statistical differences were evaluated by two-way ANOVA. *P < 0.05, ***P < 0.001. SEM is indicated. (C and D) Histopathology revealed by composite images of hematoxylin and eosin-stained 5-µm sections of infected skin xenografts at 21 dpi. Dotted lines outline the extent of virus penetration. Black boxes indicate where confocal microscopy images were captured on serial sections. Virus titers and genome copies/ng human DNA for each implant are indicated (red) in each panel. (E and F) Confocal microscopy of pOka- and Y881/920F-infected skin xenografts (21 dpi) stained for gE (cyan), capsid protein ORF23 (pink), TGN46 (brown), and nuclei (violet). Black boxes (Left) are shown in higher-magnification images (Right). (Scale bars, 100 μm.)
Typical pOka infection with penetration from the epidermis across the basal layer into the dermis was evident in hematoxylin and eosin-stained sections of skin xenografts (Fig. 6C). Replication was extensive in all cell types, especially around hair follicles, as demonstrated by the detection of ORF23 capsid protein and gE by confocal microscopy (Fig. 6E). In contrast, gB-Y881/920F infection was restricted to the epidermal layer in the only xenograft that yielded virus at 21 dpi (Fig. 6D). Whereas pOka forms moderate numbers of syncytia in the epidermis and dermis, this xenograft exhibited an extensive network of syncytia in the most superficial skin layer; gE staining outlined fractal-like structures containing nuclei positive for ORF23 capsid protein (Fig. 6E). Very small foci of fused infected cells were detected by confocal microscopy in the gB-Y881F–infected xenograft with high levels of VZV DNA (3.0 log10 genome copies/ng human DNA) but absent virus titer (Fig. S6A). In contrast, the histopathology of gB-Y920F infection resembled pOka (Fig. S6B), which was consistent with similar viral titers and genome copies in xenografts infected with these viruses (Fig. S6). Thus, regulation of syncytia formation by the capacity to phosphorylate the gBcyt ITIM through its tyrosine is essential for characteristic VZV skin pathogenesis.
Discussion
The present study identified an ITIM in the VZV gBcyt that regulates cell fusion and virus-induced syncytia formation, which is necessary for VZV pathogenesis in differentiated human skin in vivo. This process was disrupted by the Y881F substitution in the ITIM to prevent its phosphorylation, enhancing the cell–cell fusion triggered by gB/gH–gL, but did not affect the critical process of gB endocytosis. Dysregulation of ITIM function was also evident from aggressive syncytia formation by mutant viruses carrying the Y881F substitution, which resulted in reduced viral replication in vitro and severely impaired infection of skin xenografts in vivo. The Y920F mutation in the gBcyt did not affect replication or pathogenesis, indicating the specific role of the Y881 tyrosine within the ITIM in modulating cell–cell fusion.
The high degree of conservation of the gBcyt ITIM in the subfamily Alphaherpesvirinae implies that this mechanism is evolutionarily maintained, because failure to control the fusogenic properties of gB dramatically restricts viral propagation in the host. Similar to the severely deficient spread of VZV along with exaggerated syncytia formation in skin xenografts caused by the gBcyt ITIM mutation, HSV-1 gB syncytial mutants have also exhibited decreased virulence (29, 30). Whereas many HSV virions are released into extracellular spaces, VZV is highly cell-associated and infected cells must be in direct proximity to target cells (2). Therefore, less efficient production of progeny virions caused by aggressive syncytia formation would be expected to have a more severe effect on VZV pathogenesis than is observed with HSV syncytial mutants.
We propose that modulating the fusogenic potential of the herpesvirus gB/gH–gL complex is required to restrict cell–cell fusion for the vital process of glycoprotein trafficking to the cell surface, endocytosis, and TGN recruitment during secondary envelopment. Although syncytia formation is a hallmark of VZV infection, effective gBcyt control of this gB/gH–gL–dependent process is necessary to prevent extensive cytoskeletal rearrangement where endosomes and TGN become sequestered within a densely packed sphere of nuclei. Importantly, rampant syncytia formation, restricting normal herpes virion egress via the cytoskeletal networks, rendered VZV essentially avirulent in skin.
