

Abstract
Germinal centers (GC) are specialized microenvironments that generate high-affinity antibody-forming (AFCs) and memory B cells. Many B cells undergo apoptosis during B-cell clonal selection in GC. Although the factors that regulate the AFC and GC responses are not precisely understood, it is widely believed that dysregulated AFCs and GCs contribute to autoimmunity. The Mer receptor tyrosine kinase (MerTK or Mer) facilitates macrophage clearance of apoptotic cells. The TAM (Tyro-3, Axl, and Mer) receptors, including Mer, suppress Toll-like receptors (TLRs) and cytokine-mediated inflammatory responses. We report here that tingible body macrophages (TBMϕs) in GC express Mer. Compared to C57BL/6 (B6) controls, Mer deficient (Mer−/−) mice had significantly higher AFC, GC and Th1-skewed antibody (IgG2c) responses against the T-dependent Ag (4-hydroxy-3-nitrophenyl) acetyl (NP)-chicken gamma globulin (CGG). Mer−/− mice had significantly higher percentage of GC B cells on days 9, 14 and 21 post-immunization compared to B6 controls. Significantly increased numbers of apoptotic cells accumulated in Mer−/− GCs than in B6 GCs, while the number of TBMϕs remained similar in both strains. Our data are the first to demonstrate a critical role for Mer in GC apoptotic cell clearance by TBMϕs and have interesting implications for Mer in the regulation of B cell tolerance operative in the AFC and GC pathways.
Keywords: MerTK, germinal center, follicular DC, B cells
Introduction
In a T cell–dependent (TD) antigenic response, B cells either differentiate into short-lived (primary) antibody forming cells (AFCs) or become precursors destined to form germinal centers (GC). GCs are dynamic and specialized microenvironments formed in the follicles of secondary lymphoid organs by hyper-proliferative GC B cells (1). During the rapid proliferation phase of the GC response, B cell antigen receptor (BCR) or immunoglobulin (Ig) variable (V) region genes undergo somatic hypermutation (SHM) (1–3). This process generates a diverse repertoire of de novo B cell mutants with varied degrees of affinity to antigen. B cell mutants with high affinity for foreign antigen are positively selected into the long-lived AFC or memory compartments (4–8). B-cell mutants with low affinity for antigen and B cells with autoreactivity are negatively selected (9–11). Despite these insights regarding the pivotal role of GCs in TD B cell responses and the establishment of long-term humoral immunity, the factors that regulate the activation and proliferation of B cells in GCs are still poorly understood.
Immune cells are maintained in the periphery via self-renewal of mature cell populations. During this process of replenishment, in order to maintain tissue homeostasis, unwanted cells are usually purged by apoptosis (12–14). Clonal selection of high-affinity B cells in GCs also results in death of a substantial number of B-cell clones including autoreactive cells (9, 10), due to insufficient affinity to foreign antigen and to lack of sufficient survival signals. Clearance of these apoptotic cells may be essential to maintain peripheral tolerance. Dendritic cells (DCs) and macrophages (Mϕ) phagocytose these apoptotic cells and remove them from the lymphoid tissue (15, 16). Data suggest that it is the tingible body macrophages (TBMϕs) which are the most important population of phagocytic cells that clear apoptotic cells in GCs (17–19), although DCs and other phagocytic cells may be important as well.
After phagocytosis of apoptotic cells, DCs and macrophages utilize tolerogenic pathways against intracellular antigens released from the apoptotic cells (15, 20, 21). Impaired uptake of apoptotic cells by macrophages has been described in human SLE patients (22, 23) and in murine models of SLE-like disease (24–26). Disruption of this process may lead to a break in tolerance by activating inflammatory cytokine pathways which may cause autoimmune disease. A number of dual-function bridging molecules including milk fat globule EGF factor 8 (Mfge-8), growth arrest specific 6 (Gas-6), protein S and complement factor C1q facilitate the engulfment of apoptotic cells by binding both ligands exposed on the surface of apoptotic cells and receptors expressed on phagocytes. Among these molecules, Mfge-8 has been shown to play a critical role in the recognition of apoptotic cells during the process of macrophage phagocytosis (18). The receptors that mediate the phagocytosis include TAM family receptors (TAM: Tyro-3, Axl, and Mer), αvβ3-integrin, Tim4 and CD36 (27–31). TAM receptors have been shown to primarily use Gas-6 and protein S (32–34).
Mer receptor tyrosine kinase (MerTK or Mer) belongs to the Tyro-3 subfamily of TAM receptors (33). The receptors in this subfamily have important immunoregulatory roles. While TAM double (Tyro3−/−Axl−/−, Axl−/−Mer−/− and Tyro3−/−Mer−/−) and triple (Tyro3−/−Axl−/−Mer−/−) mutants suffer from more severe disease than the single mutants (35, 36), mice lacking Mer alone (Mer−/−) develop lupus-like autoimmunity (26). In addition, expression of Mer on phagocytes, i.e. Mϕs and DCs has been described to facilitate the engulfment of apoptotic cells (34, 37, 38).
We recently observed an enhanced marginal zone (MZ) B cell response to type II T-independent antigen in Mer−/− mice (W.H. S, unpublished). DNA-specific immunoglobulin heavy-chain transgenic (3H9) B cell tolerance was broken when Mer−/− mice were bred onto 3H9 knock-in mice (W.H. S, unpublished). Furthermore, strains of mice that typically develop autoimmune SLE-like illness (NZB/W F1 and MRL/lpr) and autoimmune diabetes (non-obese diabetic, NOD mice) exhibit spontaneous GC formation in the spleen by 1–2 months of age in the absence of immunization or infection (39). While Mer function has been implicated in maintaining immune tolerance, including T cells (21), the immunoregulatory role of Mer in peripheral B cell tolerance is not well defined. To understand the role of Mer in regulation of B-cell responses to exogenous antigen, we performed an in-depth analysis of the immune response of Mer−/− mice to the T-dependent antigen NP-CGG. We observed enhanced primary AFC, GC and antibody responses in Mer−/− mice compared to B6 controls. We also found that the expression of Mer on tingible body macrophages was critical for the clearance of apoptotic cells in GCs. These data indicate a vital role of Mer in clearing apoptotic cells in GCs by tingible body macrophages which may, in turn, maintain peripheral AFC and GC tolerance.
Materials and Methods
Mice
Mice deficient in Mer receptor tyrosine kinase (Mer−/−) were originally generated on a F1 hybrid background of C57BL/6 (B6) and 129 mice (40). Subsequently, these were backcrossed onto the B6 background for 10 generations. B6 WT control mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and then bred in house. All mice were 7–9 weeks old when used in experiments. All experimental procedures performed on these animals were conducted according to the guidelines of our Institutional Animal Care and Use Committee.
Antibodies and other reagents
Antibodies and other reagents used for flow cytometry and immunohistology included: FITC-GL7; alexafluor 647-anti-CD3; streptavidin-CyChrome; PE-anti-CD23 (B3B4); purified FDC-M1; FITC-anti-FcγRII/III (2.4G2, Applied Biosystems-BD Pharmingen, San Diego, CA); FITC-MOMA-1; RPE (red phycoerythrin) and alexafluor 647-anti-CD68 (AbD Serotec, Raleigh, NC); PE, FITC and biotin-anti-B220 (RA3-6B2); PE-anti-CD3; FITC and PE-anti-CD21/35 (4E3); biotin-anti-IgD (11–26) (eBioscience, San Diego, CA ); streptavidin (SA)-PE; SA-alexafluor 633; SA-alexafluor 488; biotin-anti-mouse kappa; alkaline phosphatase (AP)- anti-mouse IgG; biotin-anti-mouse IgG1 (Molecular probes, Eugene, OR); FITC- and HRP- peanut-agglutinin (PNA, Sigma Aldrich, St Louis, LO); biotin-anti-mouse IgM; biotin-mouse anti-rat; FITC-donkey anti-mouse IgM (Jackson Immunoresearch Laboratories, West Grove, PA). Alkaline phosphatase-anti-mouse IgG2c and biotin-anti-mouse IgG2b (affinity-purified goat polyclonal, Southern Biotechnology Associates, Birmingham, AL). Biotin-anti-mouse Mer (R&D, Minneapolis, MN).
Immunization procedure
T-dependent Ag (4-hydroxy-3-nitrophenyl) acetyl (NP)16-chicken gamma globulin (CGG) NP16-CGG (Biosearch Technologies, Novato, CA) was precipitated with 10% alum and injected i.p. into Mer−/− and WT mice (100 μg in 200 μl of 1×PBS per mouse). On days 9, 14 and 21-post immunization mouse spleens were removed, frozen in OCT medium and stored at −80°C. Serum samples were collected from these mice on the day (day 9, 14 and 21) animals were sacrificed.
Immunohistology and TUNEL assay
Spleen cryostat sections (5–6 microns) were prepared as described (41). Immunohistology was performed using the Abs listed above. The TUNEL (Terminal deoxynucleotidyl Transferase dUTP Nick End Labeling) assay was performed on the spleen sections using a TUNEL apoptosis detection kit (Millipore, Temecula, CA) following the manufacturer's instructions. The stained sections were analyzed using a fluorescence microscope (Leica microsystems) and images were captured as described (42). The color intensity of the image was slightly enhanced by Photoshop. This manipulation was necessary for better visualization which was carried out consistently between Mer−/− and B6 controls, while maintaining the integrity of the data. The magnification of the image was 100×, 200× or 320× as indicated in the legends. TUNEL+ and CD68+cells were counted by two individuals in randomly picked GCs from four to five B6 and Mer−/− mice.
Flow cytometry
Three and four color flow cytometric analysis was done on cell suspensions prepared from spleens of naïve and immunized mice stained with multiple combinations of the Abs listed above. Biotinylated Abs were detected with streptavidin-CyChrome. Stained cells were analyzed using a Coulter Epics XL/MCL analyzer. Data were analyzed using FlowJo software (Treestar, San Carlos, CA).
ELISpot assays
Splenocyte suspensions from NP-CGG immunized B6 and Mer−/− mice were plated at 1 × 106 cells/well and diluted serially (1:2) in NP11-BSA coated multiscreen 96-well filtration plates (Millipore, Bedford, MA) for 6 hr at 37°C. NP-specific IgM Abs produced by AFCs were detected using biotinylated anti-mouse IgM (Jackson Immunoresearch) and streptavidin (SA)-alkaline phosphatase (Vector Laboratories, Burlingame, CA). NP-specific IgG Abs produced by AFCs were detected using alkaline-phosphatase conjugated IgG (Molecular Probes). Plates were developed using the Vector Blue Alkaline-phosphatase Substrate Kit III (Vector laboratories). ELISpots were counted using a computerized imaging video system (Cellular Technology, Cleveland, OH).
ELISA
NP-specific serum Abs were measured in sera from immunized mice by solid-phase ELISA on 96-well plates (Immulon-4; Thermo Electron) as previously described (41). To measure the total serum Ab titers of different isotypes and subtypes (such as IgM, IgG, IgG1, IgG2c and 2b) ELISA plates were coated with NP11-BSA. Biotinylated antibodies were detected by streptavidin (SA)-alkaline phosphatase (Vector Laboratories, Burlingame, CA). The plates were developed by the PNPP (p-Nitrophenyl Phosphate, Disodium Salt) substrates for alkaline phosphatase. Serum samples were first diluted (1/100) in PBS and then subsequently threefold serial dilution was carried out for each sample. The dilution factor for each sample was generated in a logarithmic scale via the software named “Origin” based on the different OD values of 0.8, 0.9 or 1.0 (at 405 nm) set for different isotype-specific ELISA. The OD values of 0.8, 0.9 or 1.0 were determined based on the linear distribution of most of the samples in any given ELISA. In this way, a dilution factor of 400 for a particular sample with an OD value of 1.0 would indicate that this particular serum sample needed to be diluted 400 times to obtain an OD value of 1.0 at 405 nm. In contrast, a serum sample with a dilution factor of 150 needed to be diluted 150 times to obtain the same OD value. Therefore, the higher the dilution factor in an individual mouse the higher the Ab titers for that particular mouse.
Statistical analysis
Statistical analysis was done using Student's t-test. P values less than 0.05 (<0.05) were considered significant.
Results
Primary development of B cells is not overtly affected in Mer deficient (Mer−/−) mice
To study the role of Mer in regulating AFC, GC and antibody responses, first we evaluated whether primary development of B cells was altered in the absence of Mer using Mer deficient mice (designated Mer−/−, 40). Mer−/− mice used in the current studies were backcrossed for 10 generations onto the C57BL/6J (B6) background. By performing flow cytometric analysis on splenocytes obtained from age (8–10 wk old) and sex-matched naïve mice, we found that the total number and percentage of B220+ cells in Mer−/− mice were comparable to that observed in B6 controls (Fig. 1A). Staining with the anti-CD93 mAb specific for the C1qRp molecule expressed on the immature and transitional stages of follicular B cell development, we analyzed mature (B220+ CD93neg) and immature (B220+ CD93+) B cell populations (Fig. 1B, top panel) in the spleen. The ratios of B cells expressing and lacking this marker were comparable in both strains (data not shown).
Figure 1. Primary development of B cells in the presence or absence of Mer.
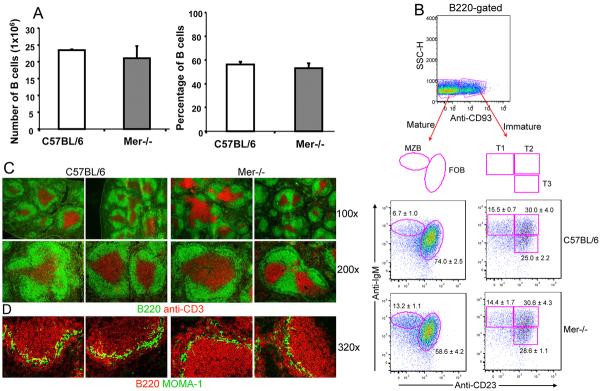
Panel (A–B): Quantitative and four color flow cytometric analysis was performed on spleen cells obtained from B6 and Mer−/− mice after staining with B220, anti-CD93, anti-IgM and anti-CD23. (A) Total number of B cells (left panel) and the percentage of B220+ cells (right panel) are shown in C57BL/6 (open bar) and Mer−/− (shaded bar) sex matched 8–10 wk old mice. (B) The percentage of mature (B220+CD93neg) and immature (B220+CD93neg) B cells are shown in the top panel. Subdivision of mature and immature B cells into marginal zone (MZB), follicular (FOB), and transitional type 1 (T1), T2 and T3 populations are shown by oval and rectangular gates, respectively, in the lower panels. The percentage of each B cell population in the spleens of B6 (middle two panels) and Mer−/− (bottom two panels) mice is indicated next to each gate. (C) Spleen sections obtained from naïve mice of the indicated genotypes were stained with B220 (green), anti-CD3 (red) and images captured by fluorescence microscopy. Original magnification of images was 100× (upper row) and 200× (lower row). (D) Similar analysis described in the legend to Figure 1C was done in which spleen sections were stained with B220 (red) and MOMA-1 (green). Original magnification of images was 320×. All data are representative of at least three mice per group.