Although prior studies did not identify the conserved alphaherpesvirus gBcyt ITIM, residues within or immediately adjacent to this motif in HSV 1 and 2, 848IRYMAL854, constitute one of the two “hot spots” for spontaneous or engineered substitutions that confer a syncytial phenotype (15). Mutations in this hot spot could deform the HSV gBcyt to inhibit tyrosine phosphorylation, resulting in a “syn” phenotype similar to the exaggerated syncytia caused by the Y881F substitution in the gBcyt ITIM. Recent investigations show that the HSV-2 gBcyt forms a protease-resistant core situated around this conserved ITIM in the presence of lipid membranes (20). The revised conformation of two independent α-helices on either side of the ITIM would leave the tyrosine prominently exposed to serve as a substrate for kinases similar to the VZV gBcyt. It has been proposed that the HSV gBcyt membrane association in its native conformation restricts fusogenic activity (20). Combined with our findings, these studies strongly support the conclusion that the ITIM is a critical factor in gBcyt-dependent fusogenicity. We propose that when ITIM phosphorylation is disrupted, membrane association of the gBcyt is perturbed and fusion progresses. These observations are consistent with the concept of an “on/off” mechanism for regulating gBcyt-dependent fusion, suggesting that cell signaling pathways that alter gBcyt ITIM phosphorylation status during herpesvirus replication warrant further study for their contribution to cell–cell fusion.
To date, platelet endothelial cell adhesion molecule 1 (PECAM-1) is the only ITIM-containing molecule for which the cytoplasmic domain structure has been determined at atomic resolution (31). PECAM-1 is a 130-kDa type I transmembrane glycoprotein with two ITIMs in the cytoplasmic domain. Similar to the herpesvirus gBcyt (32), the PECAM-1 cytoplasmic domain is disordered in aqueous solution but forms stable helices in the presence of a cell-membrane mimetic. Membrane anchoring is characteristic of functional ITIM-containing proteins. The molecular dynamics of PECAM-1 membrane interactions rely on sequential phosphorylation of serine residues at the C terminus, destabilizing the net positive charge and membrane association of the α-helix and exposing the tyrosine residue in the ITIM to tyrosine kinases. The VZV gBcyt α-helix that contains the ITIM is positively charged (pI 9.4), which fits well with predicted membrane binding, and it is plausible that phosphorylation of serine residues 886 and 893 would also expose the tyrosine in the gBcyt ITIM. Whether the predicted structure is the native form of the ITIM α-helix and how the molecular dynamics of the gBcyt regulates fusion is unknown but, for VZV, tyrosine phosphorylation at Y881 discourages cell–cell membrane mixing that is otherwise induced by gB in combination with gH–gL.
Phenylalanine substitution in the VZV gBcyt was chosen to avoid predicted structural effects and the disruption of endocytosis and ER–Golgi transport known to be caused by tyrosine-to-alanine substitutions in the YXXΦ motifs in alphaherpesviruses (33, 34). Phenylalanine can functionally replace tyrosine in endocytosis of cell-surface receptors (35). However, we investigated whether removal of the hydroxyl moiety by the Y881F substitution would adversely affect the gB structure and thereby alter its fusogenic function. The precursor protein of VZV gB becomes extensively modified by posttranslational addition of N- and O-linked glycosylation along with sialation (36). None of the gBcyt substitutions tested, with the exception of L884G, affected the molecular mass of gB, signifying that posttranslationally added glycomoieties were present and transport from the ER to the Golgi was preserved. In addition, gB cell-surface expression, endocytosis, and virus particle formation were unaffected by the Y881F or Y881/920F substitutions. Therefore, gBcyt structure was unlikely to have deformed sufficiently to generate the exaggerated cell-fusion phenotypes associated with these phenylalanine substitutions. Conversely, structural changes could result from gBcyt truncations or natural or engineered mutations described in previous herpesvirus studies, altering the gBcyt conformation or hydrophobicity and, subsequently, its attributes for membrane association (20, 23, 24). Although effects on the gB ectodomain structure cannot be discounted, studies of HSV-2 gB based on reactivity with monoclonal antibodies indicate that the ectodomain is not distorted in gBcyt mutants with syncytial phenotypes (20).