Further subdivision of the CD93+ population using peripheral B cell lineage markers CD23 and surface IgM (sIgM) revealed about a two-fold increase in the percentage of CD93negCD23neg IgMhigh marginal zone (MZB) and on average a 20% decrease in the percentage of CD93neg CD23+ IgMlow mature follicular (FOB) B cells in Mer−/− mice (bottom left panel, Fig. 1B) compared to B6 controls (middle left panel, Fig. 1B). Our finding of this increased percentage of MZB cells in Mer−/− mice is consistent with our recent observation (WH.S, unpublished). Similar analysis of the immature (B220+ CD93+) transitional B cell population revealed no differences in the percentage of CD93+CD23neg IgMhigh transitional type 1 (T1), CD93+CD23+IgMhigh T2 and CD93+ CD23+IgMlow T3 subsets (middle and bottom right two panels, Fig. 1B) between B6 and Mer−/− mice.
Additionally, we performed immunohistological analysis of spleen sections obtained from naive mice. By staining with B220 (green) and anti-CD3 (red), we observed no significant differences in the splenic architecture, organization of B (green) and T (red) cell areas in Mer−/−mice (right four panels, Fig. 1C) compared to that observed in B6 controls (left four panels, Fig. 1C). As shown in Fig. 1D, next we performed a similar analysis where we stained with B220 (red) and MOMA-1 (green). MOMA-1 stains for metallophillic macrophages located at the border of follicles and the marginal zone. Consistent with the flow cytometry data, we observed an increase in the MZ B cell population outside the MOMA-1 border in Mer−/− mice (right two panels, Fig. 1D) compared to B6 controls (left two panels, Fig. 1D). The surface levels of the activation/co-stimulatory markers CD69, CD80 and CD86 on B cells from 8–10 wks old Mer−/− naïve mice were comparable to age-matched B6 controls (data not shown). These data indicate that Mer deficiency does not overtly alter the development and maturation of B cells.
Enhanced primary (short-lived) AFC responses of mice deficient in Mer
To study whether the absence of Mer led to an enhanced primary AFC (antibody forming cell) response, we immunized Mer−/− mice with the T-dependent (TD) antigen (4-hydroxy-3-nitrophenyl) acetyl (NP)-chicken gamma globulin (CGG) in alum. We used B6 mice as controls. Spleens were harvested, and anti-NP IgM and IgG-producing AFCs were measured by ELISpot assay on days 9, 14 and 21 post-immunization. While IgM-producing AFCs remained similar between B6 and Mer−/− mice for all three time points (Fig. 2A) we found significantly higher NP-specific IgG-producing AFCs on day 14 and 21 after immunization in Mer−/− mice compared to B6 controls (Fig. 2B). A representative image from day 14 of the primary response showing significantly higher number of IgG-producing AFCs in Mer−/− mice compared to B6 controls is shown in Fig. 2C.
Figure 2. Primary IgM and IgG AFC responses in the presence or absence of Mer.

The number of splenic NP-specific IgM (A, upper panel) and IgG (B, lower panel) secreting AFCs were measured by ELISpot assay 9, 14 and 21 days after immunization of B6 and Mer−/−mice with NP-CGG. Each circle represents the number of AFCs per 1×106 splenocytes obtained from an individual mouse. Open and closed circles represent data from B6 and Mer−/− mice, respectively. Horizontal bars represent the average number of AFCs. Statistical analysis was performed by Student's t-test. (C) A representative image of an ELISpot assay obtained on day 14 of the anti-NP response in six B6 (B1–6, left) and six Mer−/− (M1–6, right) mice is shown. As described in Materials and Methods, one million (1×106) splenocytes were plated in the first row and subsequently two-fold serial dilution was carried out for each sample (indicated in the left side of the image as 1, 1/2 and 1/4). These data were obtained from age and sex-matched five to six mice of each genotype.
The anti-NP germinal center (GC) response is augmented in Mer−/− mice
To study the influence of Mer deficiency on the GC response, Mer−/− and B6 control mice were immunized with NP-CGG in alum. Flow cytometry analysis of splenocytes obtained on days 9 and 14 after immunization revealed a significant increase in the percentage of B220+PNA+ GC B cells in Mer−/− mice compared to B6 controls (Fig. 3A). Analogous results were obtained when we used a different GC B cell marker GL7 (data not shown). We next performed semi-quantitative analysis of the number of splenic GCs in Mer−/− and B6 control mice 14 days post-immunization, when mice were shown to have the peak anti-NP GC response. GC sizes were determined by counting the number of PNA+ cell diameters at 100× magnification in the largest GC dimension as we described previously (41, 42). GCs were categorized into three groups: small (10–25 cell diameters), medium (26–39) and large (40 or more). Even though the frequency of GCs in Mer−/− mice was similar to that observed in B6 controls (data not shown) the size of GCs differed. The large and medium GCs at 100× magnification field were increased in Mer−/− mice (shaded bar, Fig. 3B) compared to that observed in B6 controls (open bar, Fig. 3B). In contrast, B6 mice contained more small GCs than medium and large GCs indicating that the size of the GC response in Mer−/− mice was much greater than that observed in B6 controls.
Figure 3. Enhanced GC responses of B cells deficient in Mer.

(A) Splenocytes obtained on days 9 and 14 post-immunization from NP-CGG immunized B6 (open circle) and Mer−/− (closed circle) mice were stained with B220 and the GC B cell marker PNA. The percentage of B220+PNA+ GC B cells is shown. Horizontal bars represent the average percentage of GC B cells. (B) Spleen sections obtained from B6 and Mer−/− mice on day 14 of the NP-CGG response were stained with the GC marker PNA. The number of small, medium and large GCs per 100× magnification field was counted from randomly chosen areas on spleens from B6 (open bar) and Mer−/− (shaded bar) mice. GCs in two randomly chosen fields per mouse spleen were counted. Statistical analysis was performed by Student's t-test. These data were obtained from three to five mice of each genotype. (C) Splenocytes obtained on days 9 (left), 14 (middle) and 21 (right) post-immunization from NP-CGG immunized B6 (open) and Mer−/−(closed) mice were stained with B220 and the GC B cell marker PNA. The percentage of B220+PNA+ GC B cells for each time point is shown. These data were obtained from five to six mice of each genotype.
To examine whether the difference in the GC response between B6 and Mer−/− mice was due to the kinetic shift, we performed an additional experiment in which the GC responses were analyzed on days 9, 14 and 21 post-immunization. Flow cytometry analysis of the GC response on these three time points showed significantly higher number of B220+PNA+ GC B cells in Mer−/− mice than that observed in B6 controls (Fig. 3C).
Elevated Th1-skewed IgG2 Ab responses in Mer−/− mice
To evaluate whether the augmented anti-NP AFC and GC responses in Mer−/− mice led to an increased titer of serum Abs, we measured the levels of anti-NP Abs, 14 days after immunization with TD-Ag NP-CGG, in sera from mice described in Fig. 3A–B. The anti-NP total IgM and IgG (Fig. 4A) serum levels were significantly higher in Mer−/− mice (●) than B6 controls (◯). The T-dependent anti-NP response predominantly generates Abs of IgG1 isotype in B6 mice (43). To our surprise, we did not find any difference in IgG1 Ab response (Fig. 4A, middle). Interestingly, we found significantly higher titers of IgG2, especially IgG2c (Fig. 4A, right most) in Mer−/− mice (●) compared to B6 controls (◯).
Figure 4. Elevated levels of T-dependent (TD) Ab responses in Mer−/− mice.

(A) TD-Ag induced anti-NP total IgM, IgG, IgG1, IgG2b and IgG2c titers were measured by ELISA in B6 (◯) and Mer−/− (●) serum samples obtained on day 14 post-immunization of these mice with NP-CGG. (B) The kinetics of these Ab responses was obtained by measuring Ab levels in sera from B6 (◯) and Mer−/− (●) mice on day 9 (top), 14 (middle) and 21 (bottom). These data were obtained from five to six mice of each genotype on each time point.
Next we performed the kinetics of the anti-NP Ab responses by measuring the levels of NP-specific Abs in sera obtained from B6 and Mer−/− mice on days 9, 14 and 21 post-immunization with TD-Ag NP-CGG. While we did not observe any significant difference in anti-NP total IgM, IgG and IgG1 serum Ab levels, interestingly, we found significantly higher titers of IgG2, especially IgG2c in Mer−/− mice compared to B6 controls on days 9 (Fig. 4B, top), 14 (Fig. 4B, middle) and 21 (Fig. 4B, bottom).
Mer expressing cells are localized in both the red and white pulp areas of spleen
Having observed enhanced AFC, GC and antibody responses in Mer−/− mice, we next examined Mer expression on spleen cells that might regulate AFC, GC and antibody responses. We performed a detailed analysis of the anatomical location of cells expressing Mer in the spleen by performing three color immunofluorescence staining of spleen sections obtained from naïve B6 mice (top row, Fig. 5). By staining with MOMA-1 (green) and mAbs specific for Mer (red) and CD3 (blue), we showed that cells expressing high levels of Mer were localized in the red pulp areas of the spleen outside of MOMA-1 border (top row, 2nd and 4th column, Fig. 5). Compared to this population, cells expressing relatively low levels of Mer were localized in the white pulp area, predominantly within the T cell zone (overlay, top row, Fig. 5), while Mer expression was completely abolished in these two areas of spleen in Mer−/− mice (bottom row, Fig. 5). Consistent with previously published data by us (34) and others (35), T and B cells did not appear to express Mer as evidenced by the absence of overlap staining of Mer and CD3 in the T cell zone and the absence of Mer staining on B cells within the follicles inside the MOMA-1 border (top row, Fig. 5).
Figure 5. Localizations of Mer expressing cells in the spleen.
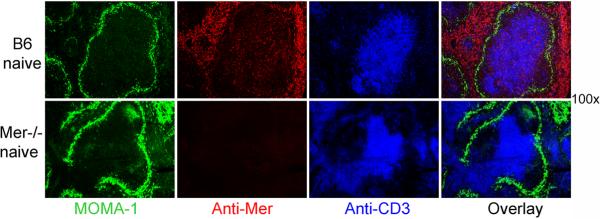
Spleen sections obtained from naïve B6 (top row) and Mer−/− (bottom row) mice were stained with MOMA-1 (green), anti-Mer (red) and anti-CD3 (blue). The images were captured by fluorescence microscopy. Original magnification of images was 100×. These data represent four to five mice of each genotype.
Mer expressing cells within GCs are not follicular dendritic cells (FDCs), and Mer deficiency does not alter the development of the FDC network in GCs
Next, we evaluated whether GCs contained any Mer expressing cells, which may regulate GC responses. Spleen sections, obtained from B6 and Mer−/− mice on day 14 after immunization, were stained with the GC B cell marker PNA (green), anti-Mer (red) and anti-CD3 (blue). While most of the Mer+ cells in the white pulp area were localized within the T cell zone, we also observed Mer+ cells within B6 GCs as evidenced by the red staining within the GC defined by the green area (top row, 2nd from left, Fig. 6A). These Mer+ cells in GCs did not co-localize with CD3+ T cells within GC and PNA+ GC B cells (top row, 4th and 5th from left, Fig 6A).
Figure 6. Mer expressing cells within GCs do not co-localize with FDCs.

(A): Immunohistological analysis of spleen sections, obtained from B6 (top row) and Mer−/− mice (bottom row) on day 14 after NP-CGG immunization, was performed using PNA (green), anti-Mer (red) and anti-CD3 (blue). In the 1st column, PNA+ (green) GC area is shown in dotted red line. In the 2nd column, the localization of Mer expressing cells within the GC marked by the dotted green line is shown. In the 3rd column, the presence of follicular helper T cells within the GC is also shown in a similar green area. In the 4th column, two color overlay image of anti-Mer (red) and anti-CD3 (blue) staining is shown. In the 5th column, the distribution of Mer expressing cells in GCs, T cell zone and in the red pulp areas are shown in the three color overlay image. Arrow indicates the accumulation of Mer+ cells at the border between GC and T cell zone. (B) Similar immunohistology analysis as in Panel A was performed in which two adjacent spleen sections were analyzed. One section was co-stained with anti-CD21/35 (green) and anti-Mer (red) and the other was stained with anti-CD21/35 (green) and FDC marker FDC-M1 (red). (C) Similar analysis as in Panel B was performed in which anti-CD21/35 (green) staining was replaced by 2.4G2 (FcγRII/III). Original magnification of images was 200×. These data were obtained from four to five mice of each genotype.
Follicular dendritic cells (FDCs) are the major stromal elements within GCs that are thought to regulate the GC response. We stained two parallel sections, obtained from B6 and Mer−/− mice on day 14 post-immunization, one with anti-CD21/35 (CR2/CR1, green) highly expressed on FDCs and anti-Mer (red) and the other with anti-CD21/35 and FDC-M1 (4C11). In this way, we showed that while CD21/35 staining overlapped with FDC-M1 (right most panel, Fig. 6B, top) CD21/35high FDC-M1+ FDCs did not express Mer as Mer staining was only restricted to individual cells near FDCs (Fig. 6B, left two panels, top). We performed similar experiments as in Fig. 6B in which we replaced CD21/35 staining with 2.4 G2 (FcγRII/III) also highly expressed on FDCs and obtained similar results (Fig. 6C) as in Fig. 6B. These data together suggest that CD21/35high 2.4G2high FDC-M1+ FDC network in the spleen do not express Mer.