To date, regulation through ITIMs has been predominantly linked to attenuation of immunological signaling processes (25), but recent studies have demonstrated that ITIM-containing transmembrane proteins have alternative functions (27, 37). The dendritic cell-specific transmembrane protein (DC-STAMP) is a pertinent example in the context of our observations about gBcyt, because it is required for cell–cell fusion during osteoclastogenesis (37). DC-STAMP inhibition prevents the formation of multinucleated osteoclasts in vitro. Apart from a few exceptions, including osteoclasts, macrophages, myoblasts, and placental trophoblasts, mammalian cells are programmed to maintain plasma membrane separation in differentiated tissues after embryogenesis. Thus, we speculate that conserved cellular mechanisms exist between ITIM-dependent signaling pathways triggered during the exceptional circumstances of mammalian cell–cell fusion and those that regulate fusion through the gBcyt in alphaherpesviruses.
Identification and evidence for the significance of the VZV gBcyt ITIM phosphorylation status in the present study, not previously reported for alphaherpesvirus gB homologs, support the hypothesis that the gBcyt regulates mechanisms of cell–cell fusion. ITIMs are becoming widely recognized in molecules not linked to immune cell function in many organisms (25). Therefore, it is clear that unexplored regulatory activity exists for these motifs. Defining the functional relationship between syncytia formation and the VZV gBcyt ITIM provides a valuable insight into alphaherpesvirus mechanisms that modulate cell fusion during infection to enable effective virus spread and pathogenesis.
Materials and Methods
Detailed methods are provided in SI Materials and Methods. These describe cells, viruses, expression vectors, cell–cell fusion, generation of recombinant viruses, electron microscopy, laser-scanning confocal microscopy, analysis of in vitro replication, and in vivo pathogenesis as well as statistical methods. Additional research materials and assays include VZV mutants that carry substitutions in the gBcyt and gB mutant expression constructs plus a flow cytometry-based endocytosis assay.
Supplementary Material
Acknowledgments
We acknowledge the following colleagues for providing reagents: Tadahiro Suenaga and Hisashi Arase (Osaka University), Yasuko Mori (National Institute of Biomedical Innovation, Osaka and Kobe University Graduate School of Medicine), Klaus Osterrieder (Freie Universität), Charles Grose (University of Iowa), and Juan Rivera [National Institutes of Health (NIH)]. Funding was provided by NIH Grants AI20459 and AI102546.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1216985110/-/DCSupplemental.
References
- 1.Ku CC, et al. Varicella-zoster virus transfer to skin by T cells and modulation of viral replication by epidermal cell interferon-alpha. J Exp Med. 2004;200(7):917–925. doi: 10.1084/jem.20040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen JI, Straus SE, Arvin A. Varicella-zoster virus replication, pathogenesis and management. In: Fields BN, Knipe DM, Howley PM, editors. Fields’ Virology. 5th Ed. Vol 2. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2007. pp. 2773–2818. [Google Scholar]
- 3.Hoggan MD, Roizman B, Roane PR., Jr Further studies of variants of herpes simplex virus that produce syncytia or pocklike lesions in cell cultures. Am J Epidemiol. 1961;73(1):114–122. doi: 10.1093/oxfordjournals.aje.a120162. [DOI] [PubMed] [Google Scholar]
- 4.Lamb RA, Parks GD. Paramyxoviridae: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fields’ Virology. 5th Ed. Vol 1. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2007. pp. 1449–1496. [Google Scholar]
- 5.Lifson JD, et al. Induction of CD4-dependent cell fusion by the HTLV-III/LAV envelope glycoprotein. Nature. 1986;323(6090):725–728. doi: 10.1038/323725a0. [DOI] [PubMed] [Google Scholar]