Mer expressing cells in GCs are tingible body macrophages
By staining spleen sections from NP-CGG immunized B6 mice with Abs against a number of myeloid cell markers such as CD11b, CD11c, F4/80, Gr-1 and CD68 we found that CD11c+DCs, and F4/80+ and CD68+ macrophages expressed Mer (Table I). Of these three myeloid cell populations expressing Mer, only CD11c+ DCs and CD68+ tingible body macrophages (TBMϕs) were localized in both the white pulp (WP) and red pulp (RP) areas of the spleen (Table I and Fig. 7). F4/80+ macrophages were only localized in the RP areas of the spleen (Table I). Tingible body macrophages (TBMϕs) are considered to play an important role in regulating germinal center (GC) reaction and tolerance by clearing apoptotic cells (17). We next examined whether Mer expressing cells within GCs were TBMϕs.
Table I.
Localization of Mer-expressing cells in the spleen of immunized mice
| Cell typesa | Mer expression in the white pulp (WP)b | Mer expression in the red pulp (RP)c | Mer expression in the germinal center (GC)d |
|---|---|---|---|
| CD11b+ leukocytes | − | − | − |
| CD11c+ DCs | + | + | − |
| F4/80+ macrophages | − | + | − |
| CD68+ TBMϕs | + | + | + |
| Gr-1+ granulocytes | − | − | − |
| B220+ B cells | − | − | − |
| CD3+ T cells | − | − | − |
Mer expression on different cell types in the spleen tissue of immunized mice was determined by immunohistology in which spleen sections obtained on day 14 post-immunization were co-stained with anti-Mer and different cell-type specific markers.
Localization of Mer expressing cell types in the white pulp (WP) area of the spleen, + and − symbols indicate the presence and absence of a particular cell type expressing Mer
Localization of Mer expressing cell types in the red pulp (RP) area of the spleen
Localization of Mer expressing cell types within the germinal centers formed in the WP area
Figure 7. Tingible body macrophages in GCs express Mer.
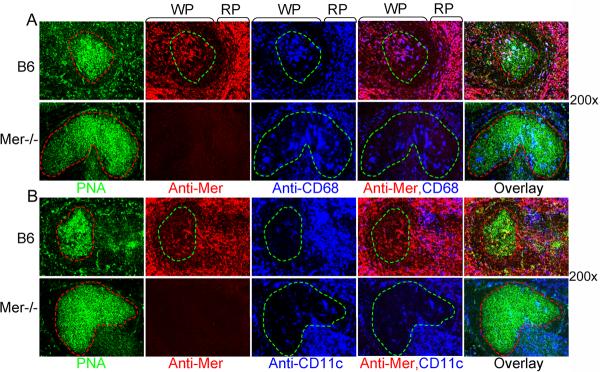
Multicolor immunohistological analysis of spleen sections, obtained from B6 (top row) and Mer−/− mice (bottom row) on day 14 after NP-CGG immunization, was performed using PNA (green), anti-Mer (red) and anti-CD68 (blue). In the 1st column, the GC areas shown in dotted red lines were defined by the presence of PNA+ cells (green). These GC areas were carried over to subsequent panels to show Mer+ (2nd column) and CD68+ (3rd column) cells within GCs. Two (anti-Mer and anti-CD68) and three (PNA, anti-Mer and anti-CD68) color overlay images are shown in the 4th and 5th column, respectively. The localization of Mer+ and CD68+ cells in the white (WP) and red (RP) pulp areas in B6 spleen (top row) are shown. Original magnification of images was 200×. Four to five mice of each genotype were used to generate these data.
As shown in Fig. 7A, we stained spleen sections from B6 and Mer−/− mice 14 days after NPCGG immunization with PNA (green), anti-Mer (red) and anti-CD68 (blue). We observed that the majority of CD68+ TBMϕs in both the red and white pulp areas expressed Mer as evidenced by the purple overlap staining of anti-Mer (red) and anti-CD68 (blue) in these areas (top row, 2nd from right, Fig 7A). Interestingly, we also found CD68+Mer+ TBMϕs within B6 GC as judged by the purple overlap staining within the GC area defined by the green dotted line (top row, 2nd from right, Fig 7A). Similar analysis on Mer−/− mice showed the presence of CD68+ cells in Mer−/− GC without the expression of Mer, indicating that the Mer expression on TBMϕs does not dictate the migration or retention of these cells within GCs.
We performed similar experiments in Fig 7B as shown in Fig 7A in which we investigated the localization of CD11c+ DCs in the spleen that expressed Mer. Similar to that shown in Fig 7A, we observed Mer+ cells in GC area defined by the green dotted line (top row, 2nd from the left, Fig 7B). In contrast to CD68+ cells a very few CD11c+ cells were observed either in B6 or Mer−/− GCs (3rd column, Fig 7B). In addition, the few Mer+ cells seen in B6 GC did not colocalize with CD11c (top row, 4th from the left, Fig 7B). Overall, a very small percentage of CD11c+ cells in B6 spleen expressed Mer, consistent with our recently published data (34). Together, these data indicate that CD68+ TBMϕs are the major phagocytes located in the GCs that express Mer.
Significantly increased number of apoptotic cells seen in the FDC areas of Mer−/− GCs
Germinal centers are microenvironment in which many B cells, including autoreactive ones, undergo negative selection via apoptosis (9, 10). As Mer expression on macrophages has been postulated to maintain tolerance by facilitating clearance of apoptotic cells, we next examined whether the enhanced GC and antibody responses in Mer−/− mice was associated with inefficient clearance of apoptotic cells generated in GCs by Mer−/− CD68+ TBMϕs. By staining the spleen sections of NP-CCG immunized B6 and Mer−/− mice with PNA (green), anti-CD68 (red) and TUNEL (blue) we showed that in the presence of Mer CD68+ cells in B6 GCs could engulf and clear the apoptotic cells efficiently as very few apoptotic cells were observed in GCs (top two rows, Fig. 8A). In addition, most of the apoptotic cells seen in B6 GCs (top two rows, middle panel, Fig 8A) were already engulfed by the TBMϕ as evidenced by the absence of apoptotic cells outside of CD68+ cells in B6 GCs (top two rows, 2nd from right, Fig 8A). Conversely, compared to B6 GCs a significantly increased number of apoptotic cells accumulated in Mer−/−GCs (bottom row, middle panel, Fig 8A). Most of these apoptotic cells were seen outside of CD68+ TBMϕs in Mer−/− GCs (bottom row, 2nd from right, Fig 8A).
Figure 8.

Accumulation of TUNEL+ cells in GCs in the absence of Mer.
(A) Three color immunohistology was performed on spleen tissues obtained on day 14 post NP-CGG immunization of B6 (top two rows) and Mer−/− (bottom row) mice. Spleen sections were stained with PNA (green, 1st column), anti-CD68 (red, 2nd column) and TUNEL (blue, 3rdcolumn). Two (anti-CD68 and TUNEL) and three (PNA, anti-CD68 and TUNEL) color overlay images are shown in the 4th and 5th column, respectively. (B) Similar analysis as in (A) was performed in which spleen sections were stained with PNA (green, 1st column), anti-CD21/35 (red, 2nd column) and TUNEL (blue, 3rd column). (C) TUNEL+ cells were counted in randomly picked GCs from four to five B6 (◯) and Mer−/− (◯) mice. (D) The number of CD68+ cells per GC was obtained by counting these cells in GCs described in panel (C). The average values for TUNEL+ and CD68+ cells per GC are shown in horizontal bars. Original magnification of images was 200×.
By immunohistological analysis of spleen sections from NP-CCG immunized B6 and Mer−/− mice with PNA (green), anti-CD21/35 (red) and TUNEL (blue) we further showed that the TUNEL+ apoptotic cells in Mer−/− mice were localized in the FDC area (CD21/35high) of GCs (bottom row, 3rd and 4th panels from left, Fig 8B), the presumed site of negative selection of GC B cells with low affinity for antigen on FDCs or B cells with autoreactivity. On the contrary, the TUNEL+ cells in B6 GCs were spread through out the GC (top row, 3rd and 4th panels from the left, Fig 8B), apparently due to the movement of TBMϕs in GCs after they engulfed the apoptotic cells. Next, we performed semi-quantitative analysis in which we counted the number of TUNEL+ cells in randomly picked small and medium GCs from 4 to 5 B6 and Mer−/− mice. Since we did not observe large GCs in B6 mice, in this analysis, we excluded the large GCs observed in Mer−/− mice. The number of TUNEL+ cells in Mer−/− GCs (red circle) was significantly higher than those in B6 GCs (blue circle, Fig 8C). These differences were much higher when we counted the number of TUNEL+ cells that were outside of CD68+ TBMϕs (data not shown) as very few un-ingested TUNEL+ cells were seen in B6 GCs. The increased number of TUNEL+ cells in Mer−/− mice was not due to the reduced number of CD68+ TBMϕs in Mer−/−GCs, as equal frequency of CD68+ cells was observed in both B6 and Mer−/− GCs (Fig. 8D).
Discussion
The germinal center (GC) reaction involves a complex mechanism of cellular proliferation, apoptosis and selection in response to foreign antigens. To ensure selection of the quality-controlled B cell clone for antibody mediated immunity, multiple molecular signals and cellular actions synchronize in the GC. We previously showed an increased marginal zone (MZ) B-cell population and spontaneous autoreactive MZ B-cell generation from Mer-deficient mice on an anti-dsDNA knockin background (W.H.S, submitted). While specific mechanisms by which MZ B cells might contribute to augmented GC response observed in Mer-−/− mice are not clear, these cells have been described to bridge the innate and adaptive immune response (44, 45). In this report, we investigated the role of Mer in regulating AFC, GC and antibody responses by clearing apoptotic cells in GCs induced by the TD-Ag NP-CGG. Our data showed an overall hyperactivity in the AFC and GC B cell responses in the absence of Mer that led to elevated anti-NP Ab responses from Mer−/− mice compared to WT controls.
The development and homeostasis of the immune system is maintained through elimination of mature immune cells that undergo apoptosis or cell death. Mer plays a pivotal role in apoptotic cell clearance by macrophage and DC engulfment and is thought to mediate immune tolerance (21, 35, 46, 47). Mer-dependent immunoregulation is mediated through inhibition of both TLR and TLR-induced inflammatory cytokine pathways (48). Recently, pretreatment of DCs with apoptotic cells in vitro was shown to induce Mer-mediated inhibition of TLR-stimulated PI3K/AKT and NF-κB activation (49). B-cell clonal selection in GC also results in an accumulation of apoptotic B cells. Rapid clearance of these apoptotic cells may be important to maintain peripheral tolerance as delayed clearance of such cells can trigger inflammation and autoimmune responses against intracellular materials (11, 29, 38, 50–53). Tingible body macrophages (TBMϕs) are the primary phagocytes in GC, which are thought to regulate GC response (17). The Nagata group has recently described a critical role of milk fat globule epidermal growth factor (EGF) 8 (MFG-E8)-mediated engulfment of apoptotic cells by TBMϕs in GCs (18). Inadequate clearance of apoptotic cells in MFG-E8-deficient (MFG-E8−/−) mice was shown to be associated with the development of autoimmune disease (18). Using the same mouse model, Kranich et al. showed that FDCs in GCs determine the engulfment of apoptotic cells by secreting MFG-E8. They proposed a model for integrin-mediated phagocytosis of apopototic bodies by TBMϕs (19). This group further showed that MFG-E8−/− mice were deficient in the development of FDCs, resulting in defective apoptotic cell clearance. While a deficiency in FDCs might play a vital role in breaking peripheral B cell tolerance leading to the development of lupus-like disease in MFG-E8−/− mice, we showed an intact FDC compartment in Mer−/− mice.
The Lemke group previously reported the expression of Mer mRNA in lymphoid tissues, including GCs (35). However, the Mer-expressing cell type in GCs was not defined. We previously reported the expression of Mer on TBMϕs in the spleen (34). We also reported expression of Mer on platelets and on a sizeable fraction of those macrophages with characteristics of splenic marginal zone (MZ) macrophages. It is likely that the MZ macrophages, as they are exposed to circulating apoptotic debris entering the spleen via the splenic artery and its branches, are equipped to deal with apoptotic debris from distant sources, and that Mer serves as an important receptor to aid them in binding to and ingesting apoptotic cells and fragments. The role of Mer in platelet remains controversial, but does not exclude a function of platelets in binding to cellular debris and subsequently adhering to mononuclear phagocytes.
In the current studies, consistent with our prior macrophage phenotype inferences, we show directly that Mer expressing TBMϕs are localized in the red pulp area, T cell zone and within GCs of B6 WT controls. Compared to B6 controls, we observed larger GCs and an accumulation of apoptotic cells outside of TBMϕs in the splenic GCs of Mer−/− mice 14 days after NP-CGG immunization. Our data indicate a GC defect in apoptotic cell clearance by TBMϕs in the absence of Mer. This apoptotic debris may differ from that ingested by MZ macrophages, and may include locally generated apoptotic cells, including B cells. While it is not clear which ligand(s) the TBMϕs might utilize in the engulfment and clearance of apoptotic cells in GCs, based on our current data, this process appears to be primarily dependent on Mer, as Mer−/− mice have an intact MFG-E8-integrin pathway. On the other hand, MFG-E8−/− mice also had a defect in apoptotic cell clearance in the presence of Mer, indicating the importance of both Mer and MFG-E8 mediated removal of apoptotic cells in GCs. The relative contributions and the mechanisms of these two pathways in clearance of apoptotic cells and the maintenance of immune tolerance can only be deduced by comparative studies between Mer−/− vs MFG-E8−/− GCs.
Multiple studies highlighted the importance of cytokines in modulating GC responses. Cytokines can affect many aspects of GC B-cell development. For instance, IL-4 induces isotype switch, IL-6 drives B-cell differentiation into antibody secreting cells, Bcl-2 and B cell activating factor (BAFF) promote B cell survival, while TNF stimulates B-cell apoptosis (54). Without Mer, Mϕ produces a greater amount of TNF-α in the presence of apoptotic cells or TLR stimuli (40). Mer-mediated Mϕ engulfment of apoptotic cells is accompanied by active production of anti-inflammatory cytokines, such as transforming growth factor-α (TGF-α) and interleukin 10 (IL-10), and by down modulation of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and IL-12 (47, 55). The anti-apoptotic effect of growth arrest protein 6 (Gas6, a ligand for Mer) involves survival pathways including Bcl-2 stimulation (56). In vitro data from the Matsushima group showed that apoptotic cells and LPS stimulated bone marrow derived DCs from Mer−/− mice contained an elevated number of BAFF-secreting cells (57). Although Mer bearing TBMϕ has the potential to affect GC B cell development through several mechanisms, elucidation of the precise pathway will require further studies.