- 6.Esiri MM, Tomlinson AH. Herpes zoster. Demonstration of virus in trigeminal nerve and ganglion by immunofluorescence and electron microscopy. J Neurol Sci. 1972;15(1):35–48. doi: 10.1016/0022-510x(72)90120-7. [DOI] [PubMed] [Google Scholar]
- 7.Reichelt M, Zerboni L, Arvin AM. Mechanisms of varicella-zoster virus neuropathogenesis in human dorsal root ganglia. J Virol. 2008;82(8):3971–3983. doi: 10.1128/JVI.02592-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moffat JF, Stein MD, Kaneshima H, Arvin AM. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J Virol. 1995;69(9):5236–5242. doi: 10.1128/jvi.69.9.5236-5242.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat Rev Microbiol. 2011;9(5):369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vleck SE, et al. Structure-function analysis of varicella-zoster virus glycoprotein H identifies domain-specific roles for fusion and skin tropism. Proc Natl Acad Sci USA. 2011;108(45):18412–18417. doi: 10.1073/pnas.1111333108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pertel PE. Human herpesvirus 8 glycoprotein B (gB), gH, and gL can mediate cell fusion. J Virol. 2002;76(9):4390–4400. doi: 10.1128/JVI.76.9.4390-4400.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Backovic M, Longnecker R, Jardetzky TS. Structure of a trimeric variant of the Epstein–Barr virus glycoprotein B. Proc Natl Acad Sci USA. 2009;106(8):2880–2885. doi: 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heldwein EE, et al. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313(5784):217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 14.Oliver SL, et al. Mutagenesis of varicella-zoster virus glycoprotein B: Putative fusion loop residues are essential for viral replication, and the furin cleavage motif contributes to pathogenesis in skin tissue in vivo. J Virol. 2009;83(15):7495–7506. doi: 10.1128/JVI.00400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gage PJ, Levine M, Glorioso JC. Syncytium-inducing mutations localize to two discrete regions within the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B. J Virol. 1993;67(4):2191–2201. doi: 10.1128/jvi.67.4.2191-2201.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruel N, Zago A, Spear PG. Alanine substitution of conserved residues in the cytoplasmic tail of herpes simplex virus gB can enhance or abolish cell fusion activity and viral entry. Virology. 2006;346(1):229–237. doi: 10.1016/j.virol.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Haan KM, Lee SK, Longnecker R. Different functional domains in the cytoplasmic tail of glycoprotein B are involved in Epstein-Barr virus-induced membrane fusion. Virology. 2001;290(1):106–114. doi: 10.1006/viro.2001.1141. [DOI] [PubMed] [Google Scholar]
- 18.Beitia Ortiz de Zarate I, Cantero-Aguilar L, Longo M, Berlioz-Torrent C, Rozenberg F. Contribution of endocytic motifs in the cytoplasmic tail of herpes simplex virus type 1 glycoprotein B to virus replication and cell-cell fusion. J Virol. 2007;81(24):13889–13903. doi: 10.1128/JVI.01231-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Minnebruggen G, Favoreel HW, Nauwynck HJ. Internalization of pseudorabies virus glycoprotein B is mediated by an interaction between the YQRL motif in its cytoplasmic domain and the clathrin-associated AP-2 adaptor complex. J Virol. 2004;78(16):8852–8859. doi: 10.1128/JVI.78.16.8852-8859.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silverman JL, Greene NG, King DS, Heldwein EE. Membrane requirement for folding of the herpes simplex virus 1 gB cytodomain suggests a unique mechanism of fusion regulation. J Virol. 2012;86(15):8171–8184. doi: 10.1128/JVI.00932-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pellett PE, Roizman B. The Herpesviridae family: A brief introduction. In: Fields BN, Knipe DM, Howley PM, editors. Fields’ Virology. 5th Ed. Vol 2. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2007. pp. 2479–2499. [Google Scholar]
- 22.Fan Z, et al. Truncation of herpes simplex virus type 2 glycoprotein B increases its cell surface expression and activity in cell-cell fusion, but these properties are unrelated. J Virol. 2002;76(18):9271–9283. doi: 10.1128/JVI.76.18.9271-9283.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heineman TC, Krudwig N, Hall SL. Cytoplasmic domain signal sequences that mediate transport of varicella-zoster virus gB from the endoplasmic reticulum to the Golgi. J Virol. 2000;74(20):9421–9430. doi: 10.1128/jvi.74.20.9421-9430.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heineman TC, Hall SL. Role of the varicella-zoster virus gB cytoplasmic domain in gB transport and viral egress. J Virol. 2002;76(2):591–599. doi: 10.1128/JVI.76.2.591-599.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daëron M, Jaeger S, Du Pasquier L, Vivier E. Immunoreceptor tyrosine-based inhibition motifs: A quest in the past and future. Immunol Rev. 2008;224(1):11–43. doi: 10.1111/j.1600-065X.2008.00666.x. [DOI] [PubMed] [Google Scholar]
- 26.Barrow AD, Trowsdale J. You say ITAM and I say ITIM, let’s call the whole thing off: The ambiguity of immunoreceptor signalling. Eur J Immunol. 2006;36(7):1646–1653. doi: 10.1002/eji.200636195. [DOI] [PubMed] [Google Scholar]
- 27.O’Brien CD, Cao G, Makrigiannakis A, DeLisser HM. Role of immunoreceptor tyrosine-based inhibitory motifs of PECAM-1 in PECAM-1-dependent cell migration. Am J Physiol Cell Physiol. 2004;287(4):C1103–C1113. doi: 10.1152/ajpcell.00573.2003. [DOI] [PubMed] [Google Scholar]
- 28.Farnsworth A, et al. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc Natl Acad Sci USA. 2007;104(24):10187–10192. doi: 10.1073/pnas.0703790104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodman JL, Engel JP. Altered pathogenesis in herpes simplex virus type 1 infection due to a syncytial mutation mapping to the carboxy terminus of glycoprotein B. J Virol. 1991;65(4):1770–1778. doi: 10.1128/jvi.65.4.1770-1778.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engel JP, Boyer EP, Goodman JL. Two novel single amino acid syncytial mutations in the carboxy terminus of glycoprotein B of herpes simplex virus type 1 confer a unique pathogenic phenotype. Virology. 1993;192(1):112–120. doi: 10.1006/viro.1993.1013. [DOI] [PubMed] [Google Scholar]
- 31.Paddock C, et al. Residues within a lipid-associated segment of the PECAM-1 cytoplasmic domain are susceptible to inducible, sequential phosphorylation. Blood. 2011;117(22):6012–6023. doi: 10.1182/blood-2010-11-317867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chowdary TK, Heldwein EE. Syncytial phenotype of C-terminally truncated herpes simplex virus type 1 gB is associated with diminished membrane interactions. J Virol. 2010;84(10):4923–4935. doi: 10.1128/JVI.00206-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Favoreel HW, Van Minnebruggen G, Nauwynck HJ, Enquist LW, Pensaert MB. A tyrosine-based motif in the cytoplasmic tail of pseudorabies virus glycoprotein B is important for both antibody-induced internalization of viral glycoproteins and efficient cell-to-cell spread. J Virol. 2002;76(13):6845–6851. doi: 10.1128/JVI.76.13.6845-6851.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imai T, et al. Role of the herpes simplex virus 1 Us3 kinase phosphorylation site and endocytosis motifs in the intracellular transport and neurovirulence of envelope glycoprotein B. J Virol. 2011;85(10):5003–5015. doi: 10.1128/JVI.02314-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trowbridge IS, Collawn JF, Hopkins CR. Signal-dependent membrane protein trafficking in the endocytic pathway. Annu Rev Cell Biol. 1993;9:129–161. doi: 10.1146/annurev.cb.09.110193.001021. [DOI] [PubMed] [Google Scholar]
- 36.Grose C. Glycoproteins encoded by varicella-zoster virus: Biosynthesis, phosphorylation, and intracellular trafficking. Annu Rev Microbiol. 1990;44:59–80. doi: 10.1146/annurev.mi.44.100190.000423. [DOI] [PubMed] [Google Scholar]
- 37.Chiu YH, et al. Regulation of human osteoclast development by dendritic cell-specific transmembrane protein (DC-STAMP) J Bone Miner Res. 2012;27(1):79–92. doi: 10.1002/jbmr.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.