The most striking observation in our current studies is the accumulation of uningested apoptotic cells in the FDC area of Mer−/− GCs apparently due to an inadequate clearance of these cells by TBMϕs in the absence of Mer. Similar features were described recently in studies analyzing lymph nodes of human SLE patients (23). While we did not explore the possibility for apoptotic cells to be bound on the surface of FDCs, the Herrmann group found TUNEL+ apopototic materials from SLE patients to be associated with the surface of FDCs (23). FDCs are the major stromal cell type in GCs that can trap Ags on their surface in the form of immune complexes (ICs) via complement receptors (58–60) and FcRs (61–63). It is not clear whether TUNEL+ cells in Mer−/− GCs are captured on the FDCs or simply localized there after B cells undergo negative selection at that site. However, the localization of TUNEL+ cells in the FDC area of Mer−/− GCs may have implications in breaking peripheral AFC and GC tolerance. Unprocessed apoptotic cells in GCs may release intracellular or nuclear materials at the late stage of apoptosis or during secondary necrosis that can potentially serve as immunogens to stimulate autoreactive B cells. This, in turn, may lead to the development of autoimmunity (11, 28). Impaired phagocytosis of apoptotic cells by peritoneal macrophages from autoimmune-prone mice has been described in vitro (24, 25). Delayed apoptotic cell clearance in Mer−/− mice was shown to be associated with lupus-like autoimmunity (26). Lupus-prone (MRL.lpr) as well as normal mice injected with irradiated apoptotic cells also develop autoantibodies (64). Whether impaired apoptotic cell clearance in spontaneously formed GCs in lupus mice and human SLE patients is associated with the defect in Mer or other Tyro3 family receptor (such as Axl and Tyro3) expression or function is not clear, and this area is under active investigation.
Acknowledgements
We thank Dr. Tim Manser for critical reading of the manuscript and comments.
Abbreviations
- MerTK
Mer receptor tyrosine kinase
- GC
germinal center
- AFC
antibody forming cell
- SHM
somatic hypermutation
- Mϕ
macrophage
- MZ
marginal zone
- FO
follicular
- TBMϕ
tingible body macrophage
- FDC
follicular dendritic cells
- DC
dendritic cells
Footnotes
These studies were supported by grants from the NIH to Z.S.M.R. (AR055701) and to P.L.C (R01DE017590).
References
- 1.MacLennan IC. Germinal centers. Annu. Rev. Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 2.Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991;354:389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- 3.Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121–1129. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- 4.MacLennan IC, Gray D. Antigen-driven selection of virgin and memory B cells. Immunol. Rev. 1986;91:61–85. doi: 10.1111/j.1600-065x.1986.tb01484.x. [DOI] [PubMed] [Google Scholar]
- 5.Tarlinton D, Radbruch A, Hiepe F, Dorner T. Plasma cell differentiation and survival. Curr. Opin. Immunol. 2008;20:162–169. doi: 10.1016/j.coi.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 6.Berek C, Jarvis JM, Milstein C. Activation of memory and virgin B cell clones in hyperimmune animals. Eur. J. Immunol. 1987;17:1121–1129. doi: 10.1002/eji.1830170808. [DOI] [PubMed] [Google Scholar]
- 7.Weiss U, Rajewsky K. The repertoire of somatic antibody mutants accumulating in the memory compartment after primary immunization is restricted through affinity maturation and mirrors that expressed in the secondary response. J. Exp. Med. 1990;172:1681–1689. doi: 10.1084/jem.172.6.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manser T, Wysocki LJ, Margolies MN, Gefter ML. Evolution of antibody variable region structure during the immune response. Immunol. Rev. 1987;96:141–162. doi: 10.1111/j.1600-065x.1987.tb00513.x. [DOI] [PubMed] [Google Scholar]
- 9.Kelsoe G. Life and death in germinal centers (redux) Immunity. 1996;4:107–111. doi: 10.1016/s1074-7613(00)80675-5. [DOI] [PubMed] [Google Scholar]
- 10.Pulendran B, van Driel R, Nossal GJ. Immunological tolerance in germinal centres. Immunol. Today. 1997;18:27–32. doi: 10.1016/s0167-5699(97)80011-4. [DOI] [PubMed] [Google Scholar]
- 11.Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat. Rev. Immunol. 2009;9:845–857. doi: 10.1038/nri2637. [DOI] [PubMed] [Google Scholar]
- 12.Green DR. Overview: apoptotic signaling pathways in the immune system. Immunol. Rev. 2003;193:5–9. doi: 10.1034/j.1600-065x.2003.00045.x. [DOI] [PubMed] [Google Scholar]
- 13.Henson PM, Hume DA. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol. 2006;27:244–250. doi: 10.1016/j.it.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 14.Cohen PL. Apoptotic cell death and lupus. Springer Semin. Immunopathol. 2006;28:145–152. doi: 10.1007/s00281-006-0038-z. [DOI] [PubMed] [Google Scholar]
- 15.Peng Y, Martin DA, Kenkel J, Zhang K, Ogden CA, Elkon KB. Innate and adaptive immune response to apoptotic cells. J. Autoimmun. 2007;29:303–309. doi: 10.1016/j.jaut.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 17.Smith JP, Burton GF, Tew JG, Szakal AK. Tingible body macrophages in regulation of germinal center reactions. Dev. Immunol. 1998;6:285–294. doi: 10.1155/1998/38923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 19.Kranich J, Krautler NJ, Heinen E, Polymenidou M, Bridel C, Schildknecht A, Huber C, Kosco-Vilbois MH, Zinkernagel R, Miele G, Aguzzi A. Follicular dendritic cells control engulfment of apoptotic bodies by secreting Mfge8. J. Exp. Med. 2008;205:1293–1302. doi: 10.1084/jem.20071019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sen P, Wallet MA, Yi Z, Huang Y, Henderson M, Mathews CE, Earp HS, Matsushima G, Baldwin AS, Jr, Tisch RM. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood. 2007;109:653–660. doi: 10.1182/blood-2006-04-017368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallet MA, Sen P, Flores RR, Wang Y, Yi Z, Huang Y, Mathews CE, Earp HS, Matsushima G, Wang B, Tisch R. MerTK is required for apoptotic cell-induced T cell tolerance. J. Exp. Med. 2008;205:219–232. doi: 10.1084/jem.20062293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaipl US, Munoz LE, Grossmayer G, Lauber K, Franz S, Sarter K, Voll RE, Winkler T, Kuhn A, Kalden J, Kern P, Herrmann M. Clearance deficiency and systemic lupus erythematosus (SLE) J. Autoimmun. 2007;28:114–121. doi: 10.1016/j.jaut.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, Kirchner T, Kalden JR, Herrmann M. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 24.Potter PK, Cortes-Hernandez J, Quartier P, Botto M, Walport MJ. Lupus-prone mice have an abnormal response to thioglycolate and an impaired clearance of apoptotic cells. J. Immunol. 2003;170:3223–3232. doi: 10.4049/jimmunol.170.6.3223. [DOI] [PubMed] [Google Scholar]
- 25.Licht R, Dieker JW, Jacobs CW, Tax WJ, Berden JH. Decreased phagocytosis of apoptotic cells in diseased SLE mice. J. Autoimmun. 2004;22:139–145. doi: 10.1016/j.jaut.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 26.Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 2002;196:135–140. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fadok VA, Warner ML, Bratton DL, Henson PM. CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (alpha v beta 3) J. Immunol. 1998;161:6250–6257. [PubMed] [Google Scholar]
- 28.Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–630. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 29.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat. Rev. Immunol. 2007;7:964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 30.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 31.Cohen PL, Caricchio R. Genetic models for the clearance of apoptotic cells. Rheum. Dis. Clin. North Am. 2004;30:473–86. viii. doi: 10.1016/j.rdc.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 32.Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008;8:327–336. doi: 10.1038/nri2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008;100:35–83. doi: 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shao WH, Zhen Y, Eisenberg RA, Cohen PL. The Mer receptor tyrosine kinase is expressed on discrete macrophage subpopulations and mainly uses Gas6 as its ligand for uptake of apoptotic cells. Clin. Immunol. 2009;133:138–144. doi: 10.1016/j.clim.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001;293:306–311. doi: 10.1126/science.1061663. [DOI] [PubMed] [Google Scholar]
- 36.Lu Q, Gore M, Zhang Q, Camenisch T, Boast S, Casagranda F, Lai C, Skinner MK, Klein R, Matsushima GK, Earp HS, Goff SP, Lemke G. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. 1999;398:723–728. doi: 10.1038/19554. [DOI] [PubMed] [Google Scholar]
- 37.D'Cruz PM, Yasumura D, Weir J, Matthes MT, Abderrahim H, LaVail MM, Vollrath D. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum. Mol. Genet. 2000;9:645–651. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- 38.Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 39.Luzina IG, Atamas SP, Storrer CE, daSilva LC, Kelsoe G, Papadimitriou JC, Handwerger BS. Spontaneous formation of germinal centers in autoimmune mice. J. Leukoc. Biol. 2001;70:578–584. [PubMed] [Google Scholar]
- 40.Camenisch TD, Koller BH, Earp HS, Matsushima GK. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J. Immunol. 1999;162:3498–3503. [PubMed] [Google Scholar]
- 41.Vora KA, Tumas-Brundage KM, Lentz VM, Cranston A, Fishel R, Manser T. Severe attenuation of the B cell immune response in Msh2-deficient mice. J. Exp. Med. 1999;189:471–482. doi: 10.1084/jem.189.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rahman ZSM, Rao SP, Kalled SL, Manser T. Normal Induction but Attenuated Progression of Germinal Center Responses in BAFF and BAFF-R Signaling-Deficient Mice. J. Exp. Med. 2003;198:1157–1169. doi: 10.1084/jem.20030495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lalor PA, Nossal GJ, Sanderson RD, McHeyzer-Williams MG. Functional and molecular characterization of single, (4-hydroxy-3-nitrophenyl)acetyl (NP)-specific, IgG1+ B cells from antibody-secreting and memory B cell pathways in the C57BL/6 immune response to NP. Eur. J. Immunol. 1992;22:3001–3011. doi: 10.1002/eji.1830221136. [DOI] [PubMed] [Google Scholar]
- 44.Ferguson AR, Youd ME, Corley RB. Marginal zone B cells transport and deposit IgM-containing immune complexes onto follicular dendritic cells. Int. Immunol. 2004;16:1411–1422. doi: 10.1093/intimm/dxh142. [DOI] [PubMed] [Google Scholar]
- 45.Cinamon G, Zachariah MA, Lam OM, Foss FW, Jr, Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat. Immunol. 2008;9:54–62. doi: 10.1038/ni1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seitz HM, Camenisch TD, Lemke G, Earp HS, Matsushima GK. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol. 2007;178:5635–5642. doi: 10.4049/jimmunol.178.9.5635. [DOI] [PubMed] [Google Scholar]
- 47.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 48.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 49.Sen P, Wallet MA, Yi Z, Huang Y, Henderson M, Mathews CE, Earp HS, Matsushima G, Baldwin AS, Jr, Tisch RM. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood. 2007;109:653–660. doi: 10.1182/blood-2006-04-017368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 51.Cocca BA, Cline AM, Radic MZ. Blebs and apoptotic bodies are B cell autoantigens. J. Immunol. 2002;169:159–166. doi: 10.4049/jimmunol.169.1.159. [DOI] [PubMed] [Google Scholar]
- 52.Radic M, Marion T, Monestier M. Nucleosomes are exposed at the cell surface in apoptosis. J. Immunol. 2004;172:6692–6700. doi: 10.4049/jimmunol.172.11.6692. [DOI] [PubMed] [Google Scholar]
- 53.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Butch AW, Chung GH, Hoffmann JW, Nahm MH. Cytokine expression by germinal center cells. J. Immunol. 1993;150:39–47. [PubMed] [Google Scholar]
- 55.Li MO, Flavell RA. Contextual regulation of inflammation: a duet by transforming growth factor-beta and interleukin-10. Immunity. 2008;28:468–476. doi: 10.1016/j.immuni.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 56.Hasanbasic I, Cuerquis J, Varnum B, Blostein MD. Intracellular signaling pathways involved in Gas6-Axl-mediated survival of endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H1207–13. doi: 10.1152/ajpheart.00020.2004. [DOI] [PubMed] [Google Scholar]
- 57.Gohlke PR, Williams JC, Vilen BJ, Dillon SR, Tisch R, Matsushima GK. The receptor tyrosine kinase MerTK regulates dendritic cell production of BAFF. Autoimmunity. 2009;42:183–197. doi: 10.1080/08916930802668586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dukor P, Bianco C, Nussenzweig V. Tissue localization of lymphocytes bearing a membrane receptor for antigen-antibody-complement complexes. Proc. Natl. Acad. Sci. U. S. A. 1970;67:991–997. doi: 10.1073/pnas.67.2.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maeda M, Muro H, Shirasawa H. C1q production and C1q-mediated immune complex retention in lymphoid follicles of rat spleen. Cell Tissue Res. 1988;254:543–551. doi: 10.1007/BF00226504. [DOI] [PubMed] [Google Scholar]
- 60.Van den Berg TK, Dopp EA, Daha MR, Kraal G, Dijkstra CD. Selective inhibition of immune complex trapping by follicular dendritic cells with monoclonal antibodies against rat C3. Eur. J. Immunol. 1992;22:957–962. doi: 10.1002/eji.1830220412. [DOI] [PubMed] [Google Scholar]
- 61.Qin D, Wu J, Vora KA, Ravetch JV, Szakal AK, Manser T, Tew JG. Fc gamma receptor IIB on follicular dendritic cells regulates the B cell recall response. J. Immunol. 2000;164:6268–6275. doi: 10.4049/jimmunol.164.12.6268. [DOI] [PubMed] [Google Scholar]
- 62.Radoux D, Kinet-Denoel C, Heinen E, Moeremans M, De Mey J, Simar LJ. Retention of immune complexes by Fc receptors on mouse follicular dendritic cells. Scand. J. Immunol. 1985;21:345–353. doi: 10.1111/j.1365-3083.1985.tb01440.x. [DOI] [PubMed] [Google Scholar]
- 63.Yoshida K, van den Berg TK, Dijkstra CD. Two different mechanisms of immune-complex trapping in the mouse spleen during immune responses. Adv. Exp. Med. Biol. 1993;329:377–382. doi: 10.1007/978-1-4615-2930-9_63. [DOI] [PubMed] [Google Scholar]
- 64.Mevorach D, Zhou JL, Song X, Elkon KB. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J. Exp. Med. 1998;188:387–392. doi: 10.1084/jem.188.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